
Abstract
Stress is one of the critical facilitators for seizure induction in patients with epilepsy. However, the neural mechanisms underlying this facilitation remain poorly understood. Here, we investigated whether noradrenaline (NA) transmission enhanced by stress exposure facilitates the induction of medial prefrontal cortex (mPFC)-originated seizures. In mPFC slices, whole-cell current-clamp recordings revealed that bath application of picrotoxin induced sporadic epileptiform activities (EAs), which consisted of depolarization with bursts of action potentials in layer 5 pyramidal cells. Addition of NA dramatically shortened the latency and increased the number of EAs. Simultaneous whole-cell and field potential recordings revealed that the EAs are synchronous in the mPFC local circuit. Terazosin, but not atipamezole or timolol, inhibited EA facilitation, indicating the involvement of α1 adrenoceptors. Intra-mPFC picrotoxin infusion induced seizures in mice in vivo. Addition of NA substantially shortened the seizure latency, while co-infusion of terazosin into the mPFC inhibited the effect of NA. Finally, acute restraint stress shortened the latency of intra-mPFC picrotoxin infusion-induced seizures, whereas prior infusion of terazosin reversed this stress-induced shortening of seizure latency. Our findings suggest that stress facilitates the induction of mPFC-originated seizures via NA stimulation of α1 adrenoceptors.
Similar content being viewed by others


Introduction
Epilepsy is a chronic neurological disorder and affects approximately 1% of the world’s population. Excessive synchronous neuronal activities induce characteristic repetitive seizures. The loss of γ-amino butyric acid (GABA)-ergic neurons and abnormal GABA receptors are observed in animal models and patients with epilepsy1,2,3. Additionally, blockade of GABAA receptors induces epileptiform activity (EA) in in vivo and in vitro4,5. Therefore, GABAergic dysfunction plays a critical role in inducing the imbalance between excitation and inhibition, leading to epileptic seizures.
Stress is one of the frequently self-reported precipitants for seizure induction in patients with epilepsy6,7,8,9. Stress hormones, such as cortisol and corticosterone, have been reported to be involved in stress-induced epileptic seizures10,11,12. However, it remains unclear how stress triggers seizures. The locus coeruleus (LC) is the source of noradrenaline (NA) in the brain and sends efferent projections to numerous areas of the brain, including the neocortex, hippocampus, and amygdala13. Stress activates LC neurons, leading to increased NA release in these brain regions14,15,16,17,18,19. The effect of NA on epileptic seizures has been mainly studied in the limbic system, such as the hippocampus and amygdala that are known for frequent seizure origin. NA is generally considered to have an anticonvulsive effect in those regions20. In contrast, the effect of NA on frontal lobe epilepsy (FLE) has not been well studied, even though FLE is the second most common type of refractory focal seizure after temporal lobe epilepsy (TLE)21. The ventromedial prefrontal cortex (vmPFC) is also a known focus of pharmacoresistant epilepsy22. Stress enhances NA release in the medial prefrontal cortex (mPFC) in rodents15,16,17, which corresponds to the caudal region of the vmPFC in humans23. In addition, we and several other researchers have shown that NA excites mPFC layer 5 (L5) pyramidal cells via α1 and β adrenoceptors in rodents24,25,26. Thus, it is hypothesized that the excitatory effect of NA may contribute to the induction of abnormal, excessive synchronous neuronal activities in epileptic seizures. Therefore, in this study, we investigated whether increased NA levels in the mPFC are involved in the induction of epileptic seizures caused by stress exposure using behavioral and electrophysiological approaches.
Methods
Animals
Male and female C57BL/6J mice (4–8 weeks of age, n = 55) were used for electrophysiological experiments, and male C57BL/6J mice (6–8 weeks of age, n = 71) were used for behavioral experiments. Mice were maintained at a constant ambient temperature (22 ± 1 °C) and under a 12-h light/dark cycle, with food and water available ad libitum as described previously26,27,28. All experiments were conducted in accordance with the National Institutes of Health guidelines and the ARRIVE guidelines and performed with the approval of the Institutional Animal Care and Use Committee at Kanazawa University. All efforts were made to minimize the suffering and number of animals used in this study.
Drugs
In electrophysiological experiments, l-noradrenaline bitartrate monohydrate (NA; Tokyo Chemical Industry, Tokyo, Japan), CNQX (Alomone Labs, Jerusalem, Israel), dl-AP-5 (AP-5; Tocris Bioscience, Bristol, UK), terazosin hydrochloride (Sigma, St. Louis, MO, USA), atipamezole hydrochloride (Wako, Osaka, Japan), timolol maleate (Sigma), and phenylephrine hydrochloride (Wako) were dissolved in distilled water. Picrotoxin (Sigma) was dissolved in dimethyl sulfoxide. These drug solutions were diluted with standard Ringer’s solution before use. Kynurenic acid hydrate (KYNA; Tokyo Chemical Industry) was dissolved in standard Ringer’s solution. In behavioral experiments, picrotoxin, NA, and terazosin (Tocris) were dissolved in phosphate-buffered saline (PBS). Picrotoxin injection into the cortex has been reported to elicit seizures29. The doses of drugs were chosen based on the previous studies26,30,31,32.
Slice preparation and electrophysiology
Procedures for preparing mPFC slices and electrophysiological recordings were similar to our previous studies26,33,34. Mice were anesthetized with isoflurane and decapitated. The brains were then dissected and submerged in ice-cold modified Ringer’s solution (containing the following [in mM]: choline chloride, 125; KCl, 4.0; NaH2PO4, 1.25; MgCl2, 7.0; CaCl2, 0.5; NaHCO3, 26; glucose, 20; ascorbate, 1.0; and pyruvate, 3.0) and bubbled with 95% O2/5% CO2 (pH 7.4). Coronal slices (250 μm thick) of the mPFC were cut using a microslicer (VT1200S; Leica, Wetzlar, Germany) and incubated at 32–34 °C for 15–30 min in standard Ringer’s solution (containing the following [in mM]: NaCl, 125; KCl, 2.5; NaH2PO4, 1.25; MgCl2, 0.9; CaCl2, 2.0; NaHCO3, 26; and glucose, 25) and bubbled with 95% O2/5% CO2 (pH 7.4). Next, the slices were transferred to standard Ringer’s solution at room temperature and then mounted in a recording chamber on an upright microscope (BX-51WI; Olympus, Tokyo, Japan). The chamber was continuously superfused with standard Ringer’s solution at a flow rate of 2–2.5 mL/min. In some experiments, mPFC-isolated slices were prepared by trimming the coronal slices.
