
Abstract
Formosan macaque (Macaca cyclopis) is the only non-human primate in Taiwan Island. We performed de novo hybrid assembly for M. cyclopis using Illumina paired-end short reads, mate-pair reads and Nanopore long reads and obtained 5065 contigs with a N50 of 2.66 megabases. M. cyclopis contigs > = 10 kb were assigned to chromosomes using Indian rhesus macaque (Macaca mulatta mulatta) genome assembly Mmul_10 as reference, resulting in a draft of M. cyclopis genome of 2,846,042,475 bases, distributed in 21 chromosomes. The draft genome contains 23,462 transcriptional origins (genes), capable of expressing 716,231 exons in 59,484 transcripts. Genome-based phylogenetic study using the assembled M. cyclopis genome together with genomes of four other macaque species, human, orangutan and chimpanzee showed similar result as previously reported. However, the M. cyclopis species was found to diverge from Chinese M. mulatta lasiota about 1.8 million years ago. Fossil gene analysis detected the presence of gap and pol endogenous viral elements of simian retrovirus in all macaques tested, including M. fascicularis, M. m. mulatta and M. cyclopis. However, M. cyclopis showed ~ 2 times less in number and more uniform in chromosomal locations. The constrain in foreign genome disturbance, presumably due to geographical isolation, should be able to simplify genomics-related investigations, making M. cyclopis an ideal primate species for medical research.
Similar content being viewed by others

Introduction
The genus Macaca constitutes the Cercopithecidae subfamily of Old World Monkeys formed by ~ 20 macaque species spreading across Asia and North Africa1. Based on male external genitalia, Fooden classified these macaques into four groups: fascicularis, silenus-sylvanus, sinica and arctoides2. As the largest group of macaques, the fascicularis group spreads across a wide geographic area3,4, suggesting a vast genetic and phenotypical diversity. This group comprises M. cyclopis (Taiwanese macaque, Formosan macaque, or Formosan rock macaque), M. fascicularis (cynomolgus/crab-eating macaque), M. fuscata (Japanese macaque) and M. mulatta which includes Indian rhesus monkey (Macaca mulatta mulatta) and Chinese rhesus monkey (M. mulatta lasiota). The silenus-sylvanus group comprises M. silenus, M. sylvanus, M. nemestrina and Sulawesi species, while the sinica group includes M. sinica, M. assamensis, M. radiate and M. thibetana and the arctoides group includes M. arctoides. Guided by the Alu element distribution pattern, Li and colleagues split sylvanus from silenus and assigned arctoides to the sinica group5. Studies based on territorial distribution pattern suggested that the silenus group diverged before the fascicularis group2,5.
Macaque species possess a strong potential for the study of primate evolution, human physiology and diseases (e.g., cancers and viral infections) due to the fact that they are very close to humans in both genetic makeup and evolution. During mid-twentieth century, millions of M. m. mulatta were exported from India to many countries, especially USA, to be used for medical and biological studies. The exporting activity resulted in a dramatic reduction of the population and was thus prohibited in 19786, causing a shortage of Indian rhesus monkey for medical and biological studies. Endemic to Taiwan, M. cyclopis is the only non-human primate in the island. Due to legislative protection act for the past few decades, the M. cyclopis population has increased. There is a need to learn more about this primate species so to prepare for its potential usage in the future.
Genome sequencing aims to provide a genetic map to facilitate the future study for the species of interest. Mainly due to the lack of a complete genome sequence, M. cyclopis is not well known and has very limited application in research. Previous phylogenetic studies solely relied on mitochondrial sequences or NRAMP1 loci7,8,9,10,11,12,13,14. We thus set out to sequence the M. cyclopis genome, in hope to facilitate its applications in disease control and the study of primate evolution.
Here, we report the M. cyclopis genome generated by de novo hybrid assembly15. This study is accomplished by using four libraries, including two Illumina paired-end (PE) short-read (SR) libraries, one Mate-Paired (MP) library, and one Nanopore long-read (LR) library, to make up a total of 115-fold coverage of the expected genome of ~ 2.9 giga base pairs (Gb). We used M. m. mulatta genome, Mmul_10 (Accession number: GCF_003339765.1), as a reference to facilitate the contig-to-chromosome assignment. MaSuRCA produced the best assembly metrics and high quality contigs and was used to build the M. cyclopis genome which contains 2,846,042,475 base pairs.
Endogenous viral elements (EVEs) represent fossil genes derived from parasitic genomes (most commonly, retroviral sequences) that integrated into the host genome during an ancient time. Most of the observed EVEs from simian virus (SIV), a retrovirus which relies on genomic insertion for replication, have undergone mutations for a long period of time. Thus, EVEs of SIV in macaques provide additional information for the study of primate evolution and medical application16. Previous studies have shown that many Old World Monkey species have either full length or near full-length simian endogenous retroviral sequences (SERSs) as a result of heterozygous integrations. As described by van der Kuyl and colleagues, most SERSs are of < 0.3 and 10 million years old16.
To further understand the geographical influence on macaque genomes, we compared the SIV gag (group specific antigen) and pol (polymerase) gene fossils in the genomes of M. cyclopis, which lives solely in Taiwan Island, M. fascicularis, which lives in the Indonesian islands, and M. m. mulatta, which lives in Asia continent. For the study, we employed TBLASTN17 to search for the EVEs of gag and pol genes. TBLASTN uses protein queries to search for the corresponding nucleotide sequences by screening all six possible reading frames to find the best alignment, whereas BLASTN uses nucleotide queries to search for similar sequences by scanning only one reading frame. As reported, TBLASTN is generally more sensitive and more accurate than BLASTN when searching for nucleotide databases18.
In parallel and to serve as a reference, we also searched for the EVEs of monkeypox viral genes encoding CL5 (a membrane protein), A15 (a core protein), P4a (another core protein), and D2 (a virion protein). Monkeypox virus is a double-stranded DNA virus which does not rely on genomic insertion for replication. The analysis revealed some interesting features of the M. cyclopis genome.
