
Abstract
Liver cancer, particularly hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC), is more common in Asians than in Caucasians. This is due, at least in part, to regional differences in the prevalence of exogenous factors such as HBV; however, endogenous factors specific to Asia might also play a role. Such endogenous factors include HLA (human leukocyte antigen) genes, which are considered candidates due to their high racial diversity. Here, we performed a pancancer association analysis of 147 alleles of HLA-class I/II genes (HLA-A, B, and C/DRB1, DQA1, DQB1, DPA1, and DPB1) in 31,727 cases of 12 cancer types, including 1684 liver cancer cases and 107,103 controls. HLA alleles comprising a haplotype prevalent in Asia were significantly associated with pancancer risk (e.g., odds ratio [OR] for a DRB1*15:02 allele = 1.12, P = 2.7 × 10–15), and the associations were particularly strong in HBV-related HCC (OR 1.95, P = 2.8 × 10–5). In silico prediction suggested that the DRB1*15:02 molecule encoded by the haplotype does not bind efficiently to HBV-derived peptides. RNA sequencing indicated that HBV-related HCC in carriers of the haplotype shows low infiltration by NK cells. These results indicate that the Asian-prevalent HLA haplotype increases the risk of HBV-related liver cancer risk by attenuating immune activity against HBV infection, and by reducing NK cell infiltration into the tumor.
Similar content being viewed by others

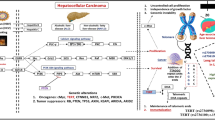

Introduction
Cancer incidence differs according to ethnicity1. Specifically, hepatocellular carcinoma (HCC) linked to hepatitis B virus (HBV) infection occurs more frequently among Asians than Caucasians2. HCC comprises 80% of all hepatic malignancies worldwide3. HBV or hepatitis C virus (HCV) infection is the primary etiology underlying persistent hepatic disease and the consequent emergence of HCC; however, there is a significant subset of HCC cases that are negative for both HBV and HCV infection, referred to as non-B non-C HCC (NBNC HCC)4. Differences in the prevalence of HBV across various regions account, at least in part, for the difference in incidence of HBV-related HCC; however, the increased incidence of HCC among Japanese Americans highlights the potential contribution of endogenous factors that are shared by Asians5. Polymorphisms in genes encoding human leukocyte antigens (HLAs) are strong candidates as endogenous determinants given that the distribution of these polymorphic alleles varies substantially across ethnic groups6. Neoantigens arising from somatic mutations in tumor cells are presented as peptides by HLA-class I molecules on tumor cells and are recognized by CD8+ (cytotoxic) T cells, which trigger killing of tumor cells. Moreover, HLA-class II molecules expressed on immune cells play a critical role in antitumor immune responses by binding to viral antigens7. Hence, it is plausible that interethnic disparities in the distribution of polymorphic HLA alleles may underpin interethnic discrepancies in susceptibility to cancer by modifying the anticancer immune response.
HLA genes, specifically HLA-A, -B, and -C (class I) and HLA-DRB1, DQA1, DQB1, DPA1, and DPB1 (class II), are highly polymorphic. These polymorphisms exist in linkage disequilibrium within the major histocompatibility complex (MHC) region, resulting in extended haplotypes8. The frequencies of these haplotypes, as well as alleles of the eight HLA genes, differ according to ethnicity. For example, A*24:02-B*52:01-C*12:02-DRB1*15:02-DQA1*01:03-DQB1*06:01-DPA1*02:01-DPB1*09:01 is the most common haplotype in Japanese individuals (8.30%)8, while it is rare in populations of European ancestry (0.34%)9. Hence, it is highly plausible that HLA alleles/haplotypes may account for the heightened susceptibility to various cancer types observed in Asian populations. However, to the best of our knowledge, no comprehensive large-scale study has been conducted to investigate the association between HLA alleles and cancer risk in Asian populations. This is due, at least partly, to the challenge of accurately imputing HLA alleles from genome-wide single nucleotide polymorphism (SNP) data, which has limited the inclusion of HLA alleles in genome-wide association studies (GWASs)10,11,12,13,14,15,16,17.