Whole-cell patch-clamp recordings were obtained from mPFC L5 pyramidal cells by visually controlling patch pipettes, which were prepared from borosilicate glass capillaries and filled with an internal solution. For whole-cell voltage- and current-clamp recordings, we used internal solutions containing (in mM) CsOH, 150; CsCl, 5.0; MgCl2, 2.0; Na2ATP, 4.0; Na3GTP, 0.3; EGTA, 10; HEPES, 10; QX-314, 3.0 (pH 7.3 with gluconic acid) and KOH, 150; KCl, 10; MgCl2, 2.0; Na2ATP, 2.0; Na3GTP, 0.3; EGTA, 0.2; HEPES, 10; spermine, 0.1 (pH 7.3 with gluconic acid), respectively. In order to identify and stain the recorded neurons, Alexa Fluor 594 (0.02 mM; Thermo Fisher Scientific, Waltham, MO, USA) and biocytin (1–3 mg/mL; Sigma) were dissolved in the corresponding internal solution. The resistance of the electrodes was 3–9 MΩ in Ringer’s solution. All recordings were performed at 32–34 °C.
For recording evoked inhibitory postsynaptic currents (eIPSCs) using the whole-cell voltage-clamp technique, the membrane potentials were held at + 10 mV in the presence of a glutamate receptor antagonist KYNA (2 mM), and electrical stimulations were applied as cathodal square-wave pulses of 200-μs duration with an intensity of up to 30 μA using a glass electrode that was located in layer 2/3 of the mPFC and filled with normal Ringer’s solution. The eIPSC amplitudes were measured in periods of 0–30 s before the end of the drug applications. The picrotoxin effects were evaluated by comparing the average values of these periods.
For whole-cell current-clamp recordings, membrane potentials were recorded without holding current injection. The generation of EAs, defined as prolonged depolarization accompanied with three or more burst firings, was recorded for 15 min (time 0 = start of drug application). EA latency was measured as the time of the induction of the first EA after drug application. If EA was not induced within 15 min, the latency was calculated as 15 min. The duration and number of burst firings of an EA were measured on the first EA in periods of 0–5 min and 10–15 min after drug application (picrotoxin, picrotoxin + NA, or picrotoxin + phenylephrine). In pharmacological experiments, EA frequency was measured in periods of 0–3 min before and 2–5 min after the application of antagonists. In some experiments, whole-cell current-clamp and field potential recordings were obtained simultaneously. The field potential recordings were obtained using a glass electrode placed in the mPFC L5 and containing 2.5 M NaCl with a resistance of 1–3 MΩ in Ringer’s solution.
Data were amplified with a Multiclamp 700B (Molecular Devices, Foster City, CA, USA) and low-pass-filtered at 3 kHz. Additionally, field potential data were high-pass-filtered at 0.1 Hz. Voltage-clamp, current-clamp, and simultaneous recording data were then digitized at 20, 6, and 10 kHz with an A/D interface (Digidata 1440A; Molecular Devices), respectively, and stored on a computer using a Clampfit 10.5 (Molecular Devices). Field potential data were further low-pass-filtered at 30 Hz with the Clampfit 10.5. The correlation between EAs and field potentials was evaluated by cross-correlation analysis. The cross-correlogram was constructed using onset time points of the first peaks of EAs and the peaks of negative shifts of field potentials. The calculation was based on the previously described methods35 and performed using the ‘scipy.signal.correlate’ function in the SciPy library in Python. A bin width of 300 ms was used.
Histochemistry
Histochemical examinations of recorded neurons were implemented as described previously26,33,34. After the recordings, the slices were fixed overnight in 4% paraformaldehyde in 0.1 M PBS (pH 7.4) at 4 °C. After three rinses with 0.05 M PBS, the slices were then incubated in 0.6% H2O2 in methanol for 30 min at room temperature to eliminate endogenous peroxidase activity, followed by 3 h of incubation in an avidin–biotin-peroxidase complex using a R.T.U. ABC Reagent (Vector laboratories, Burlingame, CA, USA) diluted fourfold in PBS containing 0.3% Triton X-100. The slices were then rinsed in 0.05 M Tris–HCl (pH 7.5) and incubated in 0.05% 3,3′-diaminobenzidine (DAB; Nacalai Tesque, Kyoto, Japan) solution. Next, the biocytin-filled cells were stained brown, and the reaction was stopped by rinsing with 0.05 M PBS. The biocytin-labeled cells were identified under a microscope (BZ-9000; Keyence, Osaka, Japan). L5 pyramidal cells were identified according to the following characteristics: the depth of the cells from the midline (350–550 μm) and shapes of the soma and apical dendrites.
Surgery and intra-mPFC infusion
Intra-mPFC infusions were performed as previously described with slight modification36,37,38. Mice were anesthetized with chloral hydrate (400 mg/kg, i.p.) and implanted with 25-gauge stainless-steel guide cannulae (o.d., 0.51 mm; i.d., 0.26 mm) above the mPFC (1.8 mm rostral, ± 1.3 mm lateral, − 1.5 mm ventral to bregma, at a 20° angle from the vertical axis in the mediolateral plane; bilateral)39. After surgery, the mice were housed individually and allowed to recover for at least 5 days. For intra-mPFC infusions, 33-gauge stainless-steel infusion cannulae (o.d., 0.2 mm; i.d., 0.08 mm) were inserted bilaterally into the guide cannula, with the infusion cannulae protruded 1.1 mm from the tip of the guide cannula to reach the mPFC. The drugs (or vehicle) were administered bilaterally in a volume of 0.2 μL/side and at a rate of 0.2 μL/min. After the infusion, the infusion cannulae were kept in place for an additional 1 min after intra-mPFC infusion to prevent backflow.
Behavioral seizure observation
All mice were handled for 2 consecutive days, followed by habituation to a transparent testing chamber (32 cm × 22 cm × 13.5 cm; Natsume Seisakusho, Tokyo, Japan) for 30 min. On day 1, each mouse was administered a bilateral injection of picrotoxin (0.1 nmol/side) into the mPFC, placed in the testing chamber for 1800s (30 min), and videotaped. On day 2, the effects of intra-mPFC infusion of NA were tested in each mouse by administering a bilateral injection of either picrotoxin (0.1 nmol/side) only, picrotoxin + terazosin (picrotoxin: 0.1 nmol/side, terazosin: 7.5 nmol/side), picrotoxin + NA (picrotoxin: 0.1 nmol/side, NA: 10 nmol/side), or picrotoxin + NA + terazosin (picrotoxin: 0.1 nmol/side, NA: 10 nmol/side, terazosin: 7.5 nmol/side) into the mPFC. The mice were then immediately placed in the testing chamber for 1800s and videotaped. On day 2, the effects of acute restraint stress were tested in each mouse by administering a bilateral injection of either vehicle (PBS) or terazosin (7.5 nmol/side) into the mPFC, followed by 20 min immobilization using a plastic bag (DecapiCones; Braintree Scientific, Braintree, MA, USA). Immediately after the restraint stress, each mouse was administered a bilateral injection of picrotoxin (0.1 nmol/side) into the mPFC, placed in the testing chamber for 1800s, and videotaped.