Results
Workflow
Workflow of M. cyclopis genome sequencing, assembly and annotation is depicted below (Fig. 1). Briefly, genomic DNA (gDNA) isolated from White Blood Cells (WBCs) of a female M.cyclopis macaque was used for genome sequencing with both Illumina and Nanopore sequencers.

Flow diagram describing sequential steps of the assembly pipeline. Genomic DNA isolated from WBCs of a female M. cyclopis monkey was processed through sequencing library preparation for Illumina short-read (SR) and Nanopore long-read (LR) sequencing. Besides regular QC, Nanopore LRs were subjected to additional sequence-level correction due to higher error rate. The high-quality reads were then assembled into contigs by the MaSuRCA hybrid assembler. These contigs were then assigned to chromosomes by aligning them to the Mmul_10 reference genome. Additional curation and annotation steps were performed to produce a final draft that was subjected to genome-based phylogenetic analysis.
Raw reads were processed to identify quality reads in each library. We used M. m. mulatta v10 assembly (Mmul_10) as the reference to facilitate the assignment of contigs to chromosomes. Breakpoints within contigs and gaps between contigs in each chromosome were curated and further assessed by BUSCO. Curated chromosomes were then subjected to annotation using MAKER pipeline19 and then used for genome-based phylogenetic analysis.
Library statistics
A total of four libraries were sequenced from M. cyclopis genomic DNA for the assembly (Table 1). A 115-fold total coverage was estimated based on the M. mulatta genome size.
Analysis of Nanopore long reads
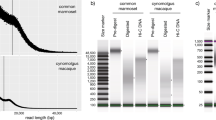
Long reads produced by ONT’s GridION sequencer had a mean read length of about 8.9 kb and N50 around 11.57 kb. Mean read quality of Nanopore reads was 12.1 and over 99% of reads were above Q5 (Table S1). Nanopore LRs were further assessed by NanoPack tools20, to shows length distribution and length-vs-average quality distribution. Both indicated that the quality of ONT LRs fell within expected range (Fig. S1).
Evaluation of assemblers using SRs and LRs
The performance of an assembler plays a critical role in genome assembly. The assembler pipelines tested included ABySS 2.0, SOAPdenovo2 (v2.04-r240), ALLPATHS-LG (v52488), and MaSuRCA (v3.2.8). To evaluate the effectiveness of these de novo assembly pipelines, either SRs or both SRs and LRs were utilized and the best assembler was selected based on, but not limited to, run time, number of contigs/scaffolds, N50 and the lengths of gaps.
With SRs alone, MaSuRCA emerged with the best result, as it produced the least number of scaffolds (55,553 in total) and the longest N50 (504 kb) compared to other assemblies tested (Table S2). It also produced the least number of gaps, with a total size of ~ 9 Mb comparing to other assemblies (gap size ≥ 60 Mb).
When tested with both Illumina SRs and ONT LRs using MaSuRCA for hybrid assembly. The N50 increased from 504 kb (with SRs alone) to almost 2.7 Mb (with both SRs and LRs). Moreover, the length of the longest contigs increased by ~ fourfold and the total number of contigs decreased by ~ tenfold in hybrid assembly with respect to short read assembly. Polishing of genome using short reads and PILON21 tool confirmed 99.24% of bases in the assembly and the N50 of the resulted assembly was slightly shorter after polishing.
We obtained 8,551,836 raw LRs from Nanopore sequencing, which were assembled to produce four different assemblies using the server in NCHC (National Center for High Performance Computing, Taiwan). Flye assembler22 was used at default parameters on raw as well as corrected long reads with target genome size of 3 Gb for macaques. Similarly, wtdbg2 algorithm was used for raw and polished reads to produces two more genome assemblies. Each of these tools run for about 2–3 days (Intel (R) Xeon, x86_64 GNU/Linux 64-bit processor, with 16 CPUs, 2-Threads per core and a 4 TB RAM) to obtain the results (Table S2). Total length of genome produced was 2,855,453,703 in 8615 contigs with N50 1,303,056 using Flye with raw reads. Length of longest contig was 17,917,662. We observed a decrease in N50 with corrected reads. Polishing of genome using short reads and PILON tool confirmed above 98% of bases in each of the assembly. Polished assemblies refer to nucleotide error correction that have been done using all short reads with PILON (Table S3), while unpolished assemblies refer to the raw assemblies obtained from the assembly pipeline.
We totally obtained 5065 contigs for hybrid assembly from MaSuRCA which is least among all other assembly pipelines we used that may involve short-reads or long reads only assemblies with different tools. Along with that we also observed N50 of this assembly was highest at contig level that is 2.66 Mb and only 277 contigs spanned around half the size of genome. 2,794,492,609 (98%) bases out of total 2,851,379,220 bases were present in 1971 (38.9%) contigs that were longer than 100 kb. Number of ambiguous bases or gaps in assembly were zero.
Further upon analysis using QUAST, we observed that 98.15% of reference genome was covered by M. cyclopis contigs. Single nucleotide mismatches and short insertions and deletions are lesser than long-reads only assemblies but slightly higher than short-reads only assemblies. BUSCO analysis revealed that assembly has similar number of BUSCOs present in MaSuRCA hybrid genome assembly (94.5%) as M. mulatta genome (94.4%).
We used a tailored approach that employed the contigs produced by hybrid assembly to construct chromosomes. Normally, scaffolding is performed prior to chromosome construction using the reads that could not be used in the process of contig assembly and these reads are supposed to be able to assist stitching the contigs into scaffolds. However, we used an alternative approach that includes mapping of contigs to the genome of closely related species (i.e., rhesus macaque) as reference to build the chromosomes based on alignment information. We observed that most reads were already used in building contigs. Therefore, upon scaffolding we observed a slight change in N50 (contig level N50: 2.66 Mb to scaffold level N50 2.85 Mb). We thus skipped scaffolding and proceeded to construct chromosomes directly from contigs to avoid false-positive merging of contigs.
Evaluation of assemblers using reference and BUSCOs
We then evaluated all the assemblies produced by long-reads only, short-reads only and hybrid strategy using QUAST using Mmul_10 assembly as reference.
