
Abstract
Current methods for assessing cell proliferation in 3D scaffolds rely on changes in metabolic activity or total DNA, however, direct quantification of cell number in 3D scaffolds remains a challenge. To address this issue, we developed an unbiased stereology approach that uses systematic-random sampling and thin focal-plane optical sectioning of the scaffolds followed by estimation of total cell number (StereoCount). This approach was validated against an indirect method for measuring the total DNA (DNA content); and the Bürker counting chamber, the current reference method for quantifying cell number. We assessed the total cell number for cell seeding density (cells per unit volume) across four values and compared the methods in terms of accuracy, ease-of-use and time demands. The accuracy of StereoCount markedly outperformed the DNA content for cases with ~ 10,000 and ~ 125,000 cells/scaffold. For cases with ~ 250,000 and ~ 375,000 cells/scaffold both StereoCount and DNA content showed lower accuracy than the Bürker but did not differ from each other. In terms of ease-of-use, there was a strong advantage for the StereoCount due to output in terms of absolute cell numbers along with the possibility for an overview of cell distribution and future use of automation for high throughput analysis. Taking together, the StereoCount method is an efficient approach for direct cell quantification in 3D collagen scaffolds. Its major benefit is that automated StereoCount could accelerate research using 3D scaffolds focused on drug discovery for a wide variety of human diseases.
Similar content being viewed by others


Introduction
Quantification of cell number in a defined volume is an essential metric for the worldwide communities of bioscientists and pre-clinical researchers. In disciplines involving dispersion of cells into 3D volumes, e.g. 3D collagen scaffolds, reliable estimates of cells are required for launch of experiments, i.e. seeding of cells and assessing cell death and proliferation over time. Enumerating cell numbers in these cases is limited to two major approaches: The Bürker chamber for direct cell counts from homogenous cell suspensions (liquid phase); and fluorescence-based approaches for total DNA estimation, e.g., CyQuant®1,2. The drawbacks of the total DNA approach are cell lysis from sample pre-processing such as scaffold digestion by collagenase and need for a calibration curve for each experiment to ensure correct read-out and correlations between linearity of the signal intensity to the cell number. Thus, there is no direct quantitative method for estimation of cell counts in 3D scaffolds.
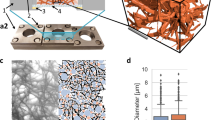
To address this issue, we introduce StereoCount™, a novel stereology-based method for direct quantification of absolute cell counts in 3D collagen scaffolds (Fig. 1).

3D collagen scaffold seeded with cells and used for the quantification of cell counts.
We demonstrate the approach using fluorescence microscopy followed by an optical sectioning and image analysis, as detailed in Materials & Methods and Fig. 2. Second, to avoid bias introduced by manual counting and improve throughput, we introduce an automated workflow using FIJI-ImageJ3 that uses two macro scripts and an intermediate, shallow-machine-learning (NL)-based step. Among the benefits are the method does not require scaffold digestion or microtome sectioning and, as a microscope-based approach, provides an overview of cell distribution in the scaffold as additional information. Here we compare and contrast StereoCount with DNA content method based on fluorescence detection of total DNA; and cell counts using the counting chamber method (Bürker), a generally reliable method for cell quantification.

StereoCount sampling procedure during microscopy. The three dimensional (3D) collagen scaffolds are sampled into 9 XY visual fields and 30 Z-stacks. Cell count is determined for each column using thin focal-plane optical scanning (optical disector method). The cell count from columns is recalculated to the whole volume of collagen scaffold.
For validation purposes, we compare the accuracy, ease-of-use, equipment requirements and time demands for sample preparation of the cell counts for the cell seeding density (cells per unit volume) across four values estimated by the three methods—StereoCount, DNA content and Bürker (reference standard). Accuracy of the methods are evaluated by these indicators (Fig. 3): (i) bias (systematic under- or over-estimation of the cell numbers relative to Bürker method mean), (ii) dispersion of the estimates (absolute/squared deviation from mean estimate of the given method, in other words how the data are close together) and (iii) overall accuracy (absolute/squared deviation from the theoretical value [i.e. the mean estimate from Bürker method], in other words the bias and dispersion taken into account mutually).

