
Abstract
Circular RNAs (circRNAs) play a role in sepsis-related autophagy. However, the role of circRNAs in autophagy after sepsis-induced cardiomyopathy (SICM) is unknown, so we explored the circRNA expression profiles associated with autophagy in an acute sepsis mouse model. At a dose of 10 mg/kg, mice were intraperitoneally administered with lipopolysaccharides. The myocardial tissue was harvested after 6 h for microarray analysis, qRT-PCR, and western blotting. Gene Ontology, Kyoto Encyclopedia of Genes and Genomes and Gene Set Enrichment Analysis were evaluated, and a competing endogenous RNA network was constructed, to evaluate the role of circRNAs related to autophagy in SICM. In total, 1,735 differently expressed circRNAs were identified in the LPS-treated group, including 990 upregulated and 745 downregulated circRNAs. The expression level of the autophagy-specific protein p62 decreased, while the ratio of LC3 II to LC3 I increased. Additionally, 309 mRNAs and 187 circRNAs were correlated with autophagy in myocardial tissue after SICM. Of these, 179 circRNAs were predicted to function as “miRNA sponges”. Some distinctive circRNAs and mRNAs found by ceRNA analysis might be involved in autophagy in SICM. These findings provide insights into circRNAs and identified new research targets that may be used to further explore the pathogenesis of SICM.
Similar content being viewed by others


Introduction
Sepsis has a high annual incidence and often causes acute dysfunction of multiple organs. In particular, sepsis-induced cardiac dysfunction maintains a high mortality1,2. The pathogenesis of sepsis is complex and involves the inflammatory cascade response, oxidative stress, mitochondrial dysfunction, calcium overload, autophagy, and apoptosis3,4,5,6,7,8,9.
Circular RNA (circRNA) molecules are a general feature in gene expression programs in human cells and were first identified in 201210. Tey et al. revealed that some circRNAs act as molecular sponges that bind and seal microRNAs11. The role of circRNAs has been studied in the pathogenesis of various diseases, e.g., cancer, spinal cord injury, and vascular diseases12,13,14,15,16. The role of circRNAs has also been reported in multiple organ damage triggered by sepsis. For example, the circRNA HIPK3 aggravates sepsis-induced acute kidney injury by modulating the microRNA-33817. However, there are few studies on the function of circRNAs in sepsis-induced cardiomyopathy (SICM).
Autophagy is a process that involves phagocytosis of the cytoplasmic protein or organelle of the cell itself, which are coated into the vesicles and fuse with the lysosome to form autolysosomes that degrade their wrapped contents. Recent studies have shown that circEXOC5-related signal cascade regulates inflammation and autophagy, and aggravates sepsis-induced acute lung injury18. It is poorly understood whether autophagy is modulated by circRNAs in SICM.
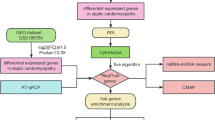
In this study, we analyzed circRNA expression profiles in SICM in a mice model to predict the autophagy-related circRNAs and mRNAs in SICM based on the circRNA-miRNA-mRNA network.
Materials and methods
Animals and study design
We conducted all animal experiments following the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (8th edition, NRC 2011). This study was approved by the Experimental Animal Ethics Committee of Zhejiang University (ZJU201305-1-02-047). Forty male C57BL/6 mice (7–8 weeks and 20–25 g, equivalent to adults at the ages of about 20 years old) were provided by the Zhejiang University Laboratory Animal Research Center. Mice had free access to food and water in cages at 23 °C and a 12-h light/dark cycle.
Mice were randomly divided into two groups and received intraperitoneal injections of lipopolysaccharides (LPS, Darmstadt) or normal saline at a dose of 10 mg/kg as described by our previous study19. According to the inclusion and exclusion criteria, the mice who died within 6 h after injection should be excluded from the study. In the present study, no mice died within 6 h after injection, so all mice treated with LPS or saline were used for further analysis in the study. After 6 h of injection, mice in each group were intraperitoneally injected with 1% pentobarbital sodium (40 mg/kg) for anesthesia and sacrificed by cervical dislocation. In total, eight mice hearts from two groups were immediately removed and placed in liquid nitrogen. The expressions of circRNAs and mRNAs in the samples were evaluated by Shanghai Biotechnology Corporation. Four additional myocardial samples per group were tested using western blotting. Sixteen mice hearts from two groups were collected to measure superoxide dismutase (SOD) activity and malondialdehyde (MDA) content. Another four left ventricle samples per group were collected to evaluate myocardial ultrastructure.