Behavioral seizures were classified on the basis of the modified Racine scale40,41: stage 1, absence‐like immobility; stage 2, hunching with facial or manual automatisms; stage 3, rearing with facial or manual automatisms and forelimb clonus; stage 4, repeated rearing with continuous forelimb clonus and falling; and stage 5, generalized tonic–clonic convulsions with lateral recumbence or jumping and wild running followed by generalized convulsions. Stage 1 and 2 were reported as non‐convulsive seizures with no clear motor component, whereas stage 3 and above were convulsive motor seizures41. Thus, we measured the latency of stage 3 seizures.
Histology
After the behavioral tests, histological analyses were performed to confirm the infusion sites in the mPFC using the methods reported in our previous studies26,27,36,37,38. Briefly, the mice were decapitated, and the brains were rapidly dissected and frozen in powdered dry ice. Coronal Sects. (50 μm thick) were prepared on a cryostat, thaw-mounted onto slides, stained with thionin, and examined under a microscope (BZ-9000). Data from mice with incorrect infusion placements (n = 22) were excluded from statistical analyses.
Statistical analyses
Data are expressed as mean ± standard error of the mean. The data were compared using Student’s t-test or paired t-test when comparing two groups, one-way repeated measures ANOVA with post hoc Holm–Sidak’s multiple comparison test or two-way repeated measures ANOVA with post hoc Bonferroni’s multiple comparison test when comparing more than two groups. All analyses were performed using Prism 6 (GraphPad Software, La Jolla, CA, USA). We also confirmed that all experiments conducted in this study had adequate statistical power (greater than 0.9) using G*Power software 3.142. Statistical significance was set at P value < 0.05.
Results
NA promotes induction of EAs in mPFC L5 pyramidal cells under the suppression of GABAA receptor-mediated inhibition in vitro
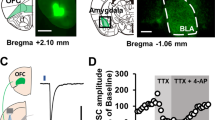
We first examined the effects of picrotoxin, a GABAA receptor antagonist, on eIPSCs in mPFC L5 pyramidal cells using in vitro whole-cell current clamp recordings (Fig. 1a). Bath application of picrotoxin (3, 10, and 30 µM) reduced eIPSC amplitude in a concentration-dependent manner, with 30 µM picrotoxin demonstrating almost complete inhibition of eIPSC (control: 340.8 ± 35.7 pA; 3 µM: 135.7 ± 21.6 pA; 10 µM: 39.1 ± 8.2 pA; and 30 µM: 5.2 ± 3.3 pA; control vs. 3 µM, P = 0.0035; control vs. 10 µM, P = 0.0034; and control vs. 30 µM, P = 0.0035, n = 5, one-way repeated measures ANOVA with post hoc Holm–Sidak’s test, Fig. 1b,c). Thus, we used 30 µM picrotoxin to reproduce an epileptiform state5,43 in the following experiments.

Noradrenaline (NA) promotes the induction of epileptiform activity (EA) in layer 5 pyramidal cells of the medial prefrontal cortex under the suppression of inhibitory synaptic transmission. (a) Representative micrograph of a recorded pyramidal cell. (b) Representative traces showing the effects of picrotoxin (Pic) on evoked inhibitory postsynaptic currents (eIPSCs). (c) Summary graph showing the effects of Pic on eIPSCs (n = 5 from 4 mice). **P < 0.01 (one-way ANOVA with post hoc Holm–Sidak’s test). (d, f) Representative traces of membrane potentials after the application of Pic (d) and Pic + NA (f). (e, g, h, i) Expanded traces of a mark in d (e), marks at short (g) and long (i) times after the application of NA in f, and a mark in g (h). (j, k) Summary graphs of EA latency (j) and number of EAs (k) after the application of Pic (n = 6 from 4 mice) or Pic + NA (n = 7 from 7 mice). (l, m) Summary graphs of changes in duration (l) and number of burst firings (m) in EAs after the application of NA (n = 6 from 6 mice). *P < 0.05, **P < 0.01, ****P < 0.0001 (Student’s t-test or paired t-test). Data are expressed as mean ± standard error of the mean.
Bath application of picrotoxin induced an EA, which consisted of a prolonged depolarization, the so-called paroxysmal depolarization shift (PDS)44, accompanied with more than 3 burst firings in 1 of 6 cells tested (Fig. 1d,e). The first EA was observed at least 7 min after the application of picrotoxin. The number of EAs generated during the 15 min recording (time 0 = start of picrotoxin application) was 1. EA was not induced during the 15 min recording in the remaining 5 cells. Picrotoxin did not significantly affect resting membrane potentials (control, − 73.15 ± 1.06 mV vs. picrotoxin, − 73.06 ± 1.10 mV, n = 7, t6 = 0.3192, P = 0.7604, paired t-test). These findings indicate that picrotoxin application alone induces EA quite sporadically.
Next, we examined the effects of NA on the generation of EA. Simultaneous bath application of picrotoxin + NA (10 µM) immediately induced depolarization (control, − 73.15 ± 1.06 mV vs. picrotoxin + NA, − 71.09 ± 1.25 mV, n = 7, t6 = 3.543, P = 0.0122, paired t-test) with/without firings as previously reported26. Soon after the depolarization, EAs were rapidly induced with a latency of 1.6 ± 0.5 min (n = 7), which was significantly shorter than that of the EAs observed in the presence of picrotoxin alone (14.2 ± 0.84 min, n = 6; picrotoxin vs. picrotoxin + NA, t11 = 13.80, P < 0.0001, Student’s t-test, Fig. 1f,j). The number of EAs after picrotoxin + NA (14.0 ± 1.9) application was significantly larger than that after picrotoxin alone (0.167 ± 0.17; picrotoxin vs. picrotoxin + NA, t11 = 6.760, P < 0.0001, Student’s t-test, Fig. 1f,k). Additionally, the duration of PDS and number of burst firings in an EA gradually increased over time (duration: 65.65 ± 14.4 ms at 0–5 min after picrotoxin + NA application vs. 1209.80 ± 207.2 ms at 10–15 min after picrotoxin + NA application, n = 6, t5 = 5.469, P = 0.0028, Fig. 1g,h,i,l; number of burst firings: 7.2 ± 1.6 at 0–5 min after picrotoxin + NA application vs. 70.7 ± 15.1 at 10–15 min after picrotoxin + NA application, t5 = 3.975, P = 0.0106, Fig. 1g,h,i,m). These findings demonstrate that NA dramatically promotes the induction and enhances the strength of EAs in mPFC L5 pyramidal cells under suppressed inhibitory synaptic transmission.