Indicators of the methods’ accuracy. These include bias (systematic under- or over-estimation of the cell counts relative to Bürker method mean), indicators reflecting dispersion around mean (absolute/squared deviation from mean estimate of given method) and finally indicators of overall accuracy (absolute/squared deviation from theoretical value [t.v.]) reflecting both the bias as well as dispersion (a). Both high dispersion as well as bias collectively decreases accuracy of the methods (b).
Results
Accuracy comparison
The Bürker method assessed the total cell count in the cell suspensions prior to infusion into the 3D collagen scaffolds. Four concentrations of cell suspensions (10,000; 125,000; 250,000 and 375,000 cells) were dispersed in the 3D scaffolds. Two methods (StereoCount and DNA content) were used to estimate total cell number one day after cell seeding from distinct scaffolds.
Accuracy of StereoCount and DNA content in 3D collagen scaffolds were compared to the counts in the cell suspensions by the Bürker method (Supplementary Table S1). Comparisons were done using generalized least squares (GLS) method as shown in Fig. 4 and Table 1. The datasets generated during the current study are available from the corresponding author on reasonable request.

Determined numbers of cells in 3D scaffolds with the cell seeding density across four values (a) 10,000, (b) 125,000, (c) 250,000 and (d) 375,000 cells/scaffold (upper part) and deviation from theoretical value (t. v., defined as Bürker method mean; shown in red dashed line), both showing in thousands of cells. Black horizontal line shows mean.
The reference values were based on mean estimates from Bürker method. The Bürker counts were 13,450 ± 2477 cells (mean ± SD) for the concentration of 10,000 cells/scaffold (Fig. 4a, Table 1a). Estimation by StereoCount showed no difference from Bürker in terms of accuracy or dispersion (variation) of the estimates. In contrast, the DNA content method lacked the sensitivity for estimation at this low cell density.
For the scaffolds infused with about 125,000 cells, Bürker estimated 125,400 ± 18,536 cells (Fig. 4b, Table 1b) and neither StereoCount, nor DNA content showed differences from Bürker. However, StereoCount method closely matched the reference method (Bürker) in terms of overall accuracy and dispersion. In contrast, DNA content showed a statistically greater difference of ~ 30,400 cells (95% CI [20,200; 40,700], p < 0.001) from the mean estimate of the reference method compared with StereoCount and higher deviation from the theoretical value by 32,200 cells (CI [24,300; 40,100], p = 0.001). Interestingly, the results from StereoCount showed even smaller dispersion than Bürker (reference method) as the deviation from mean was smaller by 7200 cells (CI [1700; 12,600], p = 0.021), i.e., closer to the initial number of cells seeded (125,000 cells/scaffold).
For the scaffolds of 250,000 cells, Bürker estimated 253,200 ± 6197 cells (Fig. 4c, Table 1c). Both StereoCount and DNA content methods showed differences from these reference values in terms of overall accuracy and dispersion. Specifically, the deviation from Bürker was higher for StereoCount by 31,600 cells [CI 14,900; 48,300] and for DNA content by 51,000 cells [CI 32,200; 69,900]. Although only DNA content underestimated the cell count by 38,000 cells [CI -72,200; -3800], p = 0.044), the dispersion and overall accuracy of both StereoCount and DNA content were not different from each other (p > 0.05).
Finally, for the highest density cell counts in the scaffolds (375,000 cells), Bürker estimated 349,200 ± 37,542 cells (Fig. 4d, Table 1d). Both StereoCount and DNA content differ from these reference values and did not differ from each other in terms of overall accuracy. However, DNA content tended to produce lower dispersion of the estimates than StereoCount: whereas the deviation from mean was higher in StereoCount by 77,500 cells [29,100; 125,800] compared to Bürker (p = 0.005), DNA content showed comparable deviation from the mean as Bürker (the difference of 2300 cells [-11,200; 15,700] p = 0.743). Although DNA content showed bias (underestimation) of -86,000 cells [CI -123,400; -49,100] (p = 0.003), the method tended toward lower dispersion of the estimates than StereoCount (DNA content deviation from mean was smaller by 79,800 cells [32,200; 127,300] compared to StereoCount (p = 0.005). Supplementary Table S2 provide further results of analyses of squared deviation from mean and squared deviation from theoretical value.
