
Abstract
Helicobacter pylori is involved in the etiology and severity of several gastroduodenal diseases; however, plasticity of the H. pylori genome makes complete genome assembly difficult. We report here the full genomes of H. pylori strains CHC155 and VN1291 isolated from a non-cardia gastric cancer patient and a duodenal ulcer patient, respectively, and their virulence demonstrated by in vitro infection. Whole-genome sequences were obtained by combining long- and short-reads with a hybrid-assembly approach. Both CHC155 and VN1291 genome possessed four kinds of genomic island: a cag pathogenicity island (cagPAI), two type 4 secretion system islands within an integrative and conjugative element (tfs ICE), and prophage. CHC155 and VN1291 carried East Asian-type cagA and vacA s1m1, and outer membrane protein genes, including two copies of oipA. Corresponded to genetic determinants of antibiotic resistance, chromosomal mutations were identified in CHC155 (rdxA, gyrA, and 23S rRNA) and VN1291 (rdxA, 23S rRNA, and pbp1A). In vitro infection of AGS cells by both strains induced the cell scattering phenotype, tyrosine phosphorylation of CagA, and promoted high levels of IL8 secretion, indicating fully intact phenotypes of the cagPAI. Virulence genes in CHC155 and VN1291 genomes are crucial for H. pylori pathogenesis and are risk factors in the development of gastric cancer and duodenal ulcer. Our in vitro studies indicate that the strains CHC155 and VN1291 carry the pathogenic potential.
Similar content being viewed by others


Introduction
Helicobacter pylori is a Gram-negative bacteria that chronically infects the human stomach and in 2015 there were approximately 4.4 billion individuals worldwide infected with H. pylori1. H. pylori infection is strongly associated with gastric neoplasm and 1–3% of such cases will develop gastric cancer compared with 0% in uninfected patients2,3,4,5. Vietnam has one of the highest H. pylori infection rates in Asia (70.3%)1 and a high age-standardized rate (ASR) of stomach cancer prevalence (15.5)1,6 according to GLOBOCAN 20227. However, there is minimal knowledge of H. pylori genomes from this region, as well as the prevalence of virulence factor (CagA variable region) and drug-resistance genes, which limits the application of molecular diagnostics, such as tests for resistance genes, and the development of vaccines and therapeutic drugs8,9,10,11.
In addition to host and environmental factors, H. pylori virulence factors play a crucial role in increasing the severity of clinical outcomes12. The H. pylori genome is approximately 1.6 Mbp and contains more than 1600 genes. Each strain harbors a unique subset of genes that help adaptation to the human gastric environment13. Virulence factor genes are protein coding. Some are essential for bacterial survival in acid environments (e.g. ure genes encode urease), some encode outer membrane proteins that promote adherence to gastric epithelial cells (hopQ, oipA and babA), and some activate the innate immune response. CagA plays a crucial role in H. pylori gastric pathogenesis by being injected into host cells and disturbing internal signaling. VacA is secreted into the gastric environment and can enter the cytosol of host cells where it forms vacuoles associated with apoptosis13,14,15. Therefore, identifying sets of virulence factor genes from strains isolated from gastric cancer and duodenal ulcer patients will help to elucidate the virulent strains of H. pylori. Virulence factors of H. pylori have recently been classified according to functional categories, including acid resistance, adherence, chemotaxis and motility, molecular mimicry, immune invasion and modulation, secretion system, proinflammatory effect, and endotoxin production16. The pathogenicity of H. pylori is dependent on virulence factors that help bacteria invade gastric epithelial cells, change cell morphology, and induce the host immune response through an inflammatory pathway. These effects can contribute to gastroduodenal diseases17. Furthermore, long-term H. pylori infection is considered a high risk for the development of gastric cancer2.
Genomic islands are clusters of genes within a bacterial genome that appear to have been acquired by horizontal gene transfer. Many genomic islands are flanked by repeat sequences and carry fragments of or complete mobile and accessory genetic elements, such as bacteriophages, plasmids, insertion sequence elements, and integrative and conjugative elements (ICEs)18,19. Genomic islands are common in both Gram-positive and Gram-negative bacteria and play diverse roles in adaptation, metabolism, fitness cost, increased virulence, and antibiotic resistance18. In H. pylori, the cag pathogenicity island (cagPAI) is a genomic island that encodes functional components of a type 4 secretion system (T4SS)20. This T4SS represents a needle-like structure protruding from the bacterial surface and is induced when it comes into contact with a host cell before injecting oncogenic CagA20. When CagA is translocated into the cytosol, it triggers proinflammatory activity and internal signaling cascades21. Other genomic islands in H. pylori include tfs3 and tfs4 ICEs, which both encode novel T4SS components22,23. Similar to cagPAI, tfs ICEs are considered gastroduodenal disease markers24,25. In addition, H. pylori can contain a prophage, a genome island that originates from a bacteriophage and is found in approximately 20% of H. pylori isolates26.
We previously performed PCR-based analyses that detected a small fraction of virulence factors10,27 but not a full gene or genomic island. Current advances in genome sequencing technology, including shotgun sequencing and long-read sequencing, make it possible to accurately obtain full bacterial genomes. There is plasticity in the H. pylori genome; some genes are duplicated (oipA in hspEAsia strains) and strains have highly similar gene families, i.e. outer membrane proteins and genome island clusters, which makes complete genome assembly difficult. Individual strain information therefore tends to be lacking. In addition, some strains possess a plasmid, which is rarely detected using only short-read assembly. To overcome these characteristics of H. pylori genomes, we used third-generation sequencing to generate long-reads to facilitate complete genome assembly. Complete genomes are useful for further characterization of virulence factors and their phenotypes. Here, we report two complete genomes of H. pylori strains, CHC155 and VN1291, isolated from a patient with gastric cancer and from a duodenal ulcer patient, respectively. We identified four genomic islands in each strain and characterized their virulence factors and genetic determinants of antibiotic resistance. These features, supported by phylogenic analysis, were representative of two Vietnamese strains.