Here, we conducted a large-scale pancancer association analysis of HLA-A, -B, and -C (class I) and HLA-DRB1, DQA1, DQB1, DPA1, and DPB1 (class II) genes to address whether specific HLA alleles contribute to cancer risk. The study subjects included 31,727 cancer cases (12 cancer types in total) and 107,103 noncancer controls; their genome-wide SNP typing data were available in the BioBank Japan database (https://biobankjp.org/), and all were genetically matched with each other for the study. The availability of the Japanese HLA reference panel enabled us to perform precise imputation of HLA alleles from the SNP data18. The outcomes of the association analysis demonstrate that HLA alleles forming a prevalent haplotype in Asian populations exhibit a substantial correlation with the overall risk of pancancer, notably displaying a markedly strong association with the risk for HBV-related HCC. Plausible molecular mechanisms underlying the association between HLA alleles and HCC risk were inferred through in silico analysis of the binding of HLA-DRB1 to HBV-derived peptides, as well as the mutational and transcriptional profiles of 160 HCC tissue samples collected from Japanese patients.
Results
Pancancer association analysis of HLA alleles
Subjects were selected carefully based on their genetic background (see “Materials and Methods”). The study examined 31,727 cases of cancer (12 different types) and 107,103 controls. The 12 types were colon cancer (n = 6,854), stomach cancer (n = 6,424), breast cancer (n = 5,476), prostate cancer (n = 5,311), lung cancer (n = 3,919), liver cancer (n = 1,684), esophageal cancer (n = 1,274), uterine cancer (n = 975), ovarian cancer (n = 704), cervical cancer (n = 523), pancreatic cancer (n = 408), and gallbladder/bile duct cancer (n = 318). Controls comprised individuals diagnosed with noncancerous diseases (Supplementary Table 1). The MHC region was comprehensively imputed with a Japanese HLA reference panel8, thereby enabling analysis of 147 alleles in total: 17 HLA-A alleles, 33 HLA-B alleles, and 18 HLA-C alleles, as well as 26 HLA-DRB1 alleles, 18 HLA-DQA1 alleles, 15 HLA-DQB1 alleles, four HLA-DPA1 alleles, and 16 HLA-DPB1 alleles. Association analysis was performed for 68 HLA-class I and 79 class II alleles.
After Bonferroni correction, we found a significant association between pancancer risk and 12 alleles (P < 0.00034 [= 0.05/147]). Notably, eight of these 12 alleles (A*24:02, B*52:01, C*12:02, DRB1*15:02, DQA1*01:03, DQB1*06:01, DPA1*02:01, and DPB1*09:01) form a haplotype that is the most prevalent in Japanese populations (Supplementary Table 2), but rare in Caucasian populations8. The eight alleles were commonly linked to an increased cancer risk (Table 1, OR of a DRB1*15:02 allele = 1.12, P = 2.7 × 10–15). In this article, we refer to this haplotype as the "Asian-prevalent HLA haplotype".
Subsequently, we investigated the association between the eight alleles comprising the Asian-prevalent haplotype and the risk for each of the 12 cancer types. After applying Bonferroni correction, we identified significant associations with four cancer types: liver, stomach, cervical, and lung (Table 2; e.g., the OR of a DRB1*15:02 allele for liver cancer risk = 1.30, P = 3.1 × 10–7). Notably, three of these cancers (liver, stomach, and cervical) are more prevalent in Asian populations than in Caucasian populations, and are linked to viral or bacterial infections1. In light of the availability of information regarding cancer subtypes and viral infection, we focused on liver cancer in the subsequent part of this study.