Ease-of-use, equipment required and time demands for sample preparation
In addition to accuracy relative to the reference method, other factors for consideration include ease-of-use, costs, equipment requirements and time demands for each method.
DNA content is a widely used fluorescence-based method that is rapid and easy to perform. Microplate reader with fluorescence signal detection is needed. The approach is well-suited to multiple-well format, whereas more scaffolds require similar processing time as cell estimation for a single scaffold. For example, analysis of one or ten scaffolds takes about three hours (Fig. 5). However, due to the requirement of scaffold digestion, information about the cell distribution is lost and no further processing or analysis is possible. Another drawback is the output is not cell count but rather a relative fluorescence unit of total DNA per well (Table 2) which requires a reference standard curve for converting sample fluorescence values into cell numbers.

Scheme of DNA content (upper panel) and StereoCount (lower panel) processes with time demands of each approach in a step-by-step manner. Process duration is not dependent on the sample size (blue). Process duration of one scaffold analysis (orange). Process duration of ten scaffolds analyses (green). Process duration when automation (script) is used for data processing (yellow). Total time demands for one or ten samples for each method are given in red frames.
The novel StereoCount approach proposed here is based on unbiased stereology principles for cell number estimation in 3D collagen scaffolds. Fluorescence microscope is required for the image acquisition. Among the advantages of StereoCount are the approach (i) provides an absolute count of cells per scaffold (Table 2); and (ii) does not require scaffold digestion, thereby conserving information about cell distributions in 3D. In terms of time, StereoCount analysis of one scaffold and ten scaffolds takes about 3 and 10 h, respectively. It is expected that automation will reduce the time of data processing to approximately 10 min or less depending on computer specifications (Fig. 5). The script for the high throughput analysis can be found in Supplementary Macro S1.
Discussion
Here we review several methods for assessing cell proliferation in 3D scaffolds. One common strategy for estimating cell proliferation is based on the conversion of various substrates by cellular enzymes to highly fluorescent products, e.g., MTT4,5, WST-16, WST-8/CCK-8, CCK-87 and AlamarBlue assay8. Since these methods depend on the activity of mitochondrial enzymes, the results reflect the metabolic activity of the cells, rather than the single cell count per se. Moreover, this process loses sensitivity as metabolic activity changes such as in the cases of senescent or slowly grown cells9.
Methods based on labeling of DNA content, e.g., CyQuant1,2, are independent of metabolic activity of the cells. However, these approaches only estimate the relative fluorescence unit, which requires DNA standard curves for conversion to DNA content per cell, i.e. a standard curve generated from the DNA content for a known number of cells seeded on a plate. Another complication is that, compared to usual 2D cultures, the estimation in 3D scaffolds requires collagen hydrogel digestion prior to detection of a fluorescent signal from the labeled cellular DNA. Although this strategy is common, the approach only provides information about the amount of DNA present, and gives no information about the absolute cell count or the cell distribution throughout the 3D scaffolds.
To address this need, we developed a novel stereology-based strategy, StereoCount, for quantifying the cell number in a 3D scaffold, without need for digestion or calibration curves for each experiment. We assessed StereoCount in comparison with a current and commonly used method, DNA content, using the counting chamber (Bürker) as a reference method. Finally, as an extension we developed an automated workflow (pipeline) to avoid the introduction of subjectivity errors from manual counting. Efficiency of the automated workflow could be potentially improved by changing the macro scripts, and further automated by combining all of the steps into one macro or switching to different platforms for each step. Our easy-to-apply workflow provides for reliable results with identical error applied to each image, avoiding inter-rater error caused by different investigators performing each task in a manual manner.