Results
Clinical characteristics of the patients and features of H. pylori strains CHC155 and VN1291
We have previously obtained over 100 H. pylori strains from Vietnamese gastric cancer and duodenal ulcer patients24. We chose strains CHC155 and VN1291 for this study, because they contain fully intact tfs3 ICEs in addition to cagPAI. H. pylori CHC155 was isolated from a gastric biopsy specimen collected by endoscopy from a 61-year-old Vietnamese male patient with non-cardia gastric cancer. The clinical isolate showed no in vitro resistance to tetracycline or amoxicillin with minimum inhibitory concentrations (MICs) of 0.12 and 0.06 mg/L, respectively. However, resistance was noted to clarithromycin, levofloxacin and metronidazole, with MICs of 4 mg/L, 2 mg/L, and ≥ 256 mg/L, respectively. Strain VN1291 was isolated from a 43-year-old female patient with duodenal ulcer at Cho Ray Hospital, Ho Chi Minh. This strain was resistant to amoxicillin, clarithromycin and metronidazole, with MICs of 0.5 mg/L, 4 mg/L, and ≥ 256 mg/L, respectively, but was susceptible to levofloxacin and tetracycline, with MICs of 0.5 mg/L and 0.125 mg/L, respectively.
The de novo assembly of the CHC155 genome resulted in a single circular contig of 1,696,601 bp. Using the CheckM v1.1.6 algorithm28, the genome assembly reached > 99% completeness with no contamination or strain heterogenicity. DFAST quality control in FastANI v.1.3329 showed the highest average nucleotide identity of the CHC155 genome to be 94.9% against H. pylori strain ATCC43504 (GCA_004295525.1) which was assigned to the same species. Strain CHC155 contained four rRNA genes, including two copies of 23S rRNA and two copies of 16S rRNA (Table 1 and Fig. 1A). We also determined the full genome of strain VN1291 (Table 1 and Fig. 1B), for comparative assessment of CHC155 virulence.

Overview of the genomes of H. pylori strain CHC155 (A) and VN1291 (B). The plot displays the genomes of CHC155 and VN1291 as circular chromosomes of 1,696,601 bp and 1,702,481 bp. The black outermost and innermost rings indicate the genes, which were predicted and annotated using PROKKA v.1.14.6 (https://github.com/tseemann/prokka/). GC skew and GC content are indicated. The genome map of strain CHC155 and VN1291 were visualized from Genbank format by Proksee (https://proksee.ca/)30.
We detected one 21-bp CRISPR-like sequence (CTTCAATCAAGGCA30CTTATAA) in both strains. This sequence was located in the vlpC gene, which encodes a putative vacuolating cytotoxin (vacA)-like protein C, an outer membrane protein toxin.
Genomic island prediction and sequence comparison
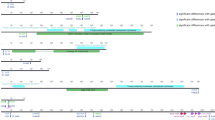
GI prediction was based on common features of genomic islands, including mobility genes, phage-related genes, direct repeats, and nucleotide composition bias. Genomic islands identified in strain CHC155 included two tfs3 ICEs, a KHP30-like prophage, and a cagPAI (Fig. 2A). Four genomic islands were identified in strain VN1291, including tfs3 ICE, hybrid tfs3_4 ICE, a KHP40-like prophage, and a cagPAI (Fig. 2B).
-
1.
cagPAI

Overview of the genomic islands (GIs) in strain CHC155 and VN1291 genomes. Locations of four genomic islands in both genomes are visualized by color scheme using SnapGene v.6.2.2. The genetic schemes of GIs in two strains (A–H) were plotted by genoplotR v.0.8.11. (A–D) GIs in strain CHC155; (A) cagPAI, (B) KHP30-like prophage, (C) tfs3 ICE_1, and (D) tfs3 ICE_2. (E–H) GIs of strain VN1291; (E) cagPAI, (F) KHP40-like prophage, (G) hybrid tfs3_4 ICE_1, and (H) tfs3 ICE_2. The black color indicates the flanking genes of GIs: sel1 (left) and murI (right) in cagPAI, comGF (left) and gene encodes DNA-binding protein (right) in KHP30-like prophage, gene encodes RloF protein (left) and hsdR (right) in tfs3 ICE_1, hsdM (left) and gene encodes RecJ protein (right) in tfs3 ICE_2. The red arrow indicates cagA, an effector protein of cagPAI, and the yellow arrow indicates ctkA (cell-translocating kinase A), an accessory gene of tfs3 ICE. Some genes of interest in KHP30-like prophage and KHP40-like prophage are marked and colored.
Strains CHC155 and VN1291 both possessed a 30-kb cagPAI, a low GC content region (less than 35%) that contained 24 cag genes, and a gene coding the effector protein, CagA (Fig. 2 and Supplementary Table S1). The cagPAI was inserted between glutamate racemase (murL/I) (right) and Sel1-like repeat protein (sel1) genes in both CHC155 (Fig. 2) and VN1291 (Supplementary Fig. S2).
-
2.
T4SS within the tfs ICE
Strains CHC155 and VN1291 both possessed two tfs ICEs (Figs. 1, 2). Based on the genetic arrangement of rlx (virD2 relaxase), xerD (integrase), virB6 (T4SS gene), and sequence identity, the two tfs ICEs in strain CHC155 were classified as tfs3 ICEs (Fig. 2). One tfs3 ICE (1,141,730–1,197,010) was flanked by the gene for RloF protein (left) and the gene for a type I restriction enzyme R protein (HsdR) (right) (Supplementary Table S1 and Fig. 2C). The flanking genes of the other tfs3 ICE (1,639,789–1,688,225) were the gene for a type I enzyme M protein (HsdM) (left) and RecJ (right), a single-stranded DNA-specific exonuclease involved in archaeal DNA replication initiation (Fig. 2D and Supplementary Table S1). Based on the genome comparison, strain VN1291 possessed a hybrid tfs3_4 ICE (1,137,628…1,209,303) that was flanked by genes for a RloF-like protein (left) and a DHH family protein (right) (Fig. 2G and Supplementary Table S1). In addition, the second tfs3 ICE of strain VN1291 (1,646,779–1,695,191) was flanked by the gene for type I restriction enzyme M protein (HsdM) (left) and a putative metal-dependent hydrolase (right) (Fig. 2H and Supplementary Table S1). All tfs ICEs in strains CHC155 and VN1291 contained genes that encode a T4SS: virB2, virB3, virB4, virB6, virB8, virB9, virB10, virB11, and virD4. In addition, they harbored the DNA processing genes, virD2 relaxase and xerT integrase/recombinase (Fig. 2). In both strains, one tfs3 ICE contained the cell-translocating kinase A (ctkA) gene, a translocation protein of tfs3 ICE. In addition, the longest tfs3 ICE gene was an 8600 bp DNA methyltransferase.
-
3.