Liver cancer association analysis of HLA alleles
HBV and HCV infections are the primary etiologies of liver cancer2,3. Hence, we investigated the differences in the association of HLA alleles in the context of viral infection. Our analysis encompassed cases of HBV-related liver cancer/HCC (n = 128/67), HCV-related liver cancer/HCC (n = 622/299), and virus-negative liver cancer/HCC (NBNC) (n = 277/130). Notably, we found that the ORs for the associations across seven HLA genes, except for HLA-A, were significantly higher for HBV-related cancers than for HCV-related and NBNC cancers (Table 3; e.g., the OR of a DRB1*15:02 allele for HBV-related liver cancer = 1.95, P = 2.8 × 10–5). Furthermore, when we limited our analysis to cases with information related to the diagnosis of carcinoma (i.e., HCC), we detected an even more substantial increase in the ORs (Table 3; e.g., the OR of a DRB1*15:02 allele for HBV-related HCC = 2.43, P = 1.7 × 10–5). Our case-case analysis comparing HBV-related cancer with NBNC cancers (i.e., NBNC cases were used as a reference) also demonstrated significant differences, suggesting that HLA alleles comprising the Asian-prevalent haplotype exhibit a stronger association with the risk for HBV-related liver cancer than for NBNC liver cancer (Supplementary Table 3).
Next, ORs were calculated according to genotype. Homozygosity for the risk alleles exhibited significantly higher ORs for HBV-related liver cancer/HCC risk than heterozygosity, suggesting that risk-associated HLA alleles have synergistic effects (Table 4; the OR of homozygotes of the DRB1*15:02 allele for HBV-related HCC = 9.82, P = 1.2 × 10–8). By contrast, the ORs for A*24:02 genotypes were not significant, which is consistent with a lack of association with the HLA-A*24:02 allele (Table 3).
Affinity of HLA-class II molecules for HBV-derived peptides
The association between HLA-class II alleles and HBV-related liver cancer prompted us to investigate whether the HLA-class II molecules encoded by the risk-associated alleles bind efficiently to HBV-derived peptides since HLA-class II molecules play a pivotal role in immune responses to viral infection19,20. Using the antigen-prediction algorithm MARIA21, we estimated the fraction of peptides efficiently captured by polymorphic HLA-DRB1 proteins among 1551 HBV-derived peptides deposited in the IEDB. Interestingly, we found that the DRB1*15:02 molecule bound fewer neoantigens than other HLA-DRB1 molecules (Fig. 1).

Potential inefficient binding of the DRB1*15:02 molecule.
Peptides derived from HBV that were predicted to bind with high affinity (i.e., predicted score > 0.95) to HBV peptides cataloged in the IEDB were identified by the MARIA program. The results for all deposited peptides (left panel, n = 1551) and for peptides that elicited a reaction in a T cell assay (right panel, n = 465) are shown. Colors indicate the protein type from which the binder peptides are derived.
Immune profile of HBV-related HCC in risk allele carriers
Often, HBV-associated HCCs exhibit reduced intratumoral infiltration by activated NK cells22, which represents an immune profile that could potentially promote tumor development and progression23. Therefore, to investigate whether the risk-associated Asian-prevalent HLA haplotype plays a role in this phenotype, we analyzed RNA sequencing and whole-exome sequencing data from 160 Japanese HCC samples24. HCC cases with the HLA-class I and -class II haplotypes were identified from noncancerous tissue whole-exome sequencing data, while intratumoral immune cell fractions were estimated from tumor tissue RNA sequencing data using the CIBERSORTx program125. Consistent with a previous report22, we found that the fraction of activated NK cells in HBV-related HCCs was lower than that in NBNC-HCCs (Supplementary Fig. 1), although the difference was not statistically significant (P > 0.05; Mann–Whitney U test). HCCs with the Asian-prevalent HLA haplotype had a lower percentage of activated NK cells than those without (Fig. 2), suggesting that intratumor infiltration by activated NK cells is lower in carriers of the HCC risk allele. By contrast, there was no significant difference in intratumor infiltration by CD8+, CD4+, and dendritic cells (P > 0.05; Mann–Whitney U test; Supplementary Fig. 2).

Proportion of activated NK cells infiltrating HCC tissues.
Fractions of activated NK cells within HCC tissues was determined using the CIBERSORTx algorithm using RNA sequencing data obtained from 160 Japanese patients with HCC. Fractions of activated NK cells was stratified according to the HLA-class I, -class II, and -class I/II genotypes. Statistical significance was determined by the Mann–Whitney U test, and P values < 0.05 were considered significant.