Among all assemblers tested, MaSuRCA produced the highest genome fraction. Compared to short reads, long reads assemblies contained significantly higher number of indels per 100 kb. On the other hand, hybrid assembly approach resulted in similar number of indels per 100 kb as short reads assemblies.
Furthermore, no additional gaps were introduced by the hybrid approach. Overall, the result suggested the hybrid approach produced better assembly than using SRs or LRs alone. From this step onwards, we retained MaSuRCA assembly as final assembly for M. cyclopis. Overall more than 98.3% genes and 98.3% exons were covered in the assembly. Number of partial genes were high (25.04%) in M. cyclopis assembly because of fragmented contigs. Number of complete genes tends to increase or decrease with N50. Also, most exons are shorter in size and hence the number of complete exons is much higher than those of genes.
BUSCO analysis with eukaryotic lineage produced similar results to M. m. mulatta Mmul_10 assembly. For, mammalian and vertebral lineages, MaSuRCA hybrid performed best among all other assemblies and it was also comparable to Mmul_10 assembly.
Chromosome assembly
A total of 2395 contigs were used in chromosome construction, while contigs shorter than 10 kb or mapped within another longer contigs, were excluded. Additionally, 19 other contigs could not be used in building the draft due to complex structural variations.
When compared to the Mmul_10 genome, we found 1236 regions in Mmul_10 were missing in the M. cyclopis assembly. We devised a local assembly strategy to address this issue. All sequence reads were mapped to each of the Mmul_10 regions with an extension of 1 kb in either direction to identify the best matched sequences to fill the gap in the M. cyclopis assembly. Out of the 1236 regions, 287 were improved—either completely (e.g., Fig. S2A) or partially (e.g., Fig. S2B), and 40 regions were improved using scaffolds from Flye long read assembly. The remaining 909 regions might not present in the M. cyclopis genome.
Ninety-eight unplaced contigs that contained gene/exons/coding sequence (CDS) were also included in the final assembly. When compared to the initial assembly, we observed increases in number of genes, CDS and exons by 287, 2151 and 2195, respectively, in the final assembly. Lastly, BUSCO analysis revealed that the final assembly of M. cyclopis contains 3893 (94.9%) complete, 101 (2.5%) fragmented and 110 (2.6%) missing mammalian BUSCOs.
With these efforts, the M. cyclopis genome was found to comprise 2,846,042,475 base pairs. Based on this draft, we continue to perform annotation.
Annotation
Repeat sequences in M. cyclopis
With Dfam mammalian repeats, RepeatMasker masked about 49.72% of the draft M. cyclopis genome (Table 2). Most repeats are SINEs (13.78%), LINEs (20.76%), LTR elements (8.99%), and interspersed repeats (47.28%). We observed high repeat content in the unplaced contigs, which could be the reason why these contigs could not be assembled into the chromosomes.
On the other hand, RepeatModeler2 predicted 937 repeat families in M. cyclopis, which covered about 39.46% of M. cyclopis genome. Similarly, most repeats are SINEs (12.36%), LINEs (15.54%), LTR elements (7.18%), and interspersed repeats (37.71%). Of the repeat families identified, 709 repeat families contributed to novel 32,754 regions (5,240,159 bases) that could not be identified by Dfam mammalian repeats. This information is highly valuable to enhance the existing annotation of mammalian repeats.
Genome annotation
We used 86,716 transcripts and 67,976 proteins from M. mulatta genomes as the input for MAKER to annotate the M. cyclopis genome. Genes in the M. cyclopis genome were predicted based on homology-based method and ab-initio method incorporated in the MAKER pipeline. The analysis revealed 23,462 protein-coding genes with 716,231 exons and 59,484 transcripts. By comparing the genome annotation of seven other primate species, we observed a high degree of similarity in gene, exon, CDS and intron lengths of M. cyclopis with that of other species (Fig. 2). Several non-coding RNA species have been identified including 609 miRNAs, 600 tRNAs, 4217 lncRNAs and 580 rRNAs. Genomic locations of these non-coding genes are provided in the supplementary files (Supplementary information).

Whole-genome comparison of basic genomic features among different primate species. Gene lengths (a), CDS lengths (b), exon lengths (c), intron lengths (d) and number of exons (e) of five macaque species (i.e., M. cyclopis, M. m. mulatta, M. mulatta lasiota, M. fascicularis, and M. nemestrina), Pongo abelii (Sumatran orangutan), Pan troglodytes (chimpanzee) and H. sapiens (humans) are compared to depict the differences in the structure of protein-coding genes. X axis represents the length in bp; Y axis represent the corresponding numbers.
Analysis of genomic translocations
Alterations in genomic sequences among M. cyclopis, M. m. mulatta, M. mulatta lasiota and M. fascicularis were depicted with Circular plot (Fig. 3). We observed significant degree of genomic translocations between M. cyclopis and M. fascicularis, much higher than that between M. cyclopis and M. m. mulatta. This result is of high degree of concordance with previous reports8, showing that with respect to M. cyclopis, M. fascicularis is more distal in evolution than M. m. mulatta.

Circular plot showing translocations of chromosomal fragments with sizes ≥ 10 kb between M. cyclopis and M. mulatta or between M. cyclopis and M. fascicularis. Chromosomes are radially aligned in a circle (red, M. m. mulatta; green, M. fascicularis; blue, M. cyclopis; purple, M. mulatta lasiota followed by chromosome number). Arcs within the circle indicate interchromosomal translocations of fragments with sizes ≥ 10 kb. There are 358 translocations between M. cyclopis and M. fascicularis (green), 512 translocations between M. cyclopis and M. mulatta lasiota (purple) and 60 translocations between M. cyclopis and M. m. mulatta (red).