Prophage
We identified a KHP30-like prophage (encoding 34 phage proteins) in strain CHC155 and a KHP40-like prophage (encoding 35 phage proteins) in strain VN1291 (Table 2). The 5′-attR/attL-3′ repeat region (ATTTTTAAAATA) was identified in the prophages of both CHC155 and VN1291. The KHP30-like and KHP40-like prophages were integrated between the competence protein, comGF (left), and a putative outer membrane protein (right) (Fig. 2). These prophages encoded proteins that play roles in host cell invasion, including integrase, tail, portal, holin, and terminase proteins. Other proteins were phage-related hypothetical proteins (Supplementary Table S1).
-
4.
Phylogenomic analysis of strains CHC155 and VN1291
The phylogenomics of strains CHC155 and VN1291 were analyzed according to the flow diagram in Fig. 3. Pangenome analysis using Roary identified 3330 genes among 37 H. pylori strains, which included 1036 conserved genes (32.8%) present in 99% of all strains (Supplementary Table S7). IQ-TREE identified 975,924 sites from the core gene alignment. The total number of sites divided into 130,962 parsimony-informative (13.4%); 77,543 singleton (8.0%), and 767,419 (78.6%) constant sites. ModelFinder selected the best fit model of substitution (GTR + F + I + I + R7) according to a Bayesian information criterion (BIC) score of 8,944,815.825 and an Akaike information criterion (AIC) score of 8,943,731.058 to build the phylogenetic tree. The phylogeny of 37 reference strains is shown in Fig. 4 and represented seven H. pylori populations: hspEAsia (F16, F30, F32, F57, 51, 52, 83, XZ274, and 35A), hspAmerind (Puno135, Puno120, v225d, Shi112, Cuz20, Shi470, Sat464, Shi417, Shi169), hybrid hspAmerind/hspEAsia (PeCan4), hpAsia2 (SNT49, India7), hpEurope (HPAG1, 26695, B8, Lithuania75, G27, P12, B38, HUP-B14, SJM180, and ELS37), hpAfrica2 (SouthAfrica7), and hpAfrica1 (Gambia94/24, J99, and Pecan18). We assigned strains CHC155 and VN1291 to the hspEAsia population according to clustering with the hspEAsia group strains (Fig. 4).

Flow diagram for phylogenomic analysis of H. pylori CHC155 and VN1291. Whole-genome sequences of strains CHC155, VN1291, and 35 reference strains were annotated by Prokka. Subsequently, the genome feature (.gff) pangenome was analyzed by Roary and exported as a core gene alignment. Using this alignment, the phylogenetic analysis was performed and exported as a Newick tree, which was visualized in a radical image. The flow diagram was drown in PowerPoint (Office 365, https://www.microsoft.com/vi-vn/microsoft-365).

Phylogenetic tree constructed from core single nucleotide polymorphisms of H. pylori CHC155, VN1291, and 35 reference strains, which were representative of different geographical populations. The tree map was visualized by iTOL v.6.7.3 (https://itol.embl.de/). The Newick format was generated from IQTREE2, H. pylori population was annotated into different colors.
Virulome and antimicrobial resistance genes of strains CHC155 and VN1291.
-
1.
Virulome
Virulence genes were identified in strains CHC155 and VN1291 by screening the virulence factor database using ABRICATE (Supplementary Table S2). These virulence factors were diverse and classified into eight categories: acid resistance, adherence, endotoxin, molecular mimicry, chemotaxis and motility, proinflammatory effects, toxins, and secretion systems (Supplementary Table S2). Strains CHC155 and VN1291 possessed 19 and 20 kinds of the outer membrane protein genes, hopA-Z, among which were hopH (oipA), hopS (babA)/hopT (babB), and hopU (babC). Interestingly, for some outer membrane proteins, two copies (hopD, hopH, hopJ, hopZ) or multiple copies (hopN, hopO) were found in one or both genomes (Supplementary Table S3). There was a slight difference in the number of outer membrane proteins (hopD, hopMN, and hopO) between the two strains. Strain VN1291 carried two genes encoding the HopD protein while strain CHC155 carried only one. Meanwhile, there were three genes encoding HopO proteins in strain VN1291 but only two in strain CHC155.
CagA, an effector protein, and VacA, which encodes a vacuolating and pore-forming protein, were also identified in the two strains. The C-terminus of CagA contained three Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs with neighboring sequences corresponding to ABD-type and East Asian specific CagA-type strains (Fig. 5A), which may undergo tyrosine phosphorylation to hijack internal signaling pathways. In addition, the vacA gene motif of CHC155 and VN1291 was assigned to s1m1 (Fig. 5B), which induces a high level of vacuolation and cytotoxicity and is associated with gastric inflammation, peptic ulcer, and gastric cancer33,34.

CagA type and VacA type CHC155 and VN1291 strains. (A) The EPIYA motifs (three black boxes) and neighboring regions in the C-terminal region of the CagA protein from four strains: Shi470, F57, CHC155, and VN1291. CagA of either strain CHC155 or VN1291 is an ABD type. (B) Characterization of VacA in strain CHC155. The black boxes in the figure display the alignment of VacA protein sequences of strain CHC155 and VN1291 compared with the signal region, s1 (ACB41503.1)/s2(BAV60359) (upper box), and intermediate region, m1(BAV60412.1)/m2 (BAB87691.1) (bottom box). The VacA alleles are characterized by deletions in the s- and m-regions corresponding to s1 and m1 types.
-
2.
Antimicrobial resistance gene profiling
Strain CHC155 was phenotypically resistant to clarithromycin, levofloxacin, and metronidazole but susceptible to amoxicillin and tetracycline (Table 3). Consistently, corresponding resistance mutations in the genes, gyrA (HP0701), 23S rRNA, and rdxA (HP0954) were identified in its genome, whereas no relevant mutations were noted in genes, pbp1a (HP0597) or 16S rRNA (Table 3). In strain VN1291, we identified resistance mutations in pbp1A (HP0597), rdxA (HP0954), and 23S rRNA. The high sequence coverage of the VN1291 and CHC155 genomes makes variant calling of antimicrobial resistance mutations reliable. To predict antibiotic resistance, we screened the CHC155 and VN1291 genomes using several antimicrobial resistance databases. However, no acquired antibiotic resistance genes were predicted using CARD, Plasmidfinder, ARG-ANNOT, and ResFinder (Supplementary Table S4). However, MEGARes detected a multidrug efflux transporter gene (HP0313) (Supplementary Table S4–6). This gene belongs to the YbfB/YjiJ family of major facilitator superfamily transporters, which is widely distributed in microbial genomes and exhibits a large spectrum of substrate specificities35.