Discussion
Here, we conducted a comprehensive association analysis of HLA alleles to determine their role in pancancer risk and in the risk of developing 12 specific cancer types. The results clearly indicate that HLA-class I and -class II alleles comprising an Asian-prevalent haplotype play a role in the risk of developing Asian-prevalent cancers such as liver, cervical, and stomach cancers. To the best of our knowledge, this is the first investigation that has provided evidence that HLA polymorphisms are an endogenous factor contributing to the risk of Asian-prevalent cancers. Of the 12 cancer types, we focused on liver cancer because it is prevalent in Asia, and information regarding viral infections is readily available. Remarkably, HLA alleles comprising the Asian-prevalent haplotype showed a more pronounced association with the risk of HBV-related HCC than HCV-related and NBNC-HCCs, a finding in line with the high incidence rate of HBV-related HCC in Asian countries, including Japan26,27. These HLA alleles did not deviate significantly from Hardy–Weinberg equilibrium (Supplementary Table 4), and showed linkage disequilibrium (Supplementary Fig. 3) in our study population. Therefore, these alleles, and the haplotype, are a common genetic risk factor shared by the Japanese population.
Two particular findings provide insight into the molecular mechanisms underlying the way in which HLA polymorphisms contribute to the risk of HBV-related HCC. First, in silico deductions suggest that the HLA-DRB1*15:02 molecule encoded by the Asian haplotype does not bind HBV-derived peptides efficiently. In particular, inefficient binding of the HLA-DRB1*15:02 molecule to large envelope protein-derived peptides is plausible, as these proteins comprise the hepatitis B surface antigen (HBsAg). We then used whole-exome sequencing data from 160 Japanese HCC cases to investigate the ability of HLA-class I molecules, specifically those encoded by alleles associated with increased risk (i.e., B*52:01 and C*12:02), to bind efficiently to neoantigens arising from somatic mutations in tumor tissues. We deduced that the number of neoantigens that bound to these HLA-class I molecules was not lower than the number that bound to other HLA-class I molecules (Supplementary Fig. 4A). Notably, a previous study showed that HLA-class II alleles, but not class I alleles, comprising the Asian-prevalent haplotype are associated with an increased risk for chronic hepatitis B infection in Japanese individuals20. As such, it is possible that this haplotype contributes to the risk of liver cancer by increasing the likelihood of developing a chronic hepatitis B infection, rather than by promoting evasion of immune surveillance by established tumor cells.
The transcriptome data from the 160 HCCs enabled us to investigate the impact of the haplotype on the immunogenic properties of HCC. Chronic HBV infection, the primary risk factor for HCC, modulates expression of inhibitory and activating receptors on NK cells within tumor tissues, leading to a decrease in NK cell activation23. In fact, HBV-associated HCCs frequently exhibit reduced infiltration by activated NK cells22, which is an observation made in the present study. Moreover, HCCs carrying the risk haplotype had a lower fraction of activated NK cells than those without the haplotype. The mechanisms underlying these observations remain unclear. Previous studies suggest that HLA-class I molecules expressed on tumor cells mediate NK cell suppression28. Intriguingly, we observed infrequent LOH of the B*52:01 and C*12:02 alleles (which are associated with increased risk) compared with other class I alleles in HCCs of individuals heterozygous for these alleles (Supplementary Fig. 4B). This suggests that retention of these HLA-class I molecules may contribute to NK cell suppression. Nevertheless, further functional investigations are needed to establish a definitive conclusion.
In summary, our investigation highlights that the Asian-prevalent haplotype encompassing HLA-class I and -class II alleles increases susceptibility to Asian-prevalent malignancies, specifically HBV-related HCC, by suppressing antiviral and antitumor immune responses. Nonetheless, certain limitations must be acknowledged. First, although our study was a large-scale association analysis encompassing 12 major cancer types, the sample size for each cancer type, including HBV-related HCC, was small. Additionally, only Japanese patients were included in our analysis; thus, the associations should be confirmed in larger and more diverse cohorts of HCC patients. Second, the transcriptome analysis of HCC focusing on NK cells was performed using only publicly available data, thereby limiting our ability to assess detailed pathological characteristics such as immune cell distribution. Therefore, the relationship between HLA alleles and the intratumoral distribution of immune cells should be evaluated further using immunohistochemical methods coupled with HLA genotype data.