Assessment of the myocardial injury
According to the commercial assay kits (Haimen), SOD activity and MDA content in the heart samples which had been homogenized and lysed in lysis buffer were measured.
Left ventricular samples were fixed in 2.5% glutaraldehyde solution and 1% osmic acid. After being dyed with 4% uranyl acetate solution, the myocardial tissues were dehydrated with alcohol and acetone. Then, the samples were embedded and sliced, stained with 5% Uranium acetate staining and lead citrate. Changes of myocardial ultrastructure were observed under a transmission electron microscopy.
RNA extraction and purification
According to the method mentioned in our previous work, RNA was extracted, identified, and purified from the left ventricle of all mice heart samples19. The reagents used included Takara RNAiso Plus (Mountain View), NucleoSpin RNA Clean-up XS kit (Düren), and RNase-Free DNase Set (QIAGEN).
Microarray analysis
Similar to our earlier work, we used Cy-3 to label the amplified total RNA19. The labeled cRNAs were purified and hybridized. Agilent Microarray Scanner was used to scan the array slides of circRNAs and mRNAs. We used Quantile algorithm and limma packages in R software to normalize the raw data. The reagents used included Low Input Quick Amp Labeling Kit, Gene Expression Hybridization Kit and Wash Buffer Kit (Santa Clara), and RNeasy mini kit (QIAGEN).
Quantitative real-time polymerase chain reaction
Similar to the previously reported method, qRT-PCR was performed to confirm circRNA expression19. The reagents used included ReverTra Ace qPCR kit (Tokyo) and Capacity cDNA Reverse Transcription Kit (ABI).
Bioinformatics analysis
Normalized signal values were calculated using the log2 method. The circRNAs (fold change > 2, p < 0.05) after SICM damage were evaluated. To explore the potential function of circRNAs, especially of the autophagy-related circRNAs, we performed relevant functional and bioinformatics analyses using the method reported by Yao Ying20. We used websites for correlation analyses, including Gene Ontology (http://www.geneontology.org), Kyoto Encyclopedia of Genes and Genomes (https://www.genome.jp/kegg/), miRDB (http://www.mirdb.org/), and Cytoscape (https://cytoscape.org/)21,22,23.
Western blot (WB) analysis
The left ventricles from mice myocardial tissue (LPS or control group, n = 4 per group) were homogenized in RIPA lysis solution (Beyotime) to prepare the sample for western blotting. We examined the contents of the autophagy landmark proteins p62 and microtubule-associated protein 1 light chain 3 (LC3) using the method reported by Lou Junsheng24. The reagents used included cocktail (Sigma-Aldrich), BCA Protein Assay Kit (Thermo Fisher Scientific), p62 (Abcam), LC3 (Cell Signaling Technology), β-actin, and secondary antibodies (Santa Cruz Biotechnology).
Gene set enrichment analysis (GSEA)
GSEA was performed to identify the markedly enriched gene set clusters in myocardial tissue. The enrichment score (ES) curve was built using the GSEA4.3.1 software. The gene set with False Discovery Rate (FDR) < 0.25, |normalized enrichment scores (NES)|> 1, and nominal (NOM) p value < 0.05 was recognized as statistically significant.
Statistical analysis
All data were shown as mean ± standard deviation. GraphPad Prism 9.0 was used for analysis. Normality test was performed by using Shapiro–Wilk test. Student’s t test was used to compare significance between two groups. P < 0.05 was considered statistically significant.
Results
CircRNA expression profiles in myocardium of septic mice
After LPS injection, mouse hearts displayed an increased MDA content and a decreased SOD activity (Fig S1). Meanwhile, the mitochondrion bloated and bubbled, and cristae was disrupted in the mice myocardial treated with LPS (Fig. S1). All the above results indicated that the animal model of sepsis-induced cardiomyopathy was established.
We performed RNA sequencing on mouse hearts (Fig. 1A). The distribution of circRNA expression profiles in all samples showed good symmetry and dispersion (Fig. 1B). Differentially expressed circRNAs (DE circRNAs) were illustrated in red or blue color (Fig. 1C, D). Red color represents twofold upregulation of circRNAs, while blue color represents twofold downregulation of circRNAs in Fig. 1C. In Fig. 1D, circRNAs with fold change ≥ 2 (p < 0.05) and those with fold change ≤ 0.05 (p < 0.05) are shown in red or blue color, respectively. The expression features of dysregulated circRNAs were evaluated (Fig. 1E). In the LPS group vis-à-vis the control group, 1,735 circRNAs were differentially expressed (fold change > 2, p < 0.05), including 990 upregulated (57.06%) and 745 downregulated (42.94%) circRNAs. Depending on the degree of the fold change, the top 20 DE circRNAs are listed in Table 1. The genomic locations of the 1735 dysregulated circRNAs transcribed from all chromosomes, except chromosomes X and Y, are shown in Fig. 2A.