Genome-based phylogenetic analysis
The complete M. cyclopis genome allowed us to perform a phylogenetic analysis based on amino acid sequences of proteins derived from the assembled chromosomes. Phylogenetic tree was constructed using single copy orthologs in M. cyclopis and seven other primate species (i.e., H. sapiens (GCF_000001405.39), Pan troglodytes (GCF_002880755.1), Pongo abelii (GCF_002880775.1), Macaca nemestrina (GCF_000956065.1), Macaca fascicularis (GCF_000364345.1), Macaca m. mulatta (GCF_003339765.1) and Macaca m. lasiota (GCF_000230795.1) to calculate the divergence time for M. cyclopis (Fig. 4). Further analysis of the results indicated that the Formosan macaque diverged from Chinese rhesus macaque (M. m. lasiota) about 1.8 million years ago.

Phylogenetic tree of eight different species with divergence time. Nodes are labelled with their putative divergence time in million years and the range with 95% confidence interval is provided in parenthesis.
The topology of our tree is consistent with those based on previous phylogenetic studies of primate species and estimates of dates of inter-species divergence. These include studies of electrophoretically defined protein coding loci, mtDNA RFLPs, sequences and whole genomes, STRs, SNPs and nuclear gene sequences. The first split in our tree separates the families Hominidae and Cercopithecidae which diverged from each other approximately 29 million years ago (mya)23,24. Of living species of Hominidae, Pongo is the most distant, as in our own tree, having separated from a clade shared with Pan, Homo and Gorilla several million years prior to the split between Pan/Homo and Gorilla, between 8 and 10 mya, and that between Pan and Homo, some 6.5 mya23,24,25. Genus Macaca includes approximately 20 species which represent what most scholars26,27 regard as four monophyletic groups of species. One of these four species groups, the silenus group, is that from which all other groups derive, and includes M. nemestrina, the oldest and most divergent cercopithecoid species in our tree. Another of the four groups, the fascicularis group, includes the remaining three cercopithecoid species (M. fascicularis, M. mulatta and M. cyclopis) comprising, with M. fuscata of Japan, the fascicularis group of macaque species28. M. mulatta is believed to have diverged from a fascicularis-like ancestor as early as 3.24 mya29 after the split of M. nemestrina, the earliest split from M. sylvannus, the ancestor of all macaques, about 7 mya. M. cyclopis is believed to have derived from a M. mulatta population that colonized Taiwan from southern China and Vietnam between 0.38 and 0.44 mya4. Therefore, our tree comports perfectly with previous phylogenetic studies.
SIV gag and pol EVEs were detected in M. fascicularis, M. m. mulatta and M. cyclopis, while none of these macaques harbour EVE-like sequences of the monkeypox viral genes tested
Fossil gene analysis by TBLASTN revealed the presence of EVEs of SIV gap and pol genes in all macaques tested, including M. fascicularis, M. m. mulatta and M. cyclopis. Among these three species, M. fascicularis genome harbours the highest number of both genes (56 sites for gag; 76 sites for pol), followed by M. m. mulatta genome (44 sites for gag; 64 sites for pol), and then the M. cyclopis genome (24 sites for gag; 23 sites for pol) (Table S4). On the other hand, all monkeypox genes tested showed no significant matches by TBLASTN under the same TBLASTN parameters. Taken together, the above fossil gene search indicates that M. cyclopis is unique in both number and distribution pattern compared to that of M. fascicularis and M. mulatta. In contrast, fossil gene analysis using same parameters showed no significant EVEs for all the monkeypox genes tested across all these macaques.
Discussion
Whole genome sequencing enhances medical research and evolutionary study. Previous studies showed that macaques shared a common ancestor with human about 25–32 mya and diverged from each other about 5–6 mya30,31. Here, whole-genome phylogenetic analysis suggests that M. cyclopis diverged from M. m. lasiota about 1.8 mya, which was much more recent compared to the onset of divergence of macaque species.
By providing the genome sequence of M. cyclopis, we add an additional piece of information for the study of primate evolution. This sequence is also reported in NCBI as reference enabling researchers to use it in genomics-based studies of Taiwanese macaque. List of variations in M. cyclopis provided will also aid the studies on genetic differences that are responsible for the phenotypic diversity in macaques. Additionally, this work also opens up the opportunity for the usage of M. cyclopis as an animal model for medical research.
Previous phylogenetic studies indicated that M. cyclopis descended from Chinese monkey M. m. lasiota8. With a completed M. cyclopis genome, we further demonstrate that the distributions of gag and pol EVEs in M. cyclopis is relatively low in number, but with higher uniformity in distribution patterns, readily distinguishable from that of M. fascicularis and M. m. mulatta. The uniqueness, which might have resulted from geographical isolation through the evolutionary process, suggests that M. cyclopis is a macaque particularly useful in medical research for the study of infectious diseases. The higher complexity of SIV fossil gene distribution pattern found in M. m. mulatta and M. fascicularis, as compared to that of M. cyclopis, is likely to result from the fact that these two macaques live in the wide-open continental area and islands in close proximity. As horizontal gene transfer is an important factor for gene transfer of SIV sequences among macaques32, such habitats provide great opportunities for cross-species and cross-organism gene transfer, not only for SIV, but also for other retroviruses as well, causing the increase in their genome complexity. When used in medical research, the genome complexity would more or less hinder the downstream analyses such as transcriptome analysis, transcription factor binding site analysis, and many others. On the other hand, geographical isolation of M. cyclopis in Taiwan Island limited the cross-species gene transfer so to result in the relative simplicity of the M. cyclopis genome, making M. cyclopis an ideal animal, or even better than M. mulatta, for biomedical research.
In terms of the usage of short reads and long reads, short reads are high in accuracy but each covers only very limited genomic range (≤ 300 bp for PE reads). On the other hand, although long reads are lower in quality but each covers a longer genomic distance. With a combination of SRs and LRs, hybrid sequencing has been commonly employed for the assembly of large genomes. To improve the accuracy, all LRs were corrected by SRs prior to hybrid assembly. With over 100-fold coverage and the usage of a closely related species, M. mulatta, as the reference, we intended to reach a fair degree of accuracy. Transcriptome or exome sequencing data should be able to validate the results. However, we are currently short of these types of data.
In summary, this work aims to set a tone for M. cyclopis-related studies and applications in the future. Results of this work seem to agree well with the previous reports. In addition, it also provides additional valuable information for future investigations. To move one step further, detailed SNP analysis will be helpful.