Materials and methods
Patients characteristics
Details of the BBJ subjects, including liver cancer cases, were described previously29. The BBJ project enrolled participants, including cases diagnosed as liver cancer, from healthcare facilities in Japan; history of HBV or HCV infection (yes, no, or unknown) was obtained from medical records, and from interviews using a standardized questionnaire at enrolment. The histological type of liver cancer was diagnosed on the basis of tissue or cytological samples obtained from biopsies. Liver cancer histology was not available for a subset of the patients because current guidelines for liver cancer from the Japan Society of Hepatology indicate that a diagnosis of HCC is sometimes based only on imaging results.
Subjects enrolled in the association analysis of HLA alleles
Genome-wide genotype data from 174,696 individuals were analyzed for the presence of 55,225 SNPs in the MHC region. The data were obtained from blood DNA samples collected through genome-wide SNP chip analysis using Illumina Human OmniExpress v1, Human OmniExpressExome v1.0, or Human OmniExpressExome v1.2 BeadChips. The data were obtained from the BioBank Japan (BBJ) database (approval number: P0067 and P0078), which is a national project that began in 2003 to collect DNA and clinical information from a total of 200,000 patients with at least one of 47 common diseases, including 12 types of cancer (http://biobankjp.org). The 174,696 subjects included 33,471 cancer cases and 141,225 controls. Principal component analysis of the 55,225 SNPs from the BBJ subjects was performed using 1,000 Genomes Project SNP data from Japanese (n = 91), Chinese (n = 190), European (n = 280), and African populations (n = 550)8. This enabled construction of a study population comprising 138,830 individuals with matched genetic backgrounds, including 31,727 cancer cases and 107,103 noncancer controls (Supplementary Fig. 5 and Supplementary Table 1).
HLA genotyping and association analysis
SNP2HLA30 and a Japanese HLA reference panel8 were used to impute the HLA alleles of the study subjects from genotype data covering the MHC region. This enabled identification of four-digit classical class I (HLA-A, B, and C) and class II (HLA-DRB1, DQA1, DQB1, DPA1, and DPB1) HLA alleles. Chi-squared tests and logistic association analyses of case/control data were performed for each HLA allele, and the 12 cancer types, using the PLINK program (version 1.07)31; age, sex, and the top five principal component scores were used as covariates. Bonferroni correction was applied to account for multiple testing. A total of 147 HLA-class I/II alleles were studied. P < 3.4 × 10–4 was considered significant (i.e., P < 3.4 × 10–4 = 0.05/147). Statistical analyses were performed using R statistical environment version 4.1.2 (GraphPad Prism9, or PLINK1.07).
In silico prediction of the binding of polymorphic HLA-DRB1 molecules to HBV peptides
The amino acid sequences of 1551 peptides derived from HBV were obtained from the Immune Epitope Database (IEDB)32. These peptides comprised fragments of proteins encoded by the HBV genome, including the capsid protein (n = 334), external core antigen (n = 202), large envelope protein (n = 555), protein P (n = 317), and protein X (n = 143). The MARIA (https://maria.stanford.edu/index.php) program, which specifically predicts peptide binding to HLA-DRB1 molecules, was used to infer the potential binding ability of these peptides to polymorphic HLA-DRB1 molecules. Peptides with a predicted score > 0.95 were considered to have positive binding ability, consistent with previous studies21.