EAs are induced synchronously via glutamatergic transmission in the mPFC local circuit
Using simultaneous whole-cell current-clamp and field potential recordings, we next investigated whether EAs are synchronous. Clear population activities were not observed shortly after the application of picrotoxin + NA, which is the time at which small EAs were generated as described above. However, several minutes later, gradual augmentation of population activities and emergence of clear negative potentials, which were time-locked to enlarged EAs, were observed (n = 6, Fig. 2a,b,c). To investigate the temporal relationship between the onset times of EAs and negative shifts in the field potentials more clearly, we conducted cross-correlation analysis. The highest peak was detected at time 0 with a bin width of 300 ms (Fig. 2d), indicating that EAs and negative shifts of field potentials occurred within 300 ms. These results indicate that EAs are synchronous in the mPFC.

Epileptiform activities are induced synchronously in the medial prefrontal cortex. (a) Representative traces of membrane potentials and population spikes from simultaneous whole-cell and field potential recordings after the application of picrotoxin (Pic) + noradrenaline (NA). (inset) Schematic image of whole-cell and field potential recordings. (b, c) Expanded traces of marks at short (b) and long (c) times after the application of Pic + NA in (a). (d) Cross-correlogram from the traces in (a) (bin width = 300 ms).
We next addressed whether EAs are induced in the mPFC local circuits using mPFC-isolated slices (Fig. 3a). Whole-cell current-clamp recordings revealed that EAs were still induced in these slices after picrotoxin + NA application (6 of 6 cells; Fig. 3b), indicating that mPFC local circuits are sufficient for inducing EAs.

Epileptiform activities are induced in medial prefrontal cortex (mPFC)-isolated slices. (a) Representative image of whole-cell current-clamp recording of an mPFC-isolated slice. Rec, recording electrode. (b) Representative trace of membrane potentials from whole-cell current-clamp recording in an mPFC-isolated slice after the application of picrotoxin (Pic) + noradrenaline (NA) (n = 6 from 6 mice).
Previous studies reported that NA enhances glutamatergic transmission in mPFC L5 pyramidal cells25,26. Because EAs were induced synchronously in the mPFC local circuit, we hypothesized that the mutual activation of mPFC pyramidal cells through glutamatergic transmission may contribute to the NA-induced facilitation of EAs. Addition of CNQX (10 µM), an AMPA receptor antagonist, to picrotoxin + NA completely suppressed the generation of EAs (picrotoxin + NA, 1.8 ± 0.3 events/min vs. picrotoxin + NA + CNQX, 0.0 ± 0.0 events/min, n = 6, t5 = 5.219, P = 0.0034, paired t-test, supplementary Fig. S1a,b). Similarly, the addition of AP-5 (50 µM), an NMDA receptor antagonist, also significantly reduced EA generation (picrotoxin + NA, 1.8 ± 0.4 events/min vs. picrotoxin + NA + AP-5, 0.3 ± 0.2 events/min, n = 8, t7 = 4.549, P = 0.0026, paired t-test, supplementary Fig. S1c,d). These results reveal that both AMPA and NMDA receptors play a critical role in NA-induced promotion of EAs.
Alpha 1 adrenoceptors mediate NA-induced facilitation of EAs
We next investigated the adrenoceptor subtypes that are involved in the NA-induced facilitation of EAs using terazosin (5 µM), atipamezole (3 µM), and timolol (10 µM), which are α1, α2, and β adrenoceptor antagonists, respectively. Addition of terazosin, but not atipamezole or timolol, to picrotoxin + NA significantly reduced the frequency of EAs (terazosin: picrotoxin + NA, 2.0 ± 0.5 events/min vs. picrotoxin + NA + terazosin, 0.4 ± 0.2 events/min, n = 7, t6 = 3.767, P = 0.0093, paired t-test, Fig. 4a,b; atipamezole: picrotoxin + NA, 1.6 ± 0.3 events/min vs. picrotoxin + NA + atipamezole, 1.6 ± 0.3 events/min, n = 7, t6 = 0.3536, P = 0.7358, paired t-test, Fig. 4c,d; and timolol: picrotoxin + NA, 2.2 ± 0.3 events/min vs. picrotoxin + NA + timolol, 1.9 ± 0.4 events/min, n = 6, t5 = 1.348, P = 0.2354, paired t-test, Fig. 4e,f). Moreover, we found that the addition of phenylephrine (100 µM), an α1 adrenoceptor agonist, to picrotoxin markedly shortened EA latency and increased the number of EAs (latency: picrotoxin, 18.3 ± 1.7 min, n = 6, vs. picrotoxin + phenylephrine, 1.4 ± 0.3 min, n = 5, t9 = 9.047, P < 0.0001, Student’s t-test; number of EAs: picrotoxin, 0.2 ± 0.2/15 min, vs. picrotoxin + phenylephrine, 17.2 ± 1.6/15 min, t9 = 11.52, P < 0.0001, Student’s t-test, Fig. 5). These findings suggest that NA promotes the induction of EAs through the activation of α1 adrenoceptors in the mPFC.

Blockade of α1 adrenoceptors inhibits noradrenaline (NA)-induced epileptiform activities (EAs). (a, c, e) Representative traces of membrane potentials before and after the addition of terazosin (Ter, a), atipamezole (Ati, c), and timolol (Tim, e) to Pic + NA. (b, d, f) Summary graphs showing the effects of Ter (b, n = 7 from 7 mice), Ati (d, n = 7 from 6 mice), and Tim (f, n = 6 from 5 mice) on EA frequency. **P < 0.01 (paired t-test). Data are expressed as mean ± standard error of the mean.

Stimulation of α1 adrenoceptors promotes the induction of epileptiform activities in layer 5 pyramidal cells of the medial prefrontal cortex under the suppression of inhibitory synaptic transmission. Representative trace of membrane potentials after the application of picrotoxin (Pic) + phenylephrine (Phe).