In vitro assessment of CHC155 and VN1291 virulence
AGS gastric epithelial cells were infected with CHC155 and VN1291 strains to understand their virulence. A cell elongation phenotype, known as the hummingbird phenotype, is observed after CagA injection from H. pylori into AGS cells36,37,38. Moreover, induction of IL8 during H. pylori infection depends on the presence of cagPAI, both in vitro and in vivo39,40. To confirm whether such virulent phenotypes occur with these two Vietnamese strains, AGS cells were infected with CHC155 or VN1291, or with 26695 as a control. Twenty-four hours after infection, we observed hummingbird phenotypes in AGS cells (red arrows in Fig. 6A).

H. pylori CHC155 and VN1291 induced the hummingbird phenotype and IL8 secretion in human gastric epithelial AGS cells. AGS cells were incubated with or without H. pylori CHC155, VN1291, or 26695 (multiplicity of infection 100) for 6 and 24 h. Bar: 50 μm. (A) Cells 24 h after infection. Red arrows indicate cells with the hummingbird phenotype. (B) Immunoblot analysis of phosphorylated CagA (pY-CagA), CagA, UreB and β-actin. (C) Induction of IL8 during infection (n = 4).
After infection of AGS cells with each strain, CagA phosphorylation (Fig. 6B) was assessed in comparison with total CagA by immunoblotting. CagA phosphorylation was induced by strains 26695 and VN1291 at 6 h after infection and the degree of phosphorylation was increased at 24 h. This phenomenon was also observed for strain CHC155, but over a longer time scale; phosphorylation was faint at 6 h, but strong at 24 h. Bacterial counts were monitored using urease subunit UreB, an abundant H. pylori protein (Fig. 6B, and Supplementary Fig. S1).
Twenty-four hours after infection, strain CHC155 induced approximately 1.5 times and strain VN1291 approximately 2 times higher levels of IL8 than strain 26695 (Fig. 6C). The level of IL8 secretion indicates the ability to induce inflammatory activity; therefore, strains CHC155 and VN1291 are inflammatory strains.
Discussion
We report the complete genome of H. pylori strains CHC155 and VN1291, which were isolated from Vietnamese patients with non-cardia gastric cancer and duodenal ulcer, respectively. Both strains can induce the hummingbird phenotype, IL8 secretion, and CagA phosphorylation. This indicates that the strains have the potentials to initiate pathogenic changes in the gastric mucosa. Phylogenetic analysis assigned the two strains to the hspEAsia population. In addition, the outer membrane protein compositions of strains CHC155 and VN1291 contain two hopH(oipA), two hopS(babA)/hopT(babB), two hopJ, and multiple hopMN proteins, which is similar to other hspEAsia strains described by Kawai et al.41. Divergence in the number of outer membrane protein loci between hspEAsia, hspAmerind, hspWAfrica, and hpEurope populations has been shown, and the hpEurope and hspWAfrica populations possess one hopH locus and three babA/babB/babC loci41. It is interesting that we found hopU (babC) from both of Vietnamese strains in low coverage to it from strain 26695. The differences in the number of outer membrane proteins and in genetic variation between two strains of the same population but isolated from two distinct diseases, cancer and duodenal ulcer, and between H. pylori populations, may reflect a flexibility and adaptation capability of the H. pylori genome in host interaction.
Some H. pylori virulence factors are crucial for prolonging infection in the gastric mucosa via molecular mimicry. Strain CHC155 harbors futB and futC, which encode Lewis antigens expressed in the gastric mucosa (Supplementary Table S2). This antigenic mimicry suppresses an immune response against the bacteria and allows it to adhere to the gastric mucosa42. NapA promotes adhesion of human neutrophils to endothelial cells and the production of reactive oxygen radicals. In addition, the outer membrane family proteins, babA/babB and hopQ are involved in H. pylori adhesion43. The inflammatory effect of H. pylori is associated with the effect of oipA protein (hopH) on IL8 production44.
Our interest focused on important known virulence factors of H. pylori that are associated with gastroduodenal diseases. Strains CHC155 and VN1291 harbored the EastAsian-type cagA (ABD motif) and vacA s1m1. Both strains also possessed cagPAI, a genomic island that encodes the T4SS machinery for translocating CagA into host cells. In addition, strain CHC155 possessed two tfs3 ICEs that contain a complete T4SS cluster. The genetics of tfs3/4 ICE varies among H. pylori strains; they can harbor a complete or partial tfs fragment, or no tfs22,23. Moreover, tfs ICE is frequently exchanged and integrated into the genome in a hybrid tfs3-4 ICE through conjugation22,23. Compared with previous genome studies of H. pylori22,23,45,46, we found that strain VN1291 possessed one tfs3 and a hybrid tfs3-4. Furthermore, we identified a KHP30-like prophage in the genome of strain CHC155 and a KHP40-like prophage in strain VN1291. The prophage was integrated between comGF at the 5′ end and a putative outer membrane protein at the 3′ end, similar to the other hspEAsia strains47. comGF plays a role in transformation and DNA binding, which contributes to the genetic variability of H. pylori48, while outer membrane proteins mediate adherence to the gastric epithelium and are associated with the clinical outcome of the infection49. These findings indicate that the prophage genetic element is adaptable in the H. pylori population.cagA gene-positive strains affect the severity of gastroduodenal disease. The phosphorylated or non-phosphorylated form of CagA activates downstream host cell-signaling pathways by binding to adaptor proteins, such as Crk, Grab2, HSP-2, PAR1, and c-Met50,51. Crk-CagA interaction induces cell–cell dissociation and development of the hummingbird phenotype50,51. Therefore, our in vitro results of CagA phosphorylation and the hummingbird phenotype in infected AGS cells by strain CHC155 or VN1291 indicate the potential virulence of both of strains. Interestingly, we observed higher levels of CagA and phosphorylated CagA in strain VN1291 at 6 h compared with levels in other strains, and increased levels of CagA at 24 h post infection in strain CHC155 compared with that in strain 26695, even if we take into account the strain specificity of the commercial CagA antibody used in our immunoblot analysis, which was originally generated using Western-CagA epitopes of a Western H. pylori strain. Regulation of cagA transcription by NaCl, was found in strain 26695 and Colombian clinical isolates, which is mediated via two copies of a TAATGA motif in the CagA promoter region52. Moreover, a + 59 motif in the cagA-5′-untranslated region influences the levels of CagA53. Further investigations are necessary to understand this mechanism of CagA regulation in Vietnamese strains.