Computational identification of HLA-class I molecule-restricted neoantigens
Whole-exome sequencing data (Fastq files) from HCC and nontumor tissue DNA samples obtained from 160 HCC patients of Japanese descent were procured from the National Bioscience Database Center (NBDC) Human Database (research ID: hum0187.v2)24. Exome sequencing was conducted on the Illumina HiSeq2000 platform using 2 × 100 bp paired-end reads (resulting in an estimated 100-fold coverage) and the SureSelect Human All Exon Kit V4/V5 (Agilent Technologies, Santa Clara, United States) or a SeqCap EZ HGSC VCRome2.1 design1 kit (Roche, Basel, Switzerland). Basic alignment and sequence quality control were then undertaken in accordance with the GATK4 best practices pipeline33. The reads obtained were aligned to the UCSC human genome 38 (hg38) reference sequence. Somatic single nucleotide variants and insertion/deletion variants were detected by the Mutect2 program (BROAD Institute; http://www.broadinstitute.org/gatk/). Each nonsynonymous single nucleotide variant was translated into a 17-mer peptide sequence centered on the mutated amino acid. The 17-mers were then used to generate 9-mers through a sliding window approach, followed by prediction of HLA-class I binding to neopeptides by the HLAthena program34. Neoantigens were selected based on a prediction score of Msi > 0.9 for each patient-specific HLA-class I allele35,36,37,38.
Estimation of somatic HLA-class I allele loss in HCC
The HLA-class I genotypes (comprising four-digit alleles) of the HCC patients were determined from whole-exome sequencing data using the HLA-HD39 and POLYSOLVER40 programs. Somatic loss of HLA-class I alleles was estimated by the LOHHLA41 program using default settings. In short, the allele-specific copy number of each HLA-class I locus was determined by realigning sequence reads to patient-specific HLA reference sequences. Somatic loss of heterozygosity (LOH) was considered positive when the difference in the log copy ratio between the two HLA alleles was less than the Pval_unique value of 0.01, as previously described42.
RNA sequencing and immune cell profiling
The RNA sequencing data (Fastq files) for tumor tissues from the same cohort of 160 HCC patients were acquired from the NBDC (research ID: hum0187.v2). The polyadenylated RNA libraries were synthesized using the TruSeq Stranded mRNA Library Prep kit (Illumina) and sequenced using the Illumina HiSeq2000 platform, generating 2 × 100 bp paired-end reads. Read alignment was performed using STAR version 2.7.3a43, with the human genome (GRCh38) and transcriptome data (GENCODE version 3144) as reference datasets. Transcripts per million (TPM) values were calculated using the StringTie program (version 2.0.4)45. Levels of immune infiltration were calculated from TPM expression data using the LM22 gene signature and the CIBERSORTx algorithm25,46. Data were run with 1000 permutations under the LM22 signature. The fraction of CD4+ cells was calculated by summing the fractions of “T cells CD4 naïve”, “T cells CD4 memory resting”, and “T cells CD4 memory activated”. The fraction of dendritic cells was calculated by summing the fractions of “dendritic cells resting” and “dendritic cells activated”.
Statistical analysis
Logistic regression analysis of HLA alleles/haplotypes was conducted using the PLINK program (version 1.07), with age, sex, and the top five principal component scores (described in “Subjects enrolled in the association analysis of HLA alleles”) as covariates. The significance of differences in the distribution of values, such as the number of neoantigens between two groups, was assessed using the Mann–Whitney U test. The binomial distribution test was used to evaluate the presence of LOH in tumors. All statistical analyses were performed using SPSS Statistics version 27 (IBM, Tokyo, Japan), the R statistical environment version 4.1.2, GraphPad Prism 9.0 (Dotmatics, Boston, United States), or PLINK 1.07.
Data availability
The genotype and phenotype data of BBJ subjects can be accessed through procedures described on its website (research ID: hum0014.v6.158k.v1 and hum0028.v2) (https://humandbs.biosciencedbc.jp/en/hum0014-v21 and https://humandbs.biosciencedbc.jp/en/hum0028-v2). The transcriptome and whole exome data of liver cancer used in this study are publicly available at the NBDC Human Database (research ID: hum0187.v2) (https://humandbs.biosciencedbc.jp/en/hum0187-v2). Peptide sequences of HBV are obtained from public databases IEBD (http://iedb.org) without restriction.
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33. https://doi.org/10.3322/caac.21708 (2022).
El-Serag, H. B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 142, 1264–1273. https://doi.org/10.1053/j.gastro.2011.12.061 (2012).
Ren, T. M. et al. Global epidemiology and genetics of hepatocellular carcinoma. Gastroenterology https://doi.org/10.1053/j.gastro.2023.01.033 (2023).