Expression profiles of circRNAs in the mouse myocardium after LPS injection. (A) Experimental design for RNA sequencing. (B) The box plot shows the distribution of circRNA expression profiles. (C–E) The scatter plot, volcano plot, and heatmap show the differentially expressed circRNAs. Red and blue colors represent upregulated and downregulated circRNAs, respectively. LPS, lipopolysaccharides; Ctrl, control; circRNA, circular RNA.

Distribution of altered circRNAs and their validation. (A) The distributions of dysregulated circRNAs in mouse chromosomes. (B, C) qRT-PCR verification of two circRNAs (circRNA.22171 and circRNA.27393). Data are presented as mean ± SD (n = 4). **p < 0.01 vs. control group.
Altered CircRNA expression was confirmed by real-time PCR
Among the top 20 DE circRNAs, two circRNAs were randomly selected for qRT-PCR. The primers of circRNAs and GAPDH are shown in Table 2. Consistent with the microarray results, circRNA.27393 was significantly upregulated, while circRNA.22171 was significantly downregulated (vs. control group, p < 0.05; Fig. 2B–C).
DE CircRNA function analysis
The potential functions of 1735 DE circRNAs were predicted by GO and KEGG enrichment analyses, and the results are shown in bubble charts (Fig. 3A–B). Based on the size of the enriched factors, the top 30 most remarkably enriched GO items were selected (Fig. 3A). The results showed that the host genes of DE circRNAs during LPS treatment were mostly involved in the “negative regulation of metalloenzyme activity” and “I-kappa B/NF-kappa B complex” (Fig. 3A). The KEGG pathway for enrichment analysis indicated that most of the host-genes of DE circRNAs were related to glycosaminoglycan degradation and ECM-receptor interaction pathways (Fig. 3B).

GO and KEGG enrichment analyses of differentially expressed circRNAs. (A) Top 30 enriched GO terms. (B) Top 30 enriched KEGG pathways.
CeRNA network prediction and annotation
The functions of circRNA include competitive adsorption of microRNAs (miRNA), regulation of RNA-binding proteins, and modulation of variable cleavage or transcription. CircRNAs bind to the corresponding miRNAs by MREs, which act as “sponges” preventing miRNA binding to the target gene and then jointly participating in the expression regulation of the target genes. This mechanism of action is called the “ceRNA mechanism”. This is the main research concept related to circRNAs. We filtered the DE circRNAs shown in Table 1 and found four associated mRNAs using ceRNA analysis: circRNA.2982, circRNA.2983, circRNA.2986, and mmu_circ_0006655. The result is shown in Fig. 4 and shows that the ceRNA mechanism exists after SICM.

CeRNA analysis of mouse myocardium after LPS injection. CircRNAs, miRNAs, and mRNAs are showed as blue diamonds, pink ellipses, and green rectangles, respectively. ceRNA, competing-endogenous RNA; miRNA, microRNA; mRNA, messenger RNA.