Methods
Ethical statement
The animal handling procedure and experimental protocols of this study fully complied with the guidelines of IACUC (Institutional Animal Care and Use Committee) and NPUST (National Pingtung University of Science and Technology) (Please see The Guide for Animal Use and Care under http://lac.npust.edu.tw/files/15-1112-30104,c4232-1.php?Lang=zh-tw) and was approved by Pingtung Rescue Center for Endangered Wild Animals (PTRC) at NPUST. All people involved in handling the animal were well trained. Moreover, this study is also in accordance with ARRIVE guidelines, and no human subject was involved in this study.
Source of DNA
A captive female Formosan macaque in the Pingtung Rescue Center for Endangered Wild Animals (PRCEWA) of National Pingtung University of Science and Technology (NPUST) was used for the study. She (NPUST ID: 10D7) was about 22-year old when her blood was first collected after anesthesia following NPUST guidelines. Genomic DNA from white blood cells (WBCs) was isolated right after blood collection and stored at − 20 °C before use.
Short-read sequencing and data processing
To prepare sequencing library for SR sequencing, DNA was randomly sheared by sonication and 300–500 bp fraction was isolated by gel excision and used for Illumina paired-end (PE) following the instructions provided by the manufacturers. SR libraries used in this work include (1) 2 × 90 bp PE SR library, sequenced by HiSeq 2000 (BGI); (2) 2 × 150 bp PE SR library, sequenced by HiSeq X Ten (Macrogen Inc.); and (3) 2 × 150 bp mate-pair (MP) library with insert size of ~ 3 kb was also prepared and sequenced (BGI).
SR raw reads were processed by the following procedure. Raw reads were examined with FastQC33 for base quality and the presence of adapters. AdapterRemoval tool34,35 was then used to remove the adapter sequences if present. Terminal ambiguous bases were trimmed off by Ambiguity trimming module of NGS QC Toolkit, and low quality reads were filtered out using IlluQC module of NGS QC Toolkit36. Reads > = 100 bp were selected by PRINSEQ software37. Mapping to reference genome was performed by BWA38. The insert size of MP reads was estimated by BamTools-based script and reads with insert sizes of about 2600 (bp) +/− 30% were selected and used in assembly.
Nanopore long-read sequencing and data processing
DNA samples isolated from WBCs of the same female monkey were also sequenced by Health GeneTech (Taiwan) using Oxford Nanopore GridION sequencer. Three micrograms of high molecular weight genomic DNA was sheared to 8–12 kb fragments by G-tube (Covaris) based on the protocol of the company. DNA was subsequently size-selected using 0.45X volume of Ampure XP (Beckman Coulter) and eluted in 48 µl nuclease-free water. Sequencing library was prepared using ONT (Oxford Nanopore Technologies) 1D ligation sequencing kit (SQK-LSK109) following manufacturer’s protocol. Briefly, FFPE DNA repair and end repair process was completed by NEBNext Ultra II End Repair/dA-Tailing Module (New England Biolabs). Blunt/TA Ligase Master Mix (New England Biolabs) was used to ligate the sequencing adapters to library. The GridION sequencing was performed using a FLO- MIN106D flow cell till all pores were exhausted. A total of 6 flow cells were used in sequencing to generate a total of 75 billion bases. NanoPlot tool was used to generate a summary for ONT long-reads20.
Nanopore reads were corrected with short reads using proovread software under default parameters39. Untrimmed reads from the output were used as corrected reads for further steps of assembly.
Selection of MaSuRCA for hybrid assembly
A number of assemblers, including ABySS 2.040,41, ALLPATHS-LG42,43,44, SOAPdenovo245,46, MaSuRCA47, FLYE 2.4.222, wtdbg248 and Spades49, were evaluated and their performances were compared with various metrics commonly used in genome assembly. All these pipelines are capable of integrating multiple libraries of variable read lengths. In our study, machine with Intel (R) Xeon, x86_64 GNU/Linux 64-bit processor, with 16 CPUs, 2-Threads per core and 4 TB RAM was used for genome assembly with different pipelines.
A hybrid assembly was generated by MaSuRCA 3.28 on the machine using three Illumina libraries (two SR libraries and the MP library) together with a ONT LR library (configuration file was changed with “CA_PARAMETERS = cgwErrorRate = 0.15 merylMemory = 2 GB ovlStoreMemory = 2 GB” and other parameters set to default). Contigs produced by hybrid assembly were polished by mapping short reads of the 2 × 150 PE and 2 × 90 PE libraries using BWA. Errors were subsequently corrected by PILON based on the consensus generated from reads mapped to the location21.
Evaluation and correction of contig misassembly prior to chromosome assembly
Alignment filtration involves exclusion of contigs, if it mapped completely within the range of another contig or it was shorter than 10 kb. Breakpoints in contigs were identified, if (1) mapped in one alignment to one chromosome; (2) mapped in multiple segments far from each other in same chromosome; (3) mapped in multiple alignments on different chromosomes. These breakpoints were confirmed from short-reads alignment of assembly and contigs were broken, if short reads are not mapped at breakpoints in concordant manner or there were no short reads are mapped at breakpoints. Contigs were placed at a gap proportional to their mapping distance on reference chromosome and merged if their mapping is overlapping on the reference.