Our data shows that for low cell numbers (10,000 cells/scaffold) DNA content is not usable due to the background fluorescence signal exceeding the signal from the florescent-stained DNA. For low cell density, therefore, StereoCount is the preferred choice since the approach is based on simple counting of cells in a known fraction of the total volume. For higher cell numbers (125,000 and 250,000 cells/scaffold), both StereoCount and DNA content provide useful results, though StereoCount more closely reflect the loading number of cells using the reference Bürker method. For higher cell density (250,000 cells/scaffold and higher) both DNA content and StereoCount are the similarly effective. The limitation for StereoCount is the extra time and care required for avoiding overlapping cells as the cell density increases. Lack of care to this issue could lead to a systematic error (bias) that favors undercounts as the cell density increases. Similarly, our results show underestimation of the cell counts by DNA content as a direct function of increasing cell density, suggesting a bias-correction may be needed for this approach as well. The difference is that for StereoCount these undercounts may be avoided by careful attention at high cell density, whereas this strategy is not available for the DNA content method.
Finally, we consider feasibility, material, machine requirements, and time demands of the StereoCount and DNA content methods. A strong advantage of DNA content is multiple-well format option that is less time-consuming method as compared to StereoCount, although the data processing (especially the multiple sample analysis) is substantially shortened by the use of an automated workflow for StereoCount. However, the faster analysis of data for DNA content is off-set by the need for building a calibration curve to estimate cell count from relative fluorescent units for each experiment. The strongest advantage of StereoCount is the ability for direct cell counts and visualization of cell distribution in 3D using simple microscopic images, without the need for scaffold digestion or microtome sectioning. Though the StereoCount approach is illustrated here using fluorescent-stained cells, the same approach is applicable to differentiate between live and dead cells when DAPI and Calcein-AM staining is combined, as well as approaches that use immunohistochemical staining of cells in the 3D scaffolds.
Conclusions
Although hydrogel scaffolds are nowadays an ordinary type of 3D cell culture, there remains a large and growing need for quantitative tools to assess cell proliferation and other parameters following different experimental manipulations. Our novel stereology-based freeware approach, StereoCount, uses optical sectioning of stained cells in non-digested collagen hydrogels with faster throughput using the provided automated workflow. We report that StereoCount is more suitable for lower cell density (~ 10,000 and ~ 125,000 cells/scaffold), where the DNA content method lacked the sensitivity for estimation of such low cell density, while DNA content by the fluorescence-based indirect method may be preferable in for higher cell density (~ 250,000 and ~ 375,000 cells/scaffold).
Materials and methods
Cell isolation and culture
Normal human dermal fibroblasts (NHDF) were isolated from skin following plastic surgery interventions after approval by the local ethics committee of the University Hospital in Pilsen, E. Benese 13, 305 99 Pilsen, Czech Republic, decision of November 5th, 2015. The guidelines in the Declaration of Helsinki were followed. All donors gave written informed consent before intervention. The samples were washed by Hank’s balanced salt solution (HBSS) (Merck KGaA, Darmstadt, Germany) containing penicillin (100 U/ml)/streptomycin (0.1 mg/ml) (Biochrom, Cambridge, UK) and gentamicin (50 μg/ml) (Biochrom). The samples were cut and digested overnight at 37 °C in HBSS containing collagenase type I (100 U/ml, Merck KGaA). On the following day, the suspension was shaken intensively and filtered through 100 µm nylon cell strainer (Falcon™, Thermo Fisher Scientific, Waltham, Massachusetts). The cell suspension was then transferred to a cultivation flask (TPP) containing low glucose Dulbecco’s Modified Eagle’s Medium (DMEM) (Merck KGaA), 10% heat-inactivated fetal bovine serum (FBS) (Merck KGaA), penicillin (100 U/ml)/streptomycin (0.1 mg/ml) (Biochrom), 0.5% L-glutamin (Biosera, Nuaille, France) and 1.0% non-essential amino acids (Biosera). The NHDF were cultured at 37 °C, 5% CO2 up to 80% confluence and then passaged. Only those NHDF from the 2th–5th passages were used in the experiments.