The observation of high IL8 levels following infection with strains CHC155 and VN1291 compared with strain 26695 was particularly interesting, although a difference in IL8 levels between cagPAI-positive strains was previously observed54. Two main hypotheses can account for this difference. First, IL8 secretion may be induced when tfs3 ICE is present, as is the case for cagPAI55. The genome analysis showed that strains CHC155 and VN1291 possessed a complete T4SS cluster (11 T4SS core genes) in the tfs3 ICE, whereas virB8, virB9, virB10, virB11, and virD4 genes were absent in strain 26695. Second, the effector protein ctkA in tfs3 ICE might promote proinflammatory activity. Strains CHC155 and VN1291 both possess ctkA but strain 26695 does not. Recent evidence indicates that T4SS genes of tfs3 support the injection of CtkA into host cells and the induction of high levels of IL8 secretion56. Our virulence profiling showed that strain VN1291 is very similar to strain CHC155. This may be because both strains belong to the hspEAsia population, although they were isolated from two patients with different diseases, duodenal ulcer (DU: VN1291) and gastric cancer (GC: CHC155). Our previous phylogenetic analysis at the whole genome scale showed that the indicated DU and GC strains were distributed together24,57. In addition, our previous genome wide association study on hspEAsia strains indicated that single nucleotide polymorphisms between strains causing different diseases could be discovered and underlying mechanisms suggested, such as electric charge alteration at the ligand-binding pocket, change in subunit interaction, and mode-switching DNA methylation. The virulent gene components of DU and GC strains were similar and single nucleotide polymorphisms may affect host–pathogen interaction and are novel candidates for disease discrimination57.
Conclusions
Here, we report the complete genomes of strains CHC155 and VN1291, which were isolated from patients with non-cardia gastric cancer and duodenal ulcer, respectively, as two representative virulent strains from Vietnam. Both strains carry East Asian-type cagA and vacA s1m1. Furthermore, each strain possesses cagPAI, two tfs3/tfs4 ICEs, and a prophage. Strains CHC155 and VN1291 can induce proinflammatory responses and morphological changes in gastric epithelial cells, indicating their potential virulence.
Materials and methods
H. pylori and genome sequencing
Helicobacter pylori CHC155 was isolated from a 61 year-old male patient with non-cardia gastric cancer at Cho Ray Hospital, Ho Chi Minh. H. pylori VN1291 was isolated from a 43-year-old female patient with duodenal ulcer at Cho Ray Hospital, Ho Chi Minh. After the international transfer of gastric antral biopsy samples to Oita University, both strains were isolated using standard culture methods as previously described9. DNA was extracted using a DNeasy Blood & Tissue kit (Qiagen Inc., Valencia, CA, USA). DNA concentration was measured using a Quantus Fluorometer (Promega, Madison, Wiscosin, USA). The extracted genomic DNA was sheared for library construction using a Covaris g-TUBE device according to the manufacturer’s instructions. High-throughput genome sequencing was performed on a HiSeq 2500 (2 × 150 paired-end reads) for strain CHC155, and MiSeq (2 × 300 paired-end reads) system for strain VN1291, following each of the manufacturer’s instructions (Illumina, San Diego, CA, USA). Trimmomatic v. 0.35 was used to remove adapter sequences and low-quality bases from raw short-read data58.
A SMRTbell library was prepared using a SMRTbell template Prep Kit 1.0 (Pacific Bioscience, CA, USA). DNA fragments larger than 17 kb, were selected using the BluePippin system (Saga Science, MA, USA). For each H. pylori strain, one SMRT cell was run on the PacBio RS II System with P6/C4 or P6/C4v2 chemistry and 360-min movies (Pacific Biosciences, Menlo Park, CA, USA). SMRT sequencing data were analyzed using SMRT Analysis version 2.3.0 via the SMRT Portal.
De novo assembly and genome annotation
To generate a complete genome assembly, de novo assembly was performed with the hybrid-assembly method using Unicycler v.0.4.8, which combined both long and short reads with default parameters; command line: unicycler -1 SHORT1 -2 SHORT2 -s UNPAIRED -l LONG –min_fasta_length 20059. Briefly, short reads (HiSeq/MiSeq), were assembled into several contigs, and gaps between contigs were connected by long-reads (PacBio in strain CHC155 and Oxford Nanopore in strain VN1291) to generate complete circular contigs, which have one link connecting the end to the start. The assembly was subsequently polished for a maximum of 10 rounds using Pilon60 and then Unicycler software was applied. Finally, genome features were annotated using PROKKA v1.14.661, a rapid bacterial genome annotation pipeline, using default parameters.
Bioinformatics software
The core genome alignment of strains CHC155 and VN1291 and the reference strains was obtained using Roary v3.13.062 with the following parameters: minimum percentage identity of 80% in blastp and core genes in 99% of isolates; command-line: roary –i 80 –e –mafft *.gff. The H. pylori reference strains represented in seven populations (hpAfrica2, hpAfrica1, hpEurope, hpAsia2, hybrid hspAmerind/hspEAsia, hspAmerind, and hspEAsia) were assigned in previous studies63,64,65. The high-resolution phylogenetic tree was constructed via a generalized time-reversible (GTR + F + I + I + R7) model to estimate the pairwise distance of 1,036 core genome alignments from the Roary output. These core genome alignments were used to construct phylogenetic trees using the maximum-likelihood method, the GTR model, and IQ-TREE v.1.6.1266; command-line: iqtree -T AUTO -m MFP –s core_gene_alignment.fasta. Prophage was predicted using the PHASTER server67 and genomic islands were predicted using the IslandViewer4 server with the GenBank files from PROKKA v1.14.6 and strain F57 serving as a reference for genome comparison68. The circular genome was visualized using Proksee (https://proksee.ca)30 and SnapGene v.6.2.2, and the genetic scheme of genomic islands were plotted using the genoplotR v.0.8.11 package (RStudio, v. 1.3.9, PBC). The virulence and antimicrobial resistance genes of strain CHC155 and VN1291 were retrieved from public databases (VFDB, ResFinder, MEGARes, ARG-ANNOT, CARD, and PlasmidFinder) using ABRICATE v1.0.016.