Contigs from hybrid assembly were further evaluated by the following steps. We mapped the reads from each library using BWA on polished assembly as reference to estimate the correctness of assembly. (1) Contigs were mapped to the reference Mmul_10 using minimap250. (2) All alignments of a contig that were contained within a larger alignment of another contigs were removed. (3) Contigs that were mapped (a) with single map on one chromosome, b) with multiple hits on one chromosome, (c) with single/multiple hits to multiple chromosomes were identified. (4) Contigs with multiple hits were checked if the hits were in consecutive to each other, if not, then their break point were checked for the read support at breakpoint locations. (5) Breakpoints within contigs that were covered by concordant reads were not broken and rest were broken since they were not covered by any concordant reads. (6) Contigs were stitched to one another based on their alignment order with respect to reference. Same length of gap was inserted between two contigs if they are non-overlapping. In case of overlap in between pair of contigs they were merged together. (7) Contigs mapped in the regions of complex structure variations were visualized using IGV and their breakpoints were recorded. Using the support from short reads insert-size we estimated the order of contigs manually. (8) Regions from Mmul_10 chromosomes that were not covered by contigs were recorded from previous step of chromosome generation. All the reads mapped in these regions including 1000 bases upstream and downstream of uncovered region were retrieved from whole genome reads files for each library. These reads were used for local assembly of uncovered regions. Contigs produced from this procedure were mapped to M. cyclopis draft chromosomes to find the anchors for new contigs and to patch the gap regions in M. cyclopis genome. (9) We next used contigs longer than 10 kb and searched if these contigs may have sequence similarity upstream or downstream to the remaining gaps. If found similarity these contigs were merged into the genome to reduce the span of gaps in the assembly. Remaining gaps were filled with the long-reads assembly produced using Flye assembler with the similar strategy as shown above. Flye scaffolds that spanned throughout the gap on the reference genome region were used to fill in the gap in the M. cyclopis genome.
Further curation of chromosome assemblies
Chromosomes were evaluated with QUAST on default parameters with M. mulatta assembly Mmul_10 as reference51. We used gene and exon information of Mmul_10 for feature based evaluation separately to evaluate whether the genes/exons/CDS have been covered completely or partially in an assembly.
After evaluation using QUAST, we analysed the genomic features obtained. We estimated the number of missing genes, exons and CDS in the chromosome level assembly and tried to recover those contigs which could not be stitched into the chromosomes. We also found missing genes, exons and CDS in the locally assembled contigs. These contigs which might have information about gene features were retained as unplaced contigs in the final assembly. Further we analysed whether these genes were not present in the genome or could not be detected due to technical issue.
Lastly, we used BUSCO tool to estimate the number of BUSCOs present in each assembly. Three different lineages (eukaryotic, mammalian and vertebral) of BUSCO were searched in each assembly52.
Repeat masking and identification
RepeatMasker version 4.1.5 (http://www.repeatmasker.org) were used to mask the draft genome of M. cyclopis, using the curated families of Dfam53 (version 3.7). We then used RepeatModeler254 to predict repeats from M. cyclopis using RECON55 and RepeatScout56 with default parameters. The output of RepeatModeler2 was then used to mask M. cyclopis genome to identify potential de novo repeat regions.
Genome annotation
For genome annotation we employed GEAN57 to annotate the genome. We used M. mulatta transcriptome assembly and protein sequences for initial genome annotation. Annotations from GEAN were further processed with MAKER v2.31.8 pipeline58. We then used SNAP, Genemark-ES and AUGUSTUS to predict the gene and exonic boundries59,60,61. Annotation of seven species were analysed including M. cyclopis, M. m. mulatta, M. fascicularis, M. nemestrina, Pongo abeli, Pan troglodytes and Homo sapiens and gene, CDS, exon and intron lengths were compared. We used a number of software tools to identify non-coding RNAs. These include tRNAscan-SE 2.062,63 for tRNAs, LncFinder v1.1.464 for lncRNAs, RNAmmer v1.265 for rRNAs and in-house scripts (based on BLAST 2.9.0+) for miRNA. Micro-RNA sequences of M. m. mulatta reference genome were extracted and used to search homologs in M. cyclopis genome by allowing a maximum of two mismatches.
Evolutionary study among key primate species with the assembled M. cyclopis genome
To identify the gene families in Taiwanese macaque, we retrieved the protein-coding genes from NCBI for seven other primate species (Homo sapiens, Pan troglodytes, Pongo abeli, M. nemestrina, M. fascicularis, M. m. mulatta and M. m. lasiota). The longest isoforms were used as the representative genes. Orthomcl v2.0.9 pipeline was used to identify the gene families in seven species at default parameters. Protein sequences of single copy orthologs present in all eight species were used for the multiple sequence alignment using MUSCLE v3.8.1551 at default parameters and Gblocks v0.91b66 was used to remove the poorly aligned regions. These sequences were concatenated to build a phylogenetic tree using RAxML v8.2.1267. Further MrBayes v3.2.7a68 was used to maximum-likelihood phylogenetic inference using 10 million generations by providing an estimate of tree age about 28.8 mya taken from TimeTree database69. “Strap” package70 in R was used to plot the phylogenetic tree.
Fossil gene analysis
In an attempt to further understand the potential differences in genome structure between M. cyclopis and its macaque relatives with close geographic proximity, we employed TBLASTN to identify the genomic distribution patterns of SIV gag and pol EVEs in M. fascicularis, M. m. mulatta and M. cyclopis. In parallel, monkeypox virus genes OPG099 (encoding CL5 membrane protein), OPG136 (encoding A15 core protein), OPG114 (encoding P4a core protein) and OPG142 (encoding D2 virion protein) were also tested for their corresponding EVEs.
All sequences for the fossil gene tested were downloaded from NCBI. These include the M. m. mulatta genome assembly Mmul_10 (ac. no.: GCF_003339765.1), M. fascicularis genome assembly MFA1912RKSv2 (ac. no.: GCF_012559485.2). SIV gene: gag (ac. no.: AAC97563.1) and pol (ac. no.: AAC97565.1). Monkeypox genes: OPG099 (ac. no.: YP_010377081.1), OPG136 (ac. no.: YP_010377124.1), OPG114 (ac. no.: YP_010377118.1) and OPG142 (ac. no.: YP_010377096.1).
As parameter setting, TBLASTN alignments with a query coverage of more than 50% of the amino acid sequence and an E-value of less than 0.05 were considered as significant. The chromosomal locations of the EVEs sites in the tested macaques were recorded.
Data availability
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JAHZQV000000000. The version described in this paper is version JAHZQV010000000. The whole genome sequencing data of M. cyclopis including two PE short reads, one MP short reads and ONT long reads were submitted to NCBI database with accession numbers Bioproject accession PRJNA559050 and Biosample accession SAMN12512033.