Chang, T. S. et al. Non-B, Non-C hepatocellular carcinoma in an HBV- and HCV-endemic area: A community-based prospective longitudinal study. Viruses https://doi.org/10.3390/v14050984 (2022).
Jin, H., Pinheiro, P. S., Xu, J. & Amei, A. Cancer incidence among Asian American populations in the United States, 2009–2011. Int. J. Cancer 138, 2136–2145. https://doi.org/10.1002/ijc.29958 (2016).
Gonzalez-Galarza, F. F. et al. Allele frequency net database (AFND) 2020 update: Gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 48, D783–D788. https://doi.org/10.1093/nar/gkz1029 (2020).
Chen, D. S. & Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 39, 1–10. https://doi.org/10.1016/j.immuni.2013.07.012 (2013).
Okada, Y. et al. Construction of a population-specific HLA imputation reference panel and its application to Graves’ disease risk in Japanese. Nat. Genet. 47, 798–802. https://doi.org/10.1038/ng.3310 (2015).
Pingel, J. et al. High-resolution HLA haplotype frequencies of stem cell donors in Germany with foreign parentage: How can they be used to improve unrelated donor searches?. Hum. Immunol. 74, 330–340. https://doi.org/10.1016/j.humimm.2012.10.029 (2013).
McKay, J. D. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 49, 1126–1132. https://doi.org/10.1038/ng.3892 (2017).
Wang, Y. et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet. 46, 736–741. https://doi.org/10.1038/ng.3002 (2014).
Hung, R. J. et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 452, 633–637. https://doi.org/10.1038/nature06885 (2008).
McKay, J. D. et al. Lung cancer susceptibility locus at 5p15.33. Nat. Genet. 40, 1404–1406. https://doi.org/10.1038/ng.254 (2008).
Shiraishi, K. et al. A genome-wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population. Nat. Genet. 44, 900–903. https://doi.org/10.1038/ng.2353 (2012).
Lan, Q. et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat Genet 44, 1330–1335. https://doi.org/10.1038/ng.2456 (2012).
Hu, Z. et al. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat. Genet. 43, 792–796. https://doi.org/10.1038/ng.875 (2011).
Miki, D. et al. Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations. Nat. Genet. 42, 893–896. https://doi.org/10.1038/ng.667 (2010).
Hirata, J. et al. Genetic and phenotypic landscape of the major histocompatibilty complex region in the Japanese population. Nat. Genet. 51, 470–480. https://doi.org/10.1038/s41588-018-0336-0 (2019).
Matsuura, K., Isogawa, M. & Tanaka, Y. Host genetic variants influencing the clinical course of hepatitis B virus infection. J. Med. Virol. 88, 371–379. https://doi.org/10.1002/jmv.24350 (2016).
Nishida, N. et al. Understanding of HLA-conferred susceptibility to chronic hepatitis B infection requires HLA genotyping-based association analysis. Sci. Rep. 6, 24767. https://doi.org/10.1038/srep24767 (2016).
Chen, B. et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 37, 1332–1343. https://doi.org/10.1038/s41587-019-0280-2 (2019).
Lim, C. J. et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 68, 916–927. https://doi.org/10.1136/gutjnl-2018-316510 (2019).
Sun, C., Sun, H., Zhang, C. & Tian, Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol. Immunol. 12, 292–302. https://doi.org/10.1038/cmi.2014.91 (2015).
Midorikawa, Y. et al. Accumulation of molecular aberrations distinctive to hepatocellular carcinoma progression. Cancer Res. 80, 3810–3819. https://doi.org/10.1158/0008-5472.Can-20-0225 (2020).
Steen, C. B., Liu, C. L., Alizadeh, A. A. & Newman, A. M. Profiling cell type abundance and expression in bulk tissues with CIBERSORTx. Methods Mol. Biol. 2117, 135–157. https://doi.org/10.1007/978-1-0716-0301-7_7 (2020).
Singh, R., Kaul, R., Kaul, A. & Khan, K. A comparative review of HLA associations with hepatitis B and C viral infections across global populations. World J. Gastroenterol. 13, 1770–1787. https://doi.org/10.3748/wjg.v13.i12.1770 (2007).