Autophagy-related CircRNA prediction
We explored the SICM-induced autophagy-related circRNAs. First, to confirm the protein expression of autophagy markers in mouse myocardium after SICM, we performed western blotting to detect the levels of proteins p62 and LC3 (Fig. 5A). Compared to the control group, the LPS group saw a decrease in the expression of p62 protein (1.80 ± 0.12, p < 0.05, Fig. 5B) as well as a significant increase in the ratio of LC3 II to LC3 I (1.60 ± 0.38, p < 0.01, Fig. 5C), suggesting the occurrence of autophagy. GSEA indicated that the gene set related to positive regulation of autophagy was enriched in SICM damage (Fig. 5D). The results further confirmed that autophagy occurs in mouse cardiomyocytes after 6 h of LPS treatment. In the animal autophagy signaling pathway (KEGG: mmu04140), several circRNA-related host genes were altered after SICM, with some of them (e.g., REDD1, FLIP, Bcl-XL, and TBK1) upregulated and others (e.g., RAB7B and RUBCN) downregulated (Fig. 5E). Next, we predicted the pathways associated with autophagy via KEGG pathway analysis and found nine autophagy-associated endogenous signaling pathways (Fig. 7A–B). The mRNAs were predominantly enriched in the PI3K-Akt (n = 103) and MAKP (n = 79) signaling pathways (Fig. 7A). The circRNAs were also predominantly enriched in the PI3K-Akt (n = 86) and MAKP (n = 70) signaling pathways (Fig. 7B). Third, based on the nine autophagy-related signaling pathways, GO analysis revealed that 39 mRNAs (fold change > 2, p < 0.05) were correlated with biological processes of autophagy, such as “autophagy (GO: 0006914)”, “regulation of autophagy (GO: 0010506)”, “negative regulation of autophagy (GO: 0010507)”, and “positive regulation of autophagy (GO: 0010508)” (Fig. 6A). Additionally, 14 circRNAs related to these mRNAs were found and were forecasted to be associated with autophagy (Fig. 6B). KEGG analysis revealed that 279 autophagy-correlated mRNAs (fold change > 2, p < 0.05) were enriched in the same nine autophagy-related signaling pathways (Fig. 7C, Table S1). Furthermore, ceRNA network prediction showed that 183 circRNAs were correlated with these mRNAs (Fig. 7D, Table S2). Finally, among these circRNAs, 179 autophagy-related circRNAs have binding sites on miRNAs based on the prediction of MREs (Table S3). Then, we found the corresponding target genes using Forecast websites. The top 10 upregulated autophagy-related circRNAs, based on the fold-change size, were circRNA.27393, circRNA.27392, circRNA.5564, circRNA.5562, mmu_circ_0005739, circRNA.5566, circRNA.5563, circRNA.27348, circRNA.5573, and circRNA.27394. Based on the top 10 circRNAs, we applied the miRDB websites and explored the mRNAs associated with autophagy. In Table 3, we listed the autophagy-related mRNAs that bind to the corresponding miRNAs with the highest target score. Most mRNAs were predominantly involved in the Hippo, PI3K-Akt, and mTOR signaling pathways.

Detection of autophagy in mouse myocardium after LPS injection. (A–C) Western blotting analysis for p62 protein expression and the ratio of LC3 II to LC3 I. *p < 0.05, **p < 0.01 vs. control group. Data are presented as mean ± SD. n = 4. (D) Positively enriched gene sets identified by GSEA. NES, NOM p value, and FDR are shown. (E) KEGG map04140 shows the autophagy-animal pathway (https://www.kegg.jp/kegg/kegg1.html)21,22,23. Red and blue colors represent gene upregulation and downregulation, respectively. GSEA, Gene Set Enrichment Analysis; NES, Normalized Enrichment Scores; NOM, Nominal; FDR, False Discovery Rate.

GO analysis of mRNAs and circRNAs related to autophagy in mouse myocardium after LPS injection. (A) Heat map showing differentially expressed mRNAs based on the GO analysis of autophagy. (B) Heat map showing differentially expressed circRNAs based on the GO analysis of autophagy-related mRNAs.

KEGG analysis of mRNAs and circRNAs related to autophagy in mouse myocardium after LPS injection. (A, B) The number of differentially expressed mRNAs and circRNAs in the prediction of autophagy-related KEGG signaling pathways. (C) Heat map showing differentially expressed mRNAs based on the KEGG analysis of autophagy. (D) Heat map showing differentially expressed circRNAs based on the KEGG analysis of autophagy-related mRNAs.
Discussion
In the present study, a mouse model of sepsis-induced cardiomyopathy was established. A total of 1735 differently expressed circRNAs were identified in the LPS-treated mouse hearts. And 187 circRNAs were found to be related to 309 autophagy-associated mRNAs in septic myocardial tissue. Among these, 179 circRNAs were predicted to function as “miRNA sponges”.
There is a high mortality rate of SICM2. Dysregulated autophagy is one of the main pathophysiological events in SICM25,26. Some studies have found that circRNAs, such as circCDYL, circCUL2, circRNA_002581, and circEXOC5, were involved in regulating autophagy, even in an LPS-challenged mouse cell model18,27,28,29. The abovementioned reports suggest a role of circRNAs in sepsis-induced autophagy. In our study, we established an SICM mouse model and confirmed the occurrence of the autophagic response in the mouse myocardium tissue by testing the protein levels and performing GSEA for LC3 and p62. In total, 1735 dysregulated circRNAs (including 990 upregulated and 745 downregulated circRNAs) were found in septic mice myocardium tissues using microarrays. In these DE circRNAs, 187 circRNAs are related to autophagy on GO and KEGG pathway analyses.