Determination of antibiotic resistance phenotypes and genotypes
Antimicrobial susceptibility was assessed using Etest® (bioMerieux) for five antibiotics (amoxicillin, clarithromycin, levofloxacin, tetracycline, and metronidazole) following European Committee on Antimicrobial Susceptibility Testing (EUCAST) v.21.01.01 protocols on Müller–Hinton agar plates supplemented with 5% horse blood. The minimum inhibitory concentrations of the antibiotics were checked every day after incubation for 3–6 days and determined. H. pylori strain 26695 was used as a control strain. Clinical breakpoints between resistant and susceptible strains were determined following the EUCAST guidelines available at http://www.eucast.org/.
The genetic determinants of antibiotic resistance in strain 26695, gyrA (HP0701), 23S rRNA, rdxA (HP0954), pbp1 (HP0597), and 16S rRNA, were used to retrieve those in strains CHC155 and VN1291 using the blastn algorithm with a minimum coverage of 80% and a minimum identity of 90%. Nonsynonymous mutations in either Vietnamese strain versus 26695, were compared with resistance mutations described in H. pylori strains69.
Evaluation of H. pylori virulence to and IL8 secretion from AGS cells
The virulence of H. pylori strains CHC155, VN1291, and reference strain 26695, was assessed experimentally by infecting human gastric epithelial AGS cells as described previously70. AGS cells were originally isolated in 1979 from the stomach tissue of a 54-year-old, white, female patient with gastric adenocarcinoma. These cells exhibit epithelial morphology71. The experiments were performed independently twice. Briefly, AGS cells were seeded onto six-well plates, grown overnight in RPMI 1640 medium supplemented with 10% FBS, and incubated at 37 °C in 5% CO2. H. pylori strains were suspended in RPMI 1640/10% FBS from a 2-day Brucella agar plate culture supplemented with 7% horse blood and added to the 70–80% confluent AGS culture at a multiplicity of infection of 100. After co-culture for 6 and 24 h, cells were fixed with 10% paraformaldehyde for 15 min, washed with PBS, and formation of the hummingbird phenotype examined under a phase-contrast microscope in randomly chosen fields. IL8 in the supernatant of infected AGS cells in 12-well plates (n = 4) was measured using a Human IL8 Uncoated ELISA Kit (Invitrogen, USA). Western blot analysis was performed using infected cell lysates from a 12-well plate culture. The antibodies against the following were used: p-Tyr (PY-99, Santa Cruz Biotechnology, Dallas, TX, USA), CagA (Austral Biologicals, San Rarnon, CA, USA), urease B (Institute of Immunology, Tokyo, Japan), and β-actin (Sigma-Aldrich, St. Louis, MO, USA).
Ethics approval and consent to participate
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of Oita University. Informed consent was obtained from all subjects involved in the study.
Consent for publication
Written informed consent was obtained from the patients to publish their data in this paper.
Data availability
The whole genome sequence data of strains CHC155 and VN1921 have been deposited at DDBJ/ENA/GeneBank. The accessions numbers are AP026446 (strain CHC155), AP026444 (strain VN1291), and AP026445 (plasmid pVN1291 in strain VN1291).
References
Hooi, J. K. Y. et al. Global prevalence of Helicobacter pylori infection: Systematic review and meta-analysis. Gastroenterology 153, 420–429. https://doi.org/10.1053/j.gastro.2017.04.022 (2017).
Uemura, N. et al. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 345, 784–789. https://doi.org/10.1056/NEJMoa001999 (2001).
Noto, J. M. & Peek, R. M. Jr. Helicobacter pylori: An overview. Methods Mol. Biol. 921, 7–10. https://doi.org/10.1007/978-1-62703-005-2_2 (2012).
de Souza, L. et al. From Helicobacter pylori infection to gastric cancer: Current evidence on the immune response. World J. Clin. Oncol. 13, 186–199. https://doi.org/10.5306/wjco.v13.i3.186 (2022).
Wroblewski, L. E., Peek, R. M. Jr. & Wilson, K. T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 23, 713–739. https://doi.org/10.1128/CMR.00011-10 (2010).
Morgan, E. et al. The current and future incidence and mortality of gastric cancer in 185 countries, 2020–40: A population-based modelling study. EClinicalMedicine 47, 101404. https://doi.org/10.1016/j.eclinm.2022.101404 (2022).
The Global Cancer Observatory (GCO) (2022).
Binh, T. T. et al. The incidence of primary antibiotic resistance of Helicobacter pylori in Vietnam. J. Clin. Gastroenterol. 47, 233–238. https://doi.org/10.1097/MCG.0b013e3182676e2b (2013).
Nguyen, T. L. et al. Helicobacter pylori infection and gastroduodenal diseases in Vietnam: A cross-sectional, hospital-based study. BMC Gastroenterol. 10, 114. https://doi.org/10.1186/1471-230X-10-114 (2010).
Uchida, T. et al. Analysis of virulence factors of Helicobacter pylori isolated from a Vietnamese population. BMC Microbiol. 9, 175. https://doi.org/10.1186/1471-2180-9-175 (2009).
Truong, B. X. et al. Diverse characteristics of the CagA gene of Helicobacter pylori strains collected from patients from southern vietnam with gastric cancer and peptic ulcer. J. Clin. Microbiol. 47, 4021–4028. https://doi.org/10.1128/JCM.00504-09 (2009).
Yamaoka, Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 7, 629–641. https://doi.org/10.1038/nrgastro.2010.154 (2010).
Alm, R. A. & Trust, T. J. Analysis of the genetic diversity of Helicobacter pylori: The tale of two genomes. J. Mol. Med. (Berl.) 77, 834–846 (1999).
Alm, R. A. et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397, 176–180. https://doi.org/10.1038/16495 (1999).
Alm, R. A. et al. Comparative genomics of Helicobacter pylori: Analysis of the outer membrane protein families. Infect. Immun. 68, 4155–4168 (2000).
Liu, B., Zheng, D., Jin, Q., Chen, L. & Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. https://doi.org/10.1093/nar/gky1080 (2019).
Kusters, J. G., van Vliet, A. H. & Kuipers, E. J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19, 449–490. https://doi.org/10.1128/CMR.00054-05 (2006).
Dobrindt, U., Hochhut, B., Hentschel, U. & Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424. https://doi.org/10.1038/nrmicro884 (2004).