NA promotes seizure induction via α1 adrenoceptors in the mPFC in vivo
We next investigated whether NA promotes the induction of seizures via α1 adrenoceptors in the mPFC. Bilateral intra-mPFC infusion of picrotoxin (0.1 nmol /side) on day 1 induced stage 3 seizures in 28 out of 35 mice within 30 min (Fig. 6a), indicating that the disinhibition of the mPFC is sufficient to induce focal mPFC seizures. On day 2, the mice were randomly divided into four groups and each group received an injection of the following drugs into the mPFC: picrotoxin only, picrotoxin + NA, picrotoxin + NA + terazosin, and picrotoxin + terazosin (Fig. 6a,b). Two-way repeated measures ANOVA revealed a significant interaction (F3, 31 = 5.719, P = 0.0031, Fig. 6b). The latency of stage 3 seizures did not show any significant change between day 1 and 2 in the picrotoxin only-injected group (day 1, 19.25 ± 1.88 min vs. day 2, 23.70 ± 1.92 min, P = 0.290, n = 12; post hoc Bonferroni's test, Fig. 6b). In contrast, intra-mPFC infusion of picrotoxin + NA on day 2 significantly shortened the latency of seizure induction when compared to picrotoxin only infusion on day 1 (picrotoxin only, day 1, 21.84 ± 2.60 min vs. picrotoxin + NA, day 2, 13.65 ± 1.76 min, P = 0.0230, n = 9, post hoc Bonferroni's test, Fig. 6b). Additionally, the latency of seizure induction in the picrotoxin + NA + terazosin-injected group on day 2 was not significantly different from that in the picrotoxin only group on day 1 (picrotoxin only, day 1, 17.76 ± 2.30 min vs. picrotoxin + NA + terazosin, day 2, 25.02 ± 2.27 min, P = 0.109, n = 7, post hoc Bonferroni's test, Fig. 6b). Similarly, the latency of seizure induction in the picrotoxin + terazosin-injected group on day 2 was not significantly different from that in the picrotoxin only-injected group on day 1 (picrotoxin only, day 1, 18.85 ± 2.33 min vs. picrotoxin + terazosin, day 2, 20.08 ± 2.98 min, P > 0.9999, n = 7, post hoc Bonferroni's test, Fig. 6b). Furthermore, on day 2, the picrotoxin + NA-injected group exhibited significantly shorter latency than the picrotoxin only-injected group (picrotoxin, 23.70 ± 1.92 min, n = 12 vs. picrotoxin + NA, 13.65 ± 1.76 min, n = 9, P = 0.0060, post hoc Bonferroni's test, Fig. 6b). This effect of NA was blocked by co-infusion of terazosin (picrotoxin + NA, day 2, 13.65 ± 1.76 min, n = 9 vs. picrotoxin + NA + terazosin, day 2, 25.02 ± 2.27 min, n = 7, P = 0.0067; post hoc Bonferroni's test, Fig. 6b). These results show that NA promotes the induction of intra-mPFC picrotoxin-induced seizures via α1 adrenoceptors in the mPFC.

Noradrenaline (NA) promotes the induction of seizures by stimulating α1 adrenoceptors in vivo. (a) Experimental timeline, placements of infusion cannula tips, and representative micrograph: picrotoxin (Pic, light gray circles, n = 12), picrotoxin + noradrenaline (Pic + NA, red circles, n = 9), picrotoxin + noradrenaline + terazosin (Pic + NA + Ter, blue circles, n = 7), and picrotoxin + terazosin (Pic + Ter, dark gray circles, n = 8). Numbers indicate the approximate anterior–posterior distance (mm) from the bregma. (b) Latency of stage 3 seizures. Data are expressed as mean ± standard error of the mean. *P < 0.05, ##P < 0.01 (two-way repeated measures ANOVA with post hoc Bonferroni's test). NS, not significant.
Restraint stress promotes the induction of seizures via α1 adrenoceptors in the mPFC
Because previous studies demonstrated that NA levels in the mPFC increased during restraint stress exposure14,15,16,17, we finally examined whether acute restraint stress promotes the induction of seizures via α1 adrenoceptor stimulation in the mPFC. Mice received bilateral intra-mPFC infusion of picrotoxin on day 1. On day 2, the mice were randomly divided into two groups. One group received intra-mPFC infusion of vehicle, followed by a 20-min restraint stress and then intra-mPFC picrotoxin infusion (Fig. 7a). The second group received similar treatment, with the exception of intra-mPFC infusion of terazosin instead of vehicle (Fig. 7a). Two-way repeated measures ANOVA revealed a significant interaction (F1, 12 = 9.851, P = 0.0086, Fig. 7b). The latency of seizure induction in the picrotoxin + restraint stress + vehicle group on day 2 was significantly shorter than that on day 1 (day 1, 17.99 ± 2.13 min vs. day 2, 10.84 ± 1.00 min, P = 0.0450, n = 7, post hoc Bonferroni’s test, Fig. 7b). The latency of seizures in the picrotoxin + restraint stress + terazosin group was not significantly different between day 1 and 2 (day 1, 15.70 ± 2.75 min vs. day 2, 20.67 ± 3.79 min, P = 0.187, n = 7, post hoc Bonferroni's test, Fig. 7b). However, it should be noted that the shortened latency induced by restraint stress was reversed by intra-mPFC infusion of terazosin on day 2 (picrotoxin + restraint stress + vehicle, 10.84 ± 1.00 min, n = 7 vs. picrotoxin + restraint stress + terazosin, 20.67 ± 3.79 min, n = 7, P = 0.0276, post hoc Bonferroni’s test, Fig. 7b). These results indicate that restraint stress promotes the induction of mPFC-originated seizures through the activation of α1 adrenoceptors in the mPFC.

Restraint stress (RS) promotes the induction of seizures by stimulating α1 adrenoceptors in vivo. (a) Experimental timeline and placements of the infusion cannula tips: picrotoxin + restraint stress + vehicle (Pic + RS + Veh, red circles, n = 9) and picrotoxin + restraint stress + terazosin (Pic + RS + Ter, blue circles, n = 7). Numbers indicate the approximate anterior–posterior distance (mm) from the bregma. (b) Latency of stage 3 seizures. Data are expressed as mean ± standard error of the mean. *P < 0.05, #P < 0.05 (two-way repeated measures ANOVA with post hoc Bonferroni's test). NS, not significant.
Discussion
The main findings of the present study were as follows: (1) bath application of NA promotes EA induction in mPFC L5 pyramidal cells via α1 but not α2 or β adrenoceptors; (2) NA-facilitated EAs are synchronously induced in the mPFC local circuit; (3) latency of intra-mPFC picrotoxin-induced seizures are shortened by co-infusion of NA, which was recovered by α1 adrenoceptor antagonism; and (4) acute restraint stress exposure shortens the latency of intra-mPFC picrotoxin-induced seizures via α1 adrenoceptors. All these findings suggest that the activation of α1 adrenoceptors in the mPFC by NA may contribute to the generation of stress-induced seizures in patients with epilepsy.
Electrophysiological recordings revealed that NA promotes EA induction in mPFC L5 pyramidal cells via α1 adrenoceptor stimulation. This result is in contrast to the finding that α1 adrenoceptor stimulation suppressed EA in hippocampal slices45. Alpha1 adrenoceptors are expressed in not only glutamatergic pyramidal cells but also GABAergic interneurons of the mPFC46. The stimulation of α1 adrenoceptors depolarizes GABAergic interneurons, leading to an enhanced GABAergic transmission onto mPFC pyramidal cells47. Considering that GABAA receptors were inhibited by picrotoxin in our experimental condition, we did not observe the effect of augmented GABAergic transmission caused by α1 adrenoceptor stimulation. However, we have previously reported that bath application of NA induces depolarization and increases EPSCs in mPFC L5 pyramidal cells in normal ACSF26, indicating that α1 adrenoceptor stimulation in the mPFC local circuit may have an excitatory net effect even if GABAergic transmission was augmented by NA. In the hippocampal CA1, the stimulation of postsynaptic α1A adrenoceptors increases firing activity of GABAergic interneurons, leading to an increased IPSC frequency in pyramidal neurons45. This effect of NA may suppress TLE seizures45. It has been reported that NA also facilitates IPSCs via stimulating α1A adrenoceptors expressed at presynaptic terminals of GABAergic neurons in the basolateral amygdala48. However, this effect of NA is impaired by chronic stress exposure, suggesting that stress may potentiate the induction of seizures originated from the basolateral amygdala48. Thus, these findings suggest that the effects of α1 adrenoceptor stimulation on local network activity depend on the brain regions and physiological/pathophysiological conditions. Nevertheless, in the absence of GABAergic function in FLE patients, the stimulation of α1 adrenoceptors may facilitate hyperexcitability in the mPFC local circuit.