Autophagy is a fundamental cell protection pathway, and lysosomes are the site of intracellular autophagy30,31,32. As autophagy-marker proteins, LC3 and p62 are related to autophagosomal membrane formation33. In the present study, the p62 protein expression decreased, while the ratio of LC3 II to LC3 I increased after LPS treatment (vs. control group, p < 0.05). This is consistent with the findings from previous studies34,35. Although autophagy was not present in the top 30 pathways shown in Fig. 3A, B, we still found that the gene set related to “positive regulation of autophagy” was enriched in the LPS group by GSEA.
Several molecular mechanisms may participate in autophagy. In our study, we found nine autophagy-associated endogenous signaling pathways based on KEGG pathway analysis. These signaling pathways associated with autophagy are involved in various cellular and animal models36,37,38,39. For example, ER stress-induced autophagy, which was mediated by oxidative stress, decreased via the modulation of the PI3K-related cascade reaction in acute lung injury in LPS-induced mice40. In an ischemic/reperfusion-induced H9C2 cell injury model, autophagy induced by HIF-1α/BNIP3 signaling pathway protects the myocardium41. Laminar flow-induced endothelial autophagy and SIRT1 expression due to inhibited Hippo/YAP signaling pathways interrupt atherosclerotic plaque formation42. Notably, the HIF-1 and Hippo signaling pathways are involved in autophagy regulation, but the regulatory role has not been adequately explored in SICM-induced autophagy.
The role of circRNAs in SICM-induced autophagy has not been fully explored. Additionally, circRNAs that act as “sponges” are involved in the regulation of target gene expression. Depending on GO and KEGG analyses, we found 179 autophagy-related circRNAs that may bind to miRNAs. Autophagy-associated mRNA-binding sites also exist on the same miRNAs. Thus, we predicted the SICM-induced autophagy-related target genes using the ceRNA networks. For example, circRNA.27393 showed the top fold-change among the autophagy-related circRNAs and might regulate the mRNAs, such as the death-associated protein (DAP), ring-finger protein 152 (rnf152), and Ajuba by sponging mmu-miR-1933-3p, mmu-miR-448-5p, mmu-miR-125a-5p, and mmu-miR-125b-5p. These predicted mRNAs are associated with autophagy43,44,45. Other predicted autophagy-related mRNAs identified on ceRNA analysis in our research have also been proved to be related to autophagy, such as Bnip3, PPP2R2A, eEF2K, and IGF146,47,48,49.
In our previous research, we identified the mitochondrial function-associated lncRNAs in SICM19. Bnip3 and PPP2R2A are predicted to be autophagy-related mRNAs and are associated with mitophagy46,47,50. These results showed that circRNAs and lncRNAs might regulate mitochondrial function and degradation after SICM. Ajuba Tead3, Serpine1, Gli2, and Bmp2 were predicted to be related to the Hippo signaling pathway and were involved in autophagy45,51,52,53,54. Serpine1 and Bcl2 were predicted to be related to the HIF signaling pathway and autophagy51,55. Therefore, our results provide new ideas to further evaluate the role of the HIF-1 and Hippo signaling pathways in LPS-induced cardiomyocyte autophagy. The role of certain predicted circRNAs and mRNAs in SICM-induced autophagy has not been elucidated and should be evaluated in future studies.
Sepsis is a life-threatening organ dysfunction. Autophagy is a major pathogenesis of sepsis-induced cardiomyopathy. Our study identified differently expressed circRNAs in the hearts of septic mice. We also gained some specific circRNAs and their potential target mRNAs which might be involved in autophagy in septic hearts. These findings offer a fine view of circRNAs and might allow developing new treatment strategies for sepsis-induced cardiomyopathy and reducing the incidence and mortality of sepsis.
Because of the limitations of our detection methods, our experiment also has some limitations. First, the present study only focused on the acute phase of sepsis. Expression profiles of cirRNAs associated with autophagy in the chronic phase of sepsis needs to be further explored, which might provide a more panoramic view of autophagy-related circRNAs in sepsis-induced cardiomyopathy. Second, the combination of various methods is more reliable to detect circRNA, such as PCR, RNase R, and Northern blot. We explored the circRNAs using only qRT-PCR56,57. Third, autophagy-related pathways were not studied in detail, and we only screened circRNAs based on the reported autophagy pathways. Therefore, some potentially undiscovered circRNAs may be missed. Fourth, although we identified some potential autophagy-related circRNAs (such as circRNA.27393, circRNA.27392, and circRNA.5564), the mechanism of action of these circRNAs in SICM needs further in vivo and in vitro studies.