Lin, Z. H. et al. Association between HLA-DRB1 alleles polymorphism and hepatocellular carcinoma: A meta-analysis. BMC Gastroenterol. 10, 145. https://doi.org/10.1186/1471-230x-10-145 (2010).
Beelen, N. A., Ehlers, F. A. I., Bos, G. M. J. & Wieten, L. Inhibitory receptors for HLA class I as immune checkpoints for natural killer cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Cancer Immunol. Immunother. https://doi.org/10.1007/s00262-022-03299-x (2022).
Ukawa, S. et al. Characteristics of patients with liver cancer in the BioBank Japan project. J. Epidemiol. 27, S43-s48. https://doi.org/10.1016/j.je.2016.12.007 (2017).
Jia, X. et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS ONE 8, e64683. https://doi.org/10.1371/journal.pone.0064683 (2013).
Monau, P. I. et al. Population structure of indigenous southern African goats based on the Illumina Goat50K SNP panel. Trop. Anim. Health Prod. 52, 1795–1802. https://doi.org/10.1007/s11250-019-02190-9 (2020).
Ogishi, M. & Yotsuyanagi, H. Quantitative prediction of the landscape of T cell epitope immunogenicity in sequence space. Front. Immunol. 10, 827. https://doi.org/10.3389/fimmu.2019.00827 (2019).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. https://doi.org/10.1038/ng.806 (2011).
Sarkizova, S. et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 38, 199–209. https://doi.org/10.1038/s41587-019-0322-9 (2020).
Ouspenskaia, T. et al. Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat. Biotechnol. 40, 209–217. https://doi.org/10.1038/s41587-021-01021-3 (2022).
Klaeger, S. et al. Optimized liquid and gas phase fractionation increases HLA-peptidome coverage for primary cell and tissue samples. Mol. Cell Proteom. 20, 100133. https://doi.org/10.1016/j.mcpro.2021.100133 (2021).
Venema, W. J. et al. ERAP2 increases the abundance of a peptide submotif highly selective for the birdshot uveitis-associated HLA-A29. Front. Immunol. 12, 634441. https://doi.org/10.3389/fimmu.2021.634441 (2021).
Hu, Z. et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 27, 515–525. https://doi.org/10.1038/s41591-020-01206-4 (2021).
Kawaguchi, S., Higasa, K., Shimizu, M., Yamada, R. & Matsuda, F. HLA-HD: An accurate HLA typing algorithm for next-generation sequencing data. Hum. Mutat. 38, 788–797. https://doi.org/10.1002/humu.23230 (2017).
Shukla, S. A. et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 33, 1152–1158. https://doi.org/10.1038/nbt.3344 (2015).
McGranahan, N. et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 171, 1259-1271.e1211. https://doi.org/10.1016/j.cell.2017.10.001 (2017).
Lin, J. et al. An EGFR L858R mutation identified in 1862 Chinese NSCLC patients can be a promising neoantigen vaccine therapeutic strategy. Front. Immunol. 13, 1022598. https://doi.org/10.3389/fimmu.2022.1022598 (2022).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. https://doi.org/10.1093/bioinformatics/bts635 (2013).
Frankish, A. et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766-d773. https://doi.org/10.1093/nar/gky955 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. https://doi.org/10.1038/nbt.3122 (2015).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782. https://doi.org/10.1038/s41587-019-0114-2 (2019).
Acknowledgements
We thank the staff of BioBank Japan and NBDC for providing deposited data.
Funding
This work was supported by Grants-in-Aid for Scientific Research (Grant number 20H03695 to K.S.) and AMED (18km0605001 to Y.M. and K.M.).
Author information
Authors and Affiliations
Contributions
T.K., K.S., and A.K. designed the experiments. K.S. and A.K. analyzed the data and wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kawamura, A., Matsuda, K., Murakami, Y. et al. Contribution of an Asian-prevalent HLA haplotype to the risk of HBV-related hepatocellular carcinoma. Sci Rep 13, 12944 (2023). https://doi.org/10.1038/s41598-023-40000-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-40000-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