Durrant, M. G., Li, M. M., Siranosian, B. A., Montgomery, S. B. & Bhatt, A. S. A bioinformatic analysis of integrative mobile genetic elements highlights their role in bacterial adaptation. Cell Host Microbe 28, 767. https://doi.org/10.1016/j.chom.2020.09.015 (2020).
Backert, S., Tegtmeyer, N. & Fischer, W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol. 10, 955–965. https://doi.org/10.2217/fmb.15.32 (2015).
Odenbreit, S. et al. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287, 1497–1500. https://doi.org/10.1126/science.287.5457.1497 (2000).
Fischer, W. et al. A comprehensive analysis of Helicobacter pylori plasticity zones reveals that they are integrating conjugative elements with intermediate integration specificity. BMC Genom. 15, 310. https://doi.org/10.1186/1471-2164-15-310 (2014).
Delahay, R. M., Croxall, N. J. & Stephens, A. D. Phylogeographic diversity and mosaicism of the Helicobacter pylori tfs integrative and conjugative elements. Mob. DNA 9, 5. https://doi.org/10.1186/s13100-018-0109-4 (2018).
Phuc, B. H. et al. Helicobacter pylori type 4 secretion systems as gastroduodenal disease markers. Sci. Rep. 11, 4584. https://doi.org/10.1038/s41598-021-83862-1 (2021).
Mucito-Varela, E., Castillo-Rojas, G., Calva, J. J. & Lopez-Vidal, Y. Integrative and conjugative elements of Helicobacter pylori are hypothetical virulence factors associated with gastric cancer. Front. Cell Infect. Microbiol. 10, 525335. https://doi.org/10.3389/fcimb.2020.525335 (2020).
Uchiyama, J. et al. Characterization of Helicobacter pylori bacteriophage KHP30. Appl. Environ. Microbiol. 79, 3176–3184. https://doi.org/10.1128/AEM.03530-12 (2013).
Binh, T. T. et al. Advanced non-cardia gastric cancer and Helicobacter pylori infection in Vietnam. Gut Pathog. 9, 46. https://doi.org/10.1186/s13099-017-0195-8 (2017).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. https://doi.org/10.1101/gr.186072.114 (2015).
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114. https://doi.org/10.1038/s41467-018-07641-9 (2018).
Grant, J. R. et al. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkad326 (2023).
Tomb, J. F. et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547. https://doi.org/10.1038/41483 (1997).
Uchiyama, J. et al. Complete genome sequences of two Helicobacter pylori bacteriophages isolated from Japanese patients. J. Virol. 86, 11400–11401. https://doi.org/10.1128/JVI.01767-12 (2012).
Isomoto, H., Moss, J. & Hirayama, T. Pleiotropic actions of Helicobacter pylori vacuolating cytotoxin, VacA. Tohoku J. Exp. Med. 220, 3–14. https://doi.org/10.1620/tjem.220.3 (2010).
Palframan, S. L., Kwok, T. & Gabriel, K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell Infect. Microbiol. 2, 92. https://doi.org/10.3389/fcimb.2012.00092 (2012).
Lee, J., Sands, Z. A. & Biggin, P. C. A numbering system for MFS transporter proteins. Front. Mol. Biosci. 3, 21. https://doi.org/10.3389/fmolb.2016.00021 (2016).
Bourzac, K. M., Botham, C. M. & Guillemin, K. Helicobacter pylori CagA induces AGS cell elongation through a cell retraction defect that is independent of Cdc42, Rac1, and Arp2/3. Infect. Immun. 75, 1203–1213. https://doi.org/10.1128/IAI.01702-06 (2007).
Backert, S., Moese, S., Selbach, M., Brinkmann, V. & Meyer, T. F. Phosphorylation of tyrosine 972 of the Helicobacter pylori CagA protein is essential for induction of a scattering phenotype in gastric epithelial cells. Mol. Microbiol. 42, 631–644. https://doi.org/10.1046/j.1365-2958.2001.02649.x (2001).
Segal, E. D., Cha, J., Lo, J., Falkow, S. & Tompkins, L. S. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 96, 14559–14564. https://doi.org/10.1073/pnas.96.25.14559 (1999).
Boonyanugomol, W. et al. Helicobacter pylori cag pathogenicity island (cagPAI) involved in bacterial internalization and IL-8 induced responses via NOD1- and MyD88-dependent mechanisms in human biliary epithelial cells. PLoS ONE 8, e77358. https://doi.org/10.1371/journal.pone.0077358 (2013).
Nguyen, L. T. et al. Clinical relevance of cagPAI intactness in Helicobacter pylori isolates from Vietnam. Eur. J. Clin. Microbiol. Infect. Dis. 29, 651–660. https://doi.org/10.1007/s10096-010-0909-z (2010).
Kawai, M. et al. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiol. 11, 104. https://doi.org/10.1186/1471-2180-11-104 (2011).
Moran, A. P. & Prendergast, M. M. Molecular mimicry in Campylobacter jejuni and Helicobacter pylori lipopolysaccharides: Contribution of gastrointestinal infections to autoimmunity. J. Autoimmun. 16, 241–256. https://doi.org/10.1006/jaut.2000.0490 (2001).
Koniger, V. et al. Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA. Nat. Microbiol. 2, 16188. https://doi.org/10.1038/nmicrobiol.2016.188 (2016).
Yamaoka, Y., Kwon, D. H. & Graham, D. Y. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 97, 7533–7538. https://doi.org/10.1073/pnas.130079797 (2000).
Fischer, W. et al. Strain-specific genes of Helicobacter pylori: Genome evolution driven by a novel type IV secretion system and genomic island transfer. Nucleic Acids Res. 38, 6089–6101. https://doi.org/10.1093/nar/gkq378 (2010).
Kersulyte, D. et al. Helicobacter pylori’s plasticity zones are novel transposable elements. PLoS ONE 4, e6859. https://doi.org/10.1371/journal.pone.0006859 (2009).
Vale, F. F. et al. Genomic structure and insertion sites of Helicobacter pylori prophages from various geographical origins. Sci. Rep. 7, 42471. https://doi.org/10.1038/srep42471 (2017).
Baltrus, D. A., Guillemin, K. & Phillips, P. C. Natural transformation increases the rate of adaptation in the human pathogen Helicobacter pylori. Evolution 62, 39–49. https://doi.org/10.1111/j.1558-5646.2007.00271.x (2008).
Oleastro, M. et al. Disease association with two Helicobacter pylori duplicate outer membrane protein genes, homB and homA. Gut Pathog. 1, 12. https://doi.org/10.1186/1757-4749-1-12 (2009).