Conclusions
Our data indicate that the circRNAs, including circRNA.27393, may influence SICM-induced autophagy. Our research provides a new potential treatment strategy for SICM via the regulation of autophagy by circRNAs.
Data availability
The microarray data of circRNAs and mRNAs have been deposited in the GEO database (GSE142615).
References
Singer, M. et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 315, 801–810. https://doi.org/10.1001/jama.2016.0287 (2016).
Fleischmann, C. et al. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med 193, 259–272, https://doi.org/10.1164/rccm.201504-0781OC (2016).
de Padua Lucio, K. et al. Anti-Inflammatory and antioxidant properties of black mulberry (Morus nigra L.) in a model of LPS-induced sepsis. Oxid Med Cell Longev 2018, 5048031, https://doi.org/10.1155/2018/5048031 (2018).
Xu, S. et al. Melatonin attenuates sepsis-induced small-intestine injury by upregulating SIRT3-mediated oxidative-stress inhibition, mitochondrial protection, and autophagy induction. Front Immunol 12, 625627, https://doi.org/10.3389/fimmu.2021.625627 (2021).
Xin, T. & Lu, C. SirT3 activates AMPK-related mitochondrial biogenesis and ameliorates sepsis-induced myocardial injury. Aging (Albany NY) 12, 16224–16237. https://doi.org/10.18632/aging.103644 (2020).
Jiang, J. et al. Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol 36, 101638, https://doi.org/10.1016/j.redox.2020.101638 (2020).
Li, Z. et al. Methane alleviates sepsis-induced injury by inhibiting pyroptosis and apoptosis: in vivo and in vitro experiments. Aging (Albany NY) 11, 1226–1239. https://doi.org/10.18632/aging.101831 (2019).
Fu, C. Y. et al. Dimethyl fumarate attenuates lipopolysaccharide-induced mitochondrial injury by activating Nrf2 pathway in cardiomyocytes. Life Sci 235, 116863, https://doi.org/10.1016/j.lfs.2019.116863 (2019).
Zhao, H. et al. Autophagy activation improves lung injury and inflammation in sepsis. Inflammation 42, 426–439. https://doi.org/10.1007/s10753-018-00952-5 (2019).
Salzman, J., Gawad, C., Wang, P. L., Lacayo, N. & Brown, P. O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7, e30733, https://doi.org/10.1371/journal.pone.0030733 (2012).
Tay, Y., Rinn, J. & Pandolfi, P. P. The multilayered complexity of ceRNA crosstalk and competition. Nature 505, 344–352. https://doi.org/10.1038/nature12986 (2014).
Huang, M., He, Y. R., Liang, L. C., Huang, Q. & Zhu, Z. Q. Circular RNA hsa_circ_0000745 may serve as a diagnostic marker for gastric cancer. World J Gastroenterol 23, 6330–6338. https://doi.org/10.3748/wjg.v23.i34.6330 (2017).
Sunagawa, Y. et al. Genome-wide identification and characterization of circular RNA in resected hepatocellular carcinoma and background liver tissue. Sci Rep 11, 6016. https://doi.org/10.1038/s41598-021-85237-y (2021).
Gong, J. et al. Integrated analysis of circular RNA-associated ceRNA network in cervical cancer: Observational Study. Medicine (Baltimore) 98, e16922, doi:https://doi.org/10.1097/MD.0000000000016922 (2019).
Sun, Y. et al. Increased AT2R expression is induced by AT1R autoantibody via two axes, Klf-5/IRF-1 and circErbB4/miR-29a-5p, to promote VSMC migration. Cell Death Dis 11, 432. https://doi.org/10.1038/s41419-020-2643-5 (2020).
Ye, X. et al. Identification of circular RNAs related to vascular endothelial proliferation, migration, and angiogenesis after spinal cord injury using microarray analysis in female mice. Front Neurol 12, 666750, doi:https://doi.org/10.3389/fneur.2021.666750 (2021).
Lu, H. et al. Circular RNA HIPK3 aggravates sepsis-induced acute kidney injury via modulating the microRNA-338/forkhead box A1 axis. Bioengineered 13, 4798–4809. https://doi.org/10.1080/21655979.2022.2032974 (2022).
Gao, P., Wu, B., Ding, Y., Yin, B. & Gu, H. circEXOC5 promotes acute lung injury through the PTBP1/Skp2/Runx2 axis to activate autophagy. Life Sci Alliance 6, https://doi.org/10.26508/lsa.202201468 (2023).