Glutamatergic transmission has been reported to be crucial for inducing EAs49,50,51. Consistent with this, our pharmacological examinations revealed that CNQX as well as AP-5 suppressed EAs in the mPFC slices, indicating the critical role of both AMPA and NMDA receptors in EA induction. Although NMDA receptors are blocked by Mg2+ at resting membrane potential, this block is removed when the membrane is depolarized. Because NA induces depolarization in mPFC L5 pyramidal cells26, NMDA receptor-mediated excitation accompanied with AMPA receptor-mediated transmission may contribute to the facilitation of EA induction. This is supported by a previous finding showing that the hyperactivation of NMDA receptors with free extracellular Mg2+ induces EA in rat hippocampal slices52.
Simultaneous whole-cell and field potential recordings revealed that individual EAs caused by bath application of picrotoxin + NA were time-locked to individual large field potential changes, which corresponded to population spikes, indicating the synchronism of the EAs. Additionally, EAs were observed in the mPFC-isolated slices that did not include other brain regions, suggesting that mPFC local circuitry is sufficient to induce EA. Because the mPFC contains recurrent excitatory connections53, activation of pyramidal cells by NA could trigger synchronous neuronal activities under the suppression of GABAA receptor-mediated inhibition.
Systemic administration of convulsants, such as picrotoxin, pentylenetetrazol, pilocarpine, and kainic acid (KA), has frequently been used to generate animal models of seizures54,55,56,57. Additionally, the local infusion of KA into the hippocampus and amygdala, which are two major focal seizure sites, is used to generate TLE models58,59. However, there are few studies involving FLE models. A previous report showed that repeated intra-PFC infusion of bicuculline, a competitive GABAA receptor antagonist, in adolescent rats induced focal EAs60. In contrast, the present study demonstrated that single intra-mPFC infusion of picrotoxin induced stage 3 seizures in most animals examined (39 out of 49 mice). However, it should be noted that we might have missed the occurrence of stages 1–2 seizures because of the paucity of observable behavior changes in stages 1–2 of the Racine scale40,41. Thus, single intra-mPFC picrotoxin infusion-induced seizures could be a simple and useful model of FLE. Although the local circuit activity is a primarily important for the mPFC-originated seizures, other brain regions might also be involved in seizure induction. The mPFC is known to project to and receive excitatory inputs from the thalamus that is one of the critical brain regions associated with epilepsy61. Thus, it is likely that abnormal excitability generated in the mPFC via stress exposure might be transmitted to this brain region, leading to the generation of epileptic seizures.
We found that the latency of stage 3 seizures induced by intra-mPFC picrotoxin + NA infusion was considerably shorter than that induced by intra-mPFC picrotoxin infusion alone. Furthermore, this shortened latency was reversed by intra-mPFC co-infusion of terazosin, indicating the involvement of α1 adrenoceptor stimulation in the shortening of the latency. Considering the in vitro electrophysiological data, these effects of NA and terazosin infusion on seizure latency may be attributed to the facilitation of EA generation and EA suppression by NA and terazosin, respectively, in the mPFC.
Previous studies have demonstrated the involvement of α1 adrenoceptors in pro- and anti-convulsant effects of NA. For example, transgenic mice with overexpression of α1B adrenoceptors exhibited increased seizures62, whereas α1A adrenoceptor agonist inhibited seizures in rat models63. Accordingly, it is predicted that α1B adrenoceptors might have played a critical role for facilitating the seizure induction observed in the present study. However, since the function and expression of each α1 adrenoceptor subtype may be different in distinct brain regions45,48, further studies are required to elucidate the α1 adrenoceptor subtype(s) that are critical for facilitating the induction of mPFC-originated seizures.
Previous studies have reported that stress induces seizures6,7,8,9. Consistent with these findings, we demonstrated that restraint stress shortened the latency of seizure induction caused by intra-mPFC picrotoxin infusion. To our knowledge, this is the first study demonstrating that single acute restraint stress exposure affects the induction of mPFC-originated seizures. However, one study has reported that repeated restraint stress increased seizure susceptibility via the hippocampus64. Stress activates LC neurons, leading to increased NA release in seizure foci, such as the mPFC, hippocampus, and amygdala14,15,16,17,18,19. Although we did not measure the level of NA in the mPFC during the restraint stress exposure in the present study, previous studies in rats demonstrated that NA levels in the mPFC increased to approximately 150% from baseline levels during restraint stress and this increase persisted for about 30 min14,15,16,17, suggesting the critical role of NA for seizure facilitation. Consistent with this, we found that intra-mPFC terazosin infusion before stress exposure suppressed the latency shortening effect of stress. All these in vivo behavioral data combined with the in vitro electrophysiological results strongly suggest that stress facilitates seizure induction via elevated NA, which stimulates α1 adrenoceptors in the mPFC. It should be noted that restraint stress has been reported to increase the levels of not only NA but also dopamine and serotonin17,65. Thus, the possible involvement of these neurotransmitters in stress-induced seizure facilitation should be examined in future studies.
The mPFC can be divided into the prelimbic and infralimbic subregions. Because of a technical limitation, we could not selectively infuse drugs into each of the mPFC subregions. Further studies may be necessary to determine the role of each subregion on the stress-facilitated seizure induction.
In conclusion, our study demonstrates that NA released in the mPFC may contribute to stress-induced seizures via α1 adrenoceptor stimulation. Thus, α1 adrenoceptor blockers could be used as prophylactic and therapeutic drugs for stress-induced seizures in patients with focal mPFC epilepsy.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
Olsen, R. W., Wamsley, J. K., McCabe, R. T., Lee, R. J. & Lomax, P. Benzodiazepine/gamma-aminobutyric acid receptor deficit in the midbrain of the seizure-susceptible gerbil. Proc. Natl. Acad. Sci. U. S. A. 82, 6701–6705 (1985).
Houser, C. R. Do structural changes in GABA neurons give rise to the epileptic state?. Adv. Exp. Med. Biol. 813, 151–160 (2014).
McDonald, J. W. et al. Altered excitatory and inhibitory amino acid receptor binding in hippocampus of patients with temporal lobe epilepsy. Ann. Neurol. 29, 529–541 (1991).