References
Fleagle, J. G. Primate Adaptation and Evolution (Elsevier Science, 2013).
Fooden, J. Provisional classifications and key to living species of macaques (primates: Macaca). Folia Primatol. 25, 225–236. https://doi.org/10.1159/000155715 (1976).
Tosi, A. J., Morales, J. C. & Melnick, D. J. Y-chromosome and mitochondrial markers in Macaca fascicularis indicate introgression with Indochinese M. mulatta and a biogeographic barrier in the Isthmus of Kra. Int. J. Primatol. 23, 161–178. https://doi.org/10.1023/a:1013258109954 (2002).
Chu, J. H., Lin, Y. S. & Wu, H. Y. Evolution and dispersal of three closely related macaque species, Macaca mulatta, M. cyclopis, and M. fuscata, in the eastern Asia. Mol. Phylogenet. Evol. 43, 418–429. https://doi.org/10.1016/j.ympev.2006.11.022 (2007).
Li, J. et al. Phylogeny of the macaques (Cercopithecidae: Macaca) based on Alu elements. Gene 448, 242–249. https://doi.org/10.1016/j.gene.2009.05.013 (2009).
Groves, C. P. Primates of the world: Distribution, abundance, and conservation. Int. J. Primatol. 6, 323–325. https://doi.org/10.1007/BF02745502 (1985).
Hayasaka, K. et al. Phylogenetic relationships among Japanese, rhesus, Formosan, and crab-eating monkeys, inferred from restriction-enzyme analysis of mitochondrial DNAs. Mol. Biol. Evol. 5, 270–281. https://doi.org/10.1093/oxfordjournals.molbev.a040492 (1988).
Huang, Y. F. et al. Complete Taiwanese macaque (Macaca cyclopis) mitochondrial genome: Reference-assisted de novo assembly with multiple k-mer Strategy. PLoS ONE 10, e0130673. https://doi.org/10.1371/journal.pone.0130673 (2015).
Deinard, A. & Smith, D. G. Phylogenetic relationships among the macaques: Evidence from the nuclear locus NRAMP1. J. Hum. Evol. 41, 45–59. https://doi.org/10.1006/jhev.2001.0480 (2001).
Fan, Z. et al. Whole-genome sequencing of tibetan macaque (Macaca thibetana) provides new insight into the macaque evolutionary history. Mol. Biol. Evol. 31, 1475–1489. https://doi.org/10.1093/molbev/msu104 (2014).
Malhi, R. S. et al. MamuSNP: A resource for Rhesus Macaque (Macaca mulatta) genomics. PLoS ONE 2, e438. https://doi.org/10.1371/journal.pone.0000438 (2007).
Rogers, J. & Gibbs, R. A. Comparative primate genomics: Emerging patterns of genome content and dynamics. Nat. Rev. Genet. 15, 347–359. https://doi.org/10.1038/nrg3707 (2014).
Smith, D. G. Genetic characterization of Indian-origin and Chinese-origin rhesus macaques (Macaca mulatta). Comp. Med. 55, 227–230 (2005).
Smith, D. G. & McDonough, J. Mitochondrial DNA variation in Chinese and Indian rhesus macaques (Macaca mulatta). Am. J. Primatol. 65, 1–25. https://doi.org/10.1002/ajp.20094 (2005).
Ekblom, R. & Wolf, J. B. A field guide to whole-genome sequencing, assembly and annotation. Evol. Appl. 7, 1026–1042. https://doi.org/10.1111/eva.12178 (2014).
van der Kuyl, A. C. Analysis of simian endogenous retrovirus (SERV) full-length proviruses in old world monkey genomes. Genes https://doi.org/10.3390/genes13010119 (2022).
Gertz, E. M., Yu, Y. K., Agarwala, R., Schaffer, A. A. & Altschul, S. F. Composition-based statistics and translated nucleotide searches: Improving the TBLASTN module of BLAST. BMC Biol. 4, 41. https://doi.org/10.1186/1741-7007-4-41 (2006).
Geisler, C. A new approach for detecting adventitious viruses shows Sf-rhabdovirus-negative Sf-RVN cells are suitable for safe biologicals production. BMC Biotechnol. 18, 8. https://doi.org/10.1186/s12896-017-0412-z (2018).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196. https://doi.org/10.1101/gr.6743907 (2008).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. https://doi.org/10.1093/bioinformatics/bty149 (2018).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963. https://doi.org/10.1371/journal.pone.0112963 (2014).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. https://doi.org/10.1038/s41587-019-0072-8 (2019).
Sibley, C. G. & Ahlquist, J. E. The phylogeny of the hominoid primates, as indicated by DNA-DNA hybridization. J. Mol. Evol. 20, 2–15. https://doi.org/10.1007/BF02101980 (1984).
Liedigk, R. et al. Mitogenomic phylogeny of the common long-tailed macaque (Macaca fascicularis fascicularis). BMC Genomics 16, 222. https://doi.org/10.1186/s12864-015-1437-0 (2015).
Perelman, P. et al. A molecular phylogeny of living primates. PLoS Genet. 7, e1001342. https://doi.org/10.1371/journal.pgen.1001342 (2011).
Tosi, A. J., Morales, J. C. & Melnick, D. J. Paternal, maternal, and biparental molecular markers provide unique windows onto the evolutionary history of macaque monkeys. Evolution 57, 1419–1435. https://doi.org/10.1111/j.0014-3820.2003.tb00349.x (2003).
Delson, E. The Macaques: Studies in Ecology, Behavior and Evolution (Von Nostrand Rheinhold, 1980).
Smith, D. G., McDonough, J. W. & George, D. A. Mitochondrial DNA variation within and among regional populations of longtail macaques (Macaca fascicularis) in relation to other species of the fascicularis group of macaques. Am. J. Primatol. 69, 182–198. https://doi.org/10.1002/ajp.20337 (2007).
Schillaci, M. A. et al. Evolutionary relationships of Macaca fascicularis fascicularis (Raffles 1821) (Primates: Cercopithecidae) from Singapore revealed by Baysian analysis of mitochondrial DNA sequences. Raffles Bull. Zool. 65, 3–19 (2017).