Suzuki, M. et al. Interaction of CagA with Crk plays an important role in Helicobacter pylori-induced loss of gastric epithelial cell adhesion. J. Exp. Med. 202, 1235–1247. https://doi.org/10.1084/jem.20051027 (2005).
Backert, S. & Selbach, M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 10, 1573–1581. https://doi.org/10.1111/j.1462-5822.2008.01156.x (2008).
Loh, J. T. et al. Analysis of Helicobacter pylori cagA promoter elements required for salt-induced upregulation of CagA expression. Infect. Immun. 80, 3094–3106. https://doi.org/10.1128/IAI.00232-12 (2012).
Loh, J. T., Lin, A. S., Beckett, A. C., McClain, M. S. & Cover, T. L. Role of a stem-loop structure in Helicobacter pylori cagA transcript stability. Infect. Immun. https://doi.org/10.1128/IAI.00692-18 (2019).
Olbermann, P. et al. A global overview of the genetic and functional diversity in the Helicobacter pylori cag pathogenicity island. PLoS Genet. 6, e1001069. https://doi.org/10.1371/journal.pgen.1001069 (2010).
Silva, B. et al. The expression of Helicobacter pylori tfs plasticity zone cluster is regulated by pH and adherence, and its composition is associated with differential gastric IL-8 secretion. Helicobacter https://doi.org/10.1111/hel.12390 (2017).
Alandiyjany, M. N., Croxall, N. J., Grove, J. I. & Delahay, R. M. A role for the tfs3 ICE-encoded type IV secretion system in pro-inflammatory signalling by the Helicobacter pylori Ser/Thr kinase, CtkA. PLoS ONE 12, e0182144. https://doi.org/10.1371/journal.pone.0182144 (2017).
Tuan, V. P. et al. Genome-wide association study of gastric cancer- and duodenal ulcer-derived Helicobacter pylori strains reveals discriminatory genetic variations and novel oncoprotein candidates. Microb. Genom. https://doi.org/10.1099/mgen.0.000680 (2021).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595. https://doi.org/10.1371/journal.pcbi.1005595 (2017).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963. https://doi.org/10.1371/journal.pone.0112963 (2014).
Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. https://doi.org/10.1093/bioinformatics/btu153 (2014).
Page, A. J. et al. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. https://doi.org/10.1093/bioinformatics/btv421 (2015).
Falush, D. et al. Traces of human migrations in Helicobacter pylori populations. Science 299, 1582–1585. https://doi.org/10.1126/science.1080857 (2003).
Montano, V. et al. Worldwide population structure, long-term demography, and local adaptation of Helicobacter pylori. Genetics 200, 947–963. https://doi.org/10.1534/genetics.115.176404 (2015).
Yahara, K. et al. Chromosome painting in silico in a bacterial species reveals fine population structure. Mol. Biol. Evol. 30, 1454–1464. https://doi.org/10.1093/molbev/mst055 (2013).
Minh, B. Q., Hahn, M. W. & Lanfear, R. New methods to calculate concordance factors for phylogenomic datasets. Mol. Biol. Evol. 37, 2727–2733. https://doi.org/10.1093/molbev/msaa106 (2020).
Arndt, D. et al. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16-21. https://doi.org/10.1093/nar/gkw387 (2016).
Bertelli, C. et al. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. https://doi.org/10.1093/nar/gkx343 (2017).
Tshibangu-Kabamba, E. & Yamaoka, Y. Helicobacter pylori infection and antibiotic resistance—From biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. https://doi.org/10.1038/s41575-021-00449-x (2021).
Choi, I. J., Fujimoto, S., Yamauchi, K., Graham, D. Y. & Yamaoka, Y. Helicobacter pylori environmental interactions: Effect of acidic conditions on H. pylori-induced gastric mucosal interleukin-8 production. Cell Microbiol. 9, 2457–2469. https://doi.org/10.1111/j.1462-5822.2007.00973.x (2007).
Barranco, S. C. et al. Establishment and characterization of an in vitro model system for human adenocarcinoma of the stomach. Cancer Res. 43, 1703–1709 (1983).
Acknowledgements
We extend our sincere thanks to all colleagues in the Department of Environmental and Preventive Medicine, Oita University and the Faculty of Applied Technology, Van Lang University, Vietnam for contributing to this research paper. We also thank Jeremy Allen, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Funding
This research was funded by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (21K08010 to T.M., 21K07898 to J.A., 221S0002, 16H06279, 18KK0266, and 19H03473 to Y.Y.), the Special Coordination Funds for Promoting Science and Technology from the MEXT of Japan (Y.Y.), and a USA National Institutes of Health grant (DK62813 to Y.Y.). This work was also supported by the Okinawa Prefectural Government. B.H.P., K.A.F., B.S., R.I.A., E.T.K., and V.P.T are doctoral students supported by the Japanese Government: MEXT (Monbukagaku-sho) Scholarship Program for 2015 (V.P.T), 2016 (E.T.K.), 2017 (B.H.P., K.A.F.) and 2019 (B.S., R.I.A.).
Author information
Authors and Affiliations
Contributions
Conceptualization, B.H.P. and Y.Y.; methodology, B.H.P., J.A. and T.M.; software, B.H.P. and T.M.; conducting experiments, B.H.P, R.I.A., B.S. and J.A.; validation, B.H.P and J.A.; formal analysis, B.H.P. and J.A.; investigation, V.P.T., T.T.B., H.D.Q.D., N.P.M.T., T.D.T, P.H.T. and Y.Y.; resources, T.T.B., H.D.Q.D. and Y.Y.; data curation, B.H.P. and R.I.A.; writing—original draft preparation, B.H.P., K.A.F., E.T.K., J.A. and T.M.; writing—review and editing, B.H.P., K.A.F., E.T.K., R.I.A., J.A. and Y.Y.; visualization, B.H.P. and J.A.; supervision, Y.Y.; project administration, Y.Y.; funding acquisition, T.M., J.A. and Y.Y. All authors have read and agreed to the submitted version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Phuc, B.H., Tuan, V.P., Binh, T.T. et al. Comparative genomics of two Vietnamese Helicobacter pylori strains, CHC155 from a non-cardia gastric cancer patient and VN1291 from a duodenal ulcer patient. Sci Rep 13, 8869 (2023). https://doi.org/10.1038/s41598-023-35527-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-35527-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