Matsumoto, H. & Ajmone Marsan, C. Cellular mechanisms in experimental epileptic seizures. Science 144, 193–194 (1964).
Schwartzkroin, P. A. & Prince, D. A. Cellular and field potential properties of epileptogenic hippocampal slices. Brain Res. 147, 117–130 (1978).
Ferlisi, M. & Shorvon, S. Seizure precipitants (triggering factors) in patients with epilepsy. Epilepsy Behav. 33, 101–105 (2014).
van Campen, J. S., Jansen, F. E., Steinbusch, L. C., Joëls, M. & Braun, K. P. J. Stress sensitivity of childhood epilepsy is related to experienced negative life events. Epilepsia 53, 1554–1562 (2012).
Sperling, M. R., Schilling, C. A., Glosser, D., Tracy, J. I. & Asadi-Pooya, A. A. Self-perception of seizure precipitants and their relation to anxiety level, depression, and health locus of control in epilepsy. Seizure 17, 302–307 (2008).
da Silva Sousa, P., Lin, K., Garzon, E., Sakamoto, A. C. & Yacubian, E. M. T. Self-perception of factors that precipitate or inhibit seizures in juvenile myoclonic epilepsy. Seizure 14, 340–346 (2005).
Joëls, M. Stress, the hippocampus, and epilepsy. Epilepsia 50, 586–597 (2009).
Tolmacheva, E. A., Oitzl, M. S. & van Luijtelaar, G. Stress, glucocorticoids and absences in a genetic epilepsy model. Horm. Behav. 61, 706–710 (2012).
van Campen, J. S. et al. Cortisol fluctuations relate to interictal epileptiform discharges in stress sensitive epilepsy. Brain 139, 1673–1679 (2016).
Benarroch, E. E. Locus coeruleus. Cell Tissue Res. 373, 221–232 (2018).
Jett, J. D. & Morilak, D. A. Too much of a good thing: Blocking noradrenergic facilitation in medial prefrontal cortex prevents the detrimental effects of chronic stress on cognition. Neuropsychopharmacology 38, 585–595 (2013).
Nakane, H., Shimizu, N. & Hori, T. Stress-induced norepinephrine release in the rat prefrontal cortex measured by microdialysis. Am. J. Physiol. 267, R1559-1566 (1994).
Shimizu, N., Nakane, H., Hori, T. & Hayashi, Y. CRF receptor antagonist attenuates stress-induced noradrenaline release in the medial prefrontal cortex of rats. Brain Res. 654, 145–148 (1994).
Shinohara, F., Arakaki, S., Amano, T., Minami, M. & Kaneda, K. Noradrenaline enhances the excitatory effects of dopamine on medial prefrontal cortex pyramidal neurons in rats. Neuropsychopharmacol Rep. 40, 348–354 (2020).
Page, M. E. & Abercrombie, E. D. An analysis of the effects of acute and chronic fluexetine on extracellular norepinephrine in the rat hippocampus during stress. Neuropsychopharmacology 16, 419–425 (1997).
Ma, S. & Morilak, D. A. Norepinephrine release in medial amygdala facilitates activation of the hypothalamic-pituitary-adrenal axis in response to acute immobilisation stress. J. Neuroendocrinol. 17, 22–28 (2005).
Giorgi, F. S., Pizzanelli, C., Biagioni, F., Murri, L. & Fornai, F. The role of norepinephrine in epilepsy: From the bench to the bedside. Neurosci. Biobehav. Rev. 28, 507–524 (2004).
Rasmussen, T. Tailoring of cortical excisions for frontal lobe epilepsy. Can. J. Neurol. Sci. J. Can. des Sci. Neurol. 18, 606–610 (1991).
Trebuchon, A., Bartolomei, F., McGonigal, A., Laguitton, V. & Chauvel, P. Reversible antisocial behavior in ventromedial prefrontal lobe epilepsy. Epilepsy Behav. 29, 367–373 (2013).
Roberts, A. C. & Clarke, H. F. Why we need nonhuman primates to study the role of ventromedial prefrontal cortex in the regulation of threat- and reward-elicited responses. Proc. Natl. Acad. Sci. U. S. A. 116, 26297–26304 (2019).
Grzelka, K., Kurowski, P., Gawlak, M. & Szulczyk, P. Noradrenaline modulates the membrane potential and holding current of medial prefrontal cortex pyramidal neurons via beta1-adrenergic receptors and HCN channels. Front. Cell. Neurosci. 11, 1–22 (2017).
Marek, G. J. & Aghajanian, G. K. 5-HT2A receptor or alpha1-adrenoceptor activation induces excitatory postsynaptic currents in layer V pyramidal cells of the medial prefrontal cortex. Eur. J. Pharmacol. 367, 197–206 (1999).
Wada, S. et al. Acute restraint stress augments the rewarding memory of cocaine through activation of alpha1 adrenoceptors in the medial prefrontal cortex of mice. Neuropharmacology 166, 107968 (2020).
Esaki, H. et al. Nicotine enhances object recognition memory through inhibition of voltage-dependent potassium 7 channels in the medial prefrontal cortex of mice. J. Pharmacol. Sci. 147, 58–61 (2021).
Zhang, T. et al. A spatiotemporal increase of neuronal activity accompanies the motivational effect of wheel running in mice. Behav. Brain Res. 432, 113981 (2022).
Katzel, D., Nicholson, E., Schorge, S., Walker, M. C. & Kullmann, D. M. Chemical-genetic attenuation of focal neocortical seizures. Nat. Commun. 5, 3847 (2014).
Fetterly, T. L. et al. Alpha2A-adrenergic receptor activation decreases parabrachial nucleus excitatory drive onto BNST CRF neurons and reduces their activity in vivo. J. Neurosci. 39, 472–484 (2019).
Kielbinski, M. et al. Acute stress modulates noradrenergic signaling in the ventral tegmental area-amygdalar circuit. J. Neurochem. 164, 598–612 (2022).
Turski, W. A. et al. Injections of picrotoxin and bicuculline into the amygdaloid complex of the rat: An electroencephalographic, behavioural and morphological analysis. Neuroscience 14, 37–53 (1985).
Izumi, S. et al. Nicotine enhances firing activity of layer 5 pyramidal neurons in the medial prefrontal cortex through inhibition of Kv7 channels. Biol. Pharm. Bull. 44, 724–731 (2021).
Zhang, T. et al. Glutamatergic neurons in the medial prefrontal cortex mediate the formation and retrieval of cocaine-associated memories in mice. Addict. Biol. 25, 1–11 (2020).
Perkel, D. H., Gerstein, G. L. & Moore, G. P. Neuronal spike trains and stochastic point processes: II. Simultaneous spike trains. Biophys. J. 7, 419–440 (1967).