Yan, G. et al. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat. Biotechnol. 29, 1019–1023. https://doi.org/10.1038/nbt.1992 (2011).
Gibbs, R. A. et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science 316, 222–234. https://doi.org/10.1126/science.1139247 (2007).
Hirsch, V. M. et al. Characterization of a novel simian immunodeficiency virus (SIV) from L’Hoest monkeys (Cercopithecus l’hoesti): Implications for the origins of SIVmnd and other primate lentiviruses. J. Virol. 73, 1036–1045. https://doi.org/10.1128/JVI.73.2.1036-1045.1999 (1999).
Brown, J., Pirrung, M. & McCue, L. A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139. https://doi.org/10.1093/bioinformatics/btx373 (2017).
Lindgreen, S. AdapterRemoval: Easy cleaning of next-generation sequencing reads. BMC Res. Notes 5, 337. https://doi.org/10.1186/1756-0500-5-337 (2012).
Schubert, M., Lindgreen, S. & Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 88. https://doi.org/10.1186/s13104-016-1900-2 (2016).
Patel, R. K. & Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 7, e30619. https://doi.org/10.1371/journal.pone.0030619 (2012).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. https://doi.org/10.1093/bioinformatics/btr026 (2011).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. https://doi.org/10.1093/bioinformatics/btp698 (2010).
Hackl, T., Hedrich, R., Schultz, J. & Forster, F. proovread: Large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30, 3004–3011. https://doi.org/10.1093/bioinformatics/btu392 (2014).
Simpson, J. T. et al. ABySS: A parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. https://doi.org/10.1101/gr.089532.108 (2009).
Jackman, S. D. et al. ABySS 2.0: Resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 27, 768–777. https://doi.org/10.1101/gr.214346.116 (2017).
Maccallum, I. et al. ALLPATHS 2: Small genomes assembled accurately and with high continuity from short paired reads. Genome Biol. 10, R103. https://doi.org/10.1186/gb-2009-10-10-r103 (2009).
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U. S. A. 108, 1513–1518. https://doi.org/10.1073/pnas.1017351108 (2011).
Butler, J. et al. ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome Res. 18, 810–820. https://doi.org/10.1101/gr.7337908 (2008).
Luo, R. et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. https://doi.org/10.1186/2047-217X-1-18 (2012).
Luo, R. et al. Erratum: SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 4, 30. https://doi.org/10.1186/s13742-015-0069-2 (2015).
Zimin, A. V. et al. The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677. https://doi.org/10.1093/bioinformatics/btt476 (2013).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158. https://doi.org/10.1038/s41592-019-0669-3 (2020).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. https://doi.org/10.1093/bioinformatics/bty191 (2018).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. https://doi.org/10.1093/bioinformatics/btt086 (2013).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. https://doi.org/10.1093/bioinformatics/btv351 (2015).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 12, 2. https://doi.org/10.1186/s13100-020-00230-y (2021).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U. S. A. 117, 9451–9457. https://doi.org/10.1073/pnas.1921046117 (2020).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276. https://doi.org/10.1101/gr.88502 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351-358. https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Song, B. et al. Complement genome annotation lift over using a weighted sequence alignment strategy. Front. Genet. 10, 1046. https://doi.org/10.3389/fgene.2019.01046 (2019).
Yandell, M. & Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 13, 329–342. https://doi.org/10.1038/nrg3174 (2012).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59. https://doi.org/10.1186/1471-2105-5-59 (2004).
Borodovsky, M. & Lomsadze, A. Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Curr. Protoc. Bioinform. 1, 4–5. https://doi.org/10.1002/0471250953.bi0406s35 (2011).
Nachtweide, S. & Stanke, M. Multi-genome annotation with AUGUSTUS. Methods Mol. Biol. 1962, 139–160. https://doi.org/10.1007/978-1-4939-9173-0_8 (2019).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 1962, 1–14. https://doi.org/10.1007/978-1-4939-9173-0_1 (2019).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. https://doi.org/10.1093/nar/25.5.955 (1997).
Han, S. et al. LncFinder: An integrated platform for long non-coding RNA identification utilizing sequence intrinsic composition, structural information and physicochemical property. Brief Bioinform. 20, 2009–2027. https://doi.org/10.1093/bib/bby065 (2019).
Lagesen, K. et al. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. https://doi.org/10.1093/nar/gkm160 (2007).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. https://doi.org/10.1080/10635150701472164 (2007).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033 (2014).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. https://doi.org/10.1093/bioinformatics/17.8.754 (2001).
Kumar, S., Stecher, G., Suleski, M. & Hedges, S. B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. https://doi.org/10.1093/molbev/msx116 (2017).
Bell, M. A. & Lloyd, G. T. strap: An R package for plotting phylogenies against stratigraphy and assessing their stratigraphic congruence. Palaeontology 58, 379–389. https://doi.org/10.1111/pala.12142 (2015).
Acknowledgements
We want to thank CEO of Health Genetech, Dr. Mengchu Wu, for the help on Nanopore sequencing and Mohit Midha for doing some analysis of the sequence data and writing some sections of the manuscript.
Funding
Major funding was provided by Academia Sinica as the Research funding for Principal Investigator, while Top Science Biotechnologies, Inc. also provided financial support to the project.
Author information
Authors and Affiliations
Contributions
K.P.C., D.G.S., J.Y. and K.J.C.P. conceptualized M. cyclopis genome sequencing. K.J.C.P. prepared and provided the genomic DNA samples. K.P.C. supervised the analyses of the data, assembled the genome and wrote most of the manuscript. L.S. did fossil gene analysis and wrote part of the manuscript, H.S.O. did Repeat Masking, checked through the manuscript and provided critical proofreading of the manuscript. All authors also proofread the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chiu, KP., Stuart, L., Ooi, H.S. et al. Genome sequencing and application of Taiwanese macaque Macaca cyclopis. Sci Rep 13, 11545 (2023). https://doi.org/10.1038/s41598-023-38402-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-38402-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