Isolation of rat tail collagen type I
The type I collagen used for preparing the 3D collagen scaffold was isolated from rat tails. The collection of rat tails for collagen isolation was approved by the Animal Welfare Advisory Committee of the Ministry of Education, Youth and Sports of the Czech Republic (approval ID MSMT-249/2017-2) and conducted under the supervision of the Animal Welfare Advisory Committee of the Charles University Faculty of Medicine in Pilsen following the technological, hygienic, welfare and ethical standards given by Directive 2010/63/EU and FELASA Guidelines and Recommendations (the workplace accreditation number 4891/2015-MZE-17214). The strain of rats was Wistar, the gender was male, and the age was 6–7 months. The rats were used in another independent study without any effect on the tails. Thus the tails were collected after animals sacrifice approved by the protocol above (approval ID MSMT-249/2017-2). The study was carried out in compliance with the ARRIVE guidelines. The tendons were removed from the rat tails and maintained 3 × 24 h in PBS on a laboratory shaker at room temperature. PBS was exchanged every single day. Afterwards, the procedure was repeated with citrate buffer (0.08 M, pH 3.7). Subsequently, the tendons were digested in 0.1 M acetic acid for 48 h at 4 °C. The collagen digested in acetic acid was homogenized with blender and the suspension was ultracentrifuged at the maximum speed (approx. 46,000× g) for 1 h in order to remove the tissue debris. The supernatant containing the collagen type I was lyophilized and stored in -20 °C. The collagen solution for preparation of the 3D collagen scaffold was prepared by dissolving the lyophilized collagen in 0.02 M acetic acid at 4 °C at least 5 days to a concentration of 5 mg/ml and was stored at 4 °C as a stock solution for maximal 1 month.
Preparation and culture of cell-seeded collagen scaffolds
3D collagen scaffolds containing cells were prepared as follows. 600 µl of collagen stock solution (5 mg/ml) was mixed properly with 290 µl of culture medium, 10 µl of sodium bicarbonate (Merck KGaA) and 100 µl of cell suspension in medium on ice. 0.5 ml of collagen suspension with cells was plated into the 24-well plate. The final collagen concentration was 3 mg/ml and the final cell counts seeded were 10,000; 125,000; 250,000 and 375,000 cells per 0.5 ml. 0.5 ml of collagen suspension with homogeneously distributed cells was plated into the 24-well plate. The polymerization occurred at 37 °C, 5% CO2 and humidified atmosphere after 20 min. The change in pH from acidic to neutral and 37 °C caused the collagen to polymerize and self-assembly into a gel. After collagen polymerization 0.5 ml of culture medium was carefully added on the top of the hydrogels. The culture medium was exchanged 2–3 times a week.
Cell number estimation by stereology (StereoCount)
Cell staining and collagen scaffold optical sectioning by microscopy
The day after their preparation the cell-seeded collagen scaffolds (N = 25 for 10,000 cells/scaffold; N = 19 for 125,000 cells/scaffold; N = 17 for 250,000 cells/scaffold; N = 7 for 375,000 cells/scaffold) were fixed with 4% paraformaldehyde (Merck KGaA) for 30 min. After washing with PBS the collagen scaffolds were maintained in 0.2% Triton X-100 (Merck KGaA) for 30 min followed by 30 min in 5% BSA (Merck KGaA). Nuclei of NHDF in collagen scaffolds were stained by DAPI (1:1000) for 10 min in the dark at room temperature. The process of optical sectioning of cell-seeded collagen scaffolds was as follow. Nine Z-axis columns were located through the XYZ-axes of each collagen scaffold (Fig. 2). The columns included 30 separated Z stacks with 0.02 mm distance between each stack giving a total volume of each column of 1.4 mm3 = 1.4 μl. Thus, the total volume of all 9 columns was 12.6 mm3. The mean coefficient of variance (CV) for the sampling of 9 columns was in the range of 9–20% and is comparable with CV of DNA content and Bürker methods (Supplementary Table S1) and with other studies10,11. 2D images of each section were taken by Olympus UPlanFL N 10x/0.30 objective and the pictures of columns were saved as image sequence (*.stk file) by VisiView® software. The number of cells was recalculated to the whole collagen scaffold volume.
Cell number estimation by image analysis
Cell numbers were counted within columns using FIJI-ImageJ software (National Institute of Health, Bethesda, USA) and cell number in total volume of a collagen scaffold was estimated from each column using the optical disector principle12. Further details on the schema can be found in Supplementary Fig. S1.