Deyama, S. et al. Resolvin D1 and D2 reverse lipopolysaccharide-induced depression-like behaviors through the mTORC1 signaling pathway. Int. J. Neuropsychopharmacol. 20, 575–584 (2017).
Ito, S. et al. Effects of the synthetic cannabinoid 5F-AMB on anxiety and recognition memory in mice. Psychopharmacology 236, 2235–2242 (2019).
Chu, J. et al. Role of 5-HT1A receptor-mediated serotonergic transmission in the medial prefrontal cortex in acute restraint stress-induced augmentation of rewarding memory of cocaine in mice. Neurosci. Lett. 743, 135555 (2021).
Paxinos, G. & Franklin, K. B. J. The Mouse in Sterotaxic Coordinates 3rd edn. (Academic Press, 2007).
Racine, R. J. Modification of seizure activity by electrical modification of after-discharge. Electroencephalogr. Clin. Neurophysiol. 32, 281–294 (1972).
Tse, K., Puttachary, S., Beamer, E., Sills, G. J. & Thippeswamy, T. Advantages of repeated low dose against single high dose of kainate in C57BL/6J mouse model of status epilepticus: behavioral and electroencephalographic studies. PLoS ONE 9, e96622 (2014).
Faul, F., Erdfelder, E., Buchner, A. & Lang, A.-G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 41, 1149–1160 (2009).
Shu, Y., Duque, A., Yu, G., Haider, B. & McCormick, D. A. Properties of action-potential initiation in neocortical pyramidal cells: Evidence from whole cell axon recordings. J. Neurophysiol. 97, 746–760 (2007).
Hotka, M. & Kubista, H. The paroxysmal depolarization shift in epilepsy research. Int. J. Biochem. Cell Biol. 107, 77–81 (2019).
Hillman, K. L., Lei, S., Doze, V. A. & Porter, J. E. Alpha-1A adrenergic receptor activation increases inhibitory tone in CA1 hippocampus. Epilepsy Res. 84, 97–109 (2009).
Santana, N. & Artigas, F. Laminar and cellular distribution of monoamine receptors in rat medial prefrontal cortex. Front. Neuroanat. 11, 1–13 (2017).
Luo, F., Tang, H. & Cheng, Z. Y. Stimulation of alpha1-adrenoceptors facilitates GABAergic transmission onto pyramidal neurons in the medial prefrontal cortex. Neuroscience 300, 63–74 (2015).
Braga, M. F. M., Aroniadou-Anderjaska, V., Manion, S. T., Hough, C. J. & Li, H. Stress impairs alpha1A adrenoceptor-mediated noradrenergic facilitation of GABAergic transmission in the basolateral amygdala. Neuropsychopharmacology 29, 45–58 (2004).
Hwa, G. G. C. & Avoli, M. The involvement of excitatory amino acids in neocortical epileptogenesis: NMDA and non-NMDA receptors. Exp. Brain Res. 86, 248–256 (1991).
Jones, R. S. G. & Lambert, J. D. C. The role of excitatory amino acid receptors in the propagation of epileptiform discharges from the entorhinal cortex to the dentate gyrus in vitro. Exp. Brain Res. 80, 310–322 (1990).
Panuccio, G., Curia, G., Colosimo, A., Cruccu, G. & Avoli, M. Epileptiform synchronization in the cingulate cortex. Epilepsia 50, 521–536 (2009).
Anderson, W. W., Anderson, W. W., Lewis, D. V., Scott Swartzwelder, H. & Wilson, W. A. Magnesium-free medium activates seizure-like events in the rat hippocampal slice. Brain Res. 398, 215–219 (1986).
Anastasiades, P. G. & Carter, A. G. Circuit organization of the rodent medial prefrontal cortex. Trends Neurosci. 44, 550–563 (2021).
Curia, G., Longo, D., Biagini, G., Jones, R. S. G. & Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 172, 143–157 (2008).
Cymerblit-Sabba, A. & Schiller, Y. Network dynamics during development of pharmacologically induced epileptic seizures in rats in vivo. J. Neurosci. 30, 1619–1630 (2010).
Lévesque, M. & Avoli, M. The kainic acid model of temporal lobe epilepsy. Neurosci. Biobehav. Rev. 37, 2887–2899 (2013).
Shimada, T. & Yamagata, K. Pentylenetetrazole-induced kindling mouse model. J. Vis. Exp. 2018, 1–10 (2018).
Bouilleret, V. et al. Recurrent seizures and hippocampal sclerosis following intrahippocampal kainate injection in adult mice: Electroencephalography, histopathology and synaptic reorganization similar to mesial temporal lobe epilepsy. Neuroscience 89, 717–729 (1999).
Mouri, G. et al. Unilateral hippocampal CA3-predominant damage and short latency epileptogenesis after intra-amygdala microinjection of kainic acid in mice. Brain Res. 1213, 140–151 (2008).
Hernan, A. E. et al. Focal epileptiform activity in the prefrontal cortex is associated with long-term attention and sociability deficits. Neurobiol. Dis. 63, 25–34 (2014).
Kim, J. et al. Thalamic T-type Ca2+ channels mediate frontal lobe dysfunctions caused by a hypoxia-like damage in the prefrontal cortex. J. Neurosci. 31, 4063–4073 (2011).
Kunieda, T. et al. Systemic overexpression of the alpha1B-adrenergic receptor in mice: an animal model of epilepsy. Epilepsia 43, 1324–1329 (2002).
Clinckers, R. et al. Pharmacological and neurochemical characterization of the involvement of hippocampal adrenoreceptor subtypes in the modulation of acute limbic seizures. J. Neurochem. 115, 1595–1607 (2010).
Zhu, X. et al. Repeated restraint stress increases seizure susceptibility by activation of hippocampal endoplasmic reticulum stress. Neurochem. Int. 110, 25–37 (2017).
Matuszewich, L., Filon, M. E., Finn, D. A. & Yamamoto, B. K. Altered forebrain neurotransmitter responses to immobilization stress following 3,4-methylenedioxymethamphetamine. Neuroscience 110, 41–48 (2002).
Acknowledgements
This study was supported by Grant-in-Aid for Challenging Research (Exploratory) (21K18548 to K.K.) from the Japan Society for the Promotion of Science, Koyanagi Foundation (K.K.), Smoking Research Foundation (K.K.), and WISE Program for Nano-Precision Medicine, Science, and Technology.
Author information
Authors and Affiliations
Contributions
K.N., S.I., S.W. and S.I. collected and analyzed the data. K.N., N.N., S.D. and K.K. wrote the manuscript. K.K. contributed to the concept and design. All authors contributed to and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Niitani, K., Ito, S., Wada, S. et al. Noradrenergic stimulation of α1 adrenoceptors in the medial prefrontal cortex mediates acute stress-induced facilitation of seizures in mice. Sci Rep 13, 8089 (2023). https://doi.org/10.1038/s41598-023-35242-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-35242-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