Shi, Y. et al. Identification of mitochondrial function-associated lncRNAs in septic mice myocardium. J Cell Biochem 122, 53–68. https://doi.org/10.1002/jcb.29831 (2021).
Yao, Y. et al. Microarray assay of circular RNAs reveals cicRNA.7079 as a new anti-apoptotic molecule in spinal cord injury in mice. Brain Res. Bull. 164, 157–171, https://doi.org/10.1016/j.brainresbull.2020.08.004 (2020).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Lou, J. et al. Inhibition of PLA2G4E/cPLA2 promotes survival of random skin flaps by alleviating Lysosomal membrane permeabilization-Induced necroptosis. Autophagy 18, 1841–1863. https://doi.org/10.1080/15548627.2021.2002109 (2022).
Sun, Y., Cai, Y. & Zang, Q. S. Cardiac autophagy in sepsis. Cells 8. https://doi.org/10.3390/cells8020141 (2019).
Sun, Y. et al. Beclin-1-dependent autophagy protects the heart during sepsis. Circulation 138, 2247–2262. https://doi.org/10.1161/CIRCULATIONAHA.117.032821 (2018).
Liang, G. et al. Autophagy-associated circRNA circCDYL augments autophagy and promotes breast cancer progression. Mol Cancer 19, 65. https://doi.org/10.1186/s12943-020-01152-2 (2020).
Peng, L. et al. circCUL2 regulates gastric cancer malignant transformation and cisplatin resistance by modulating autophagy activation via miR-142-3p/ROCK2. Mol Cancer 19, 156. https://doi.org/10.1186/s12943-020-01270-x (2020).
Jin, X. et al. Antagonizing circRNA_002581-miR-122-CPEB1 axis alleviates NASH through restoring PTEN-AMPK-mTOR pathway regulated autophagy. Cell Death Dis 11, 123. https://doi.org/10.1038/s41419-020-2293-7 (2020).
Deter, R. L., Baudhuin, P. & De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol 35, C11-16. https://doi.org/10.1083/jcb.35.2.c11 (1967).
Deter, R. L. & De Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol 33, 437–449. https://doi.org/10.1083/jcb.33.2.437 (1967).
de Duve, C. Lysosomes revisited. Eur J Biochem 137, 391–397. https://doi.org/10.1111/j.1432-1033.1983.tb07841.x (1983).
Pugsley, H. R. Assessing autophagic flux by measuring LC3, p62, and LAMP1 Co-localization using multispectral imaging flow cytometry. J Vis Exp https://doi.org/10.3791/55637 (2017).
Pankiv, S. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282, 24131–24145. https://doi.org/10.1074/jbc.M702824200 (2007).
Deng, Z. et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 16, 917–931. https://doi.org/10.1080/15548627.2019.1644076 (2020).
Xu, Y. et al. RND2 attenuates apoptosis and autophagy in glioblastoma cells by targeting the p38 MAPK signalling pathway. J Exp Clin Cancer Res 39, 174. https://doi.org/10.1186/s13046-020-01671-2 (2020).
Deng, Z. et al. Selective autophagy of AKAP11 activates cAMP/PKA to fuel mitochondrial metabolism and tumor cell growth. Proc. Natl. Acad. Sci. USA 118. https://doi.org/10.1073/pnas.2020215118 (2021).
Liu, F. et al. MicroRNA-27a controls the intracellular survival of Mycobacterium tuberculosis by regulating calcium-associated autophagy. Nat Commun 9, 4295. https://doi.org/10.1038/s41467-018-06836-4 (2018).
Hu, Y. et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 17, 1142–1156. https://doi.org/10.1080/15548627.2020.1749490 (2021).
Huang, C. Y., Deng, J. S., Huang, W. C., Jiang, W. P. & Huang, G. J. Attenuation of lipopolysaccharide-induced acute lung injury by hispolon in mice, through regulating the TLR4/PI3K/Akt/mTOR and Keap1/Nrf2/HO-1 pathways, and suppressing oxidative stress-mediated ER stress-induced apoptosis and autophagy. Nutrients 12. https://doi.org/10.3390/nu12061742 (2020).
Zhang, Y. et al. HIF-1alpha/BNIP3 signaling pathway-induced-autophagy plays protective role during myocardial ischemia-reperfusion injury. Biomed Pharmacother 120, 109464. https://doi.org/10.1016/j.biopha.2019.109464 (2019).
Yuan, P. et al. Laminar flow inhibits the Hippo/YAP pathway via autophagy and SIRT1-mediated deacetylation against atherosclerosis. Cell Death Dis 11, 141. https://doi.org/10.1038/s41419-020-2343-1 (2020).
Yahiro, K. et al. DAP1, a negative regulator of autophagy, controls SubAB-mediated apoptosis and autophagy. Infect Immun 82, 4899–4908. https://doi.org/10.1128/IAI.02213-14 (2014).
Deng, L. et al. The ubiquitination of rag A GTPase by RNF152 negatively regulates mTORC1 activation. Mol Cell 58, 804–818. https://doi.org/10.1016/j.molcel.2015.03.033 (2015).
Sultana, M. A. et al. The SQSTM1/p62 UBA domain regulates Ajuba localisation, degradation and NF-kappaB signalling function. PLoS One 16, e0259556. https://doi.org/10.1371/journal.pone.0259556 (2021).
Fu, Z. J. et al. HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 36, 101671. https://doi.org/10.1016/j.redox.2020.101671 (2020).
Yu, L. et al. Integrating serum exosomal microRNA and liver microRNA profiles disclose the function role of autophagy and mechanisms of Fructus Meliae Toosendan-induced hepatotoxicity in mice. Biomed. Pharmacother. 123, 109709, https://doi.org/10.1016/j.biopha.2019.109709 (2020).
Pires Da Silva, J. et al. SIRT1 Protects the heart from ER stress-induced injury by promoting eEF2K/eEF2-dependent autophagy. Cells 9, https://doi.org/10.3390/cells9020426 (2020).
Wu, Q. et al. IGF1 receptor inhibition amplifies the effects of cancer drugs by autophagy and immune-dependent mechanisms. J. Immunother Cancer 9, https://doi.org/10.1136/jitc-2021-002722 (2021).
Zhang, J. & Ney, P. A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 16, 939–946. https://doi.org/10.1038/cdd.2009.16 (2009).
Fernandez, A. F. et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140. https://doi.org/10.1038/s41586-018-0162-7 (2018).
Song, Q. et al. YAP enhances autophagic flux to promote breast cancer cell survival in response to nutrient deprivation. PLoS One 10, e0120790. https://doi.org/10.1371/journal.pone.0120790 (2015).
Fan, J. et al. Regulating autophagy facilitated therapeutic efficacy of the sonic Hedgehog pathway inhibition on lung adenocarcinoma through GLI2 suppression and ROS production. Cell Death Dis 10, 626. https://doi.org/10.1038/s41419-019-1840-6 (2019).
Pang, X., Zhong, Z., Jiang, F., Yang, J. & Nie, H. Juglans regia L. extract promotes osteogenesis of human bone marrow mesenchymal stem cells through BMP2/Smad/Runx2 and Wnt/beta-catenin pathways. J. Orthop. Surg. Res. 17, 88, https://doi.org/10.1186/s13018-022-02949-1 (2022).
Vidal, R. et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight 4, https://doi.org/10.1172/jci.insight.131092 (2019).
Santer, L., Bar, C. & Thum, T. Circular RNAs: a novel class of functional RNA molecules with a Therapeutic perspective. Mol Ther 27, 1350–1363. https://doi.org/10.1016/j.ymthe.2019.07.001 (2019).
Jeck, W. R. & Sharpless, N. E. Detecting and characterizing circular RNAs. Nat Biotechnol 32, 453–461. https://doi.org/10.1038/nbt.2890 (2014).
Acknowledgements
This study was partially supported by the Basic Scientific Research Funds of Department of Education of Zhejiang Province (KYYB202005); Xinmiao Talent Plan of Zhejiang Province (2022R424A005); The Scientific Research Fund of Zhejiang Provincial Education Department (Y202249309); The grant from the Zhejiang Provincial Administration of Traditional Chinese Medicine (2023ZF083). We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Funding
The study is reported in accordance with ARRIVE guidelines.
Author information
Authors and Affiliations
Contributions
Z.L.R., C.Y.Y. and W.L.L. designed the experiment and revised the manuscript. F.C.Y., M.X.J. and Z.K. performed gene differential analysis and the qRT‐PCR experiment. Z.M.Z. and L.X. performed the statistical analysis and drafted the manuscript. L.J.S., L.L.H.Z. and F.Y.P. evaluated the autophagy. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, Mz., Lou, Js., Fan, Yp. et al. Identification of autophagy-associated circRNAs in sepsis-induced cardiomyopathy of mice. Sci Rep 13, 11807 (2023). https://doi.org/10.1038/s41598-023-38998-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-38998-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