Automated workflow for cell number estimation by image analysis
After acquisition, we first batch process images to make them easier to work with. First provided short macro (Supplementary Macro S1) automatically converts the acquired images to 8-bit images and maximum intensity Z projection (Fig. 6a). The following, intermediate, step requires significant user input in the beginning (Fig. 6b). Herein we used a LABKIT13 plugin in FIJI which uses a shallow machine learning approach to segment images, based on training. To generate a training dataset, we concatenated six sample images (pre-processed with step A of the workflow) from different cell seeding density. Afterwards a user-induced training was applied to generate a classifier. This classifier was used to batch process all pre-processed images from step A and generate segmentations. These segmentations were then analyzed in step C (Fig. 6c) of the workflow. We applied additional short macro (Supplementary Macro S2) to batch-analyze the masks and report count of detected objects per image. We apply the macros for free use without any guarantees and maintains.

Scheme of an automated workflow which consists of two macro scripts (a,c) and an intermediate (b), shallow-machine-learning-based step.
Estimation of collagen scaffold volume
Collagen scaffold volume was estimated according to Archimedes´ principle. 5-ml measuring cylinder with 0.1 ml marked lines was filled with precise 3 ml of PBS. Each collagen scaffold was put into the cylinder with PBS and the buoyed volume of PBS was read on the cylinder scale. The increased volume in the cylinder corresponded to the volume of the collagen scaffold.
Cell number calculation
The cell count in acquired image sequences (XYZ column) was recalculated to the whole cell-seeded collagen scaffold, i.e. for estimation of cell number in one collagen scaffold, cell count from 9 image sequences (i.e. 9 columns) were recalculated to the total collagen scaffold volume. This calculation was performed from the 9 columns and the mean of the values was considered as a result (total cell count) for one collagen scaffold.
Cell number estimation by Bürker chamber method (Bürker)
The NHDF-seeded culture dishes were trypsinized and resuspended in fresh culture medium. A portion of the suspension was diluted with Trypan blue (Merck KGaA) for the cell count estimation (five repetitions). The mean estimate of cell number served as an initial cell count for further dilutions.
From the initial cell suspensions 5 × 0.5 ml (containing 10,000; 125,000; 250,000 and 375,000 cells) were prepared. From these 0.5 ml-suspensions the portion was diluted with Trypan blue and the cell number was estimated. This counting was performed in 10 repetitions by two independent observers (N = 10 of each cell seeding density).
Cell number estimation by total DNA quantification (DNA content)
Cell-loaded collagen scaffolds (N = 4 for 10,000 cells/scaffold; N = 8 for 125,000 cells/scaffold; N = 9 for 250,000 cells/scaffold; N = 5 for 375,000 cells/scaffold) were prepared according to the chapter “Preparation and culture of cell-seeded collagen scaffolds”. The next day, the scaffolds were washed with PBS and placed into − 80 °C. On the day of analysis, the collagen scaffolds were thawed.
Firstly, Proteinase K (0.2 mg/ml; 2.5 DMC-U/ml, Serva, Heidelberg, Germany) in phosphate buffered EDTA (PBE) was added to scaffolds with cells in 1:1 ratio. The samples were then incubated for 90 min at 50 °C. Afterwards, the digested sample fluid was homogenized by pipetting and vortexed. 462 µl of the sample fluid (half of the sample volume) was transferred to fresh tube and 17.6 µl of RNAse A (1 mg/ml, Serva), 1 µl of EDTA (0.5 M, Thermo Fisher Scientific) and 20 µl of NaCl-Tris–EDTA (TE) buffer solution (10.5 mg NaCl per sample diluted in 50 × TE buffer) was added and incubated for 1 h at room temperature. RNAse-processed sample fluid was transferred to black 96-well plate (100 µl/well) and 50 µl of CyQuant solution (Thermo Fisher Scientific) in TE buffer was added (final TE buffer concentration 1 × , CyQuant:TE buffer 1:200). Samples were incubated at room temperature, shielded from light for 10 min and occasionally shaken. Fluorescence signal was measured by microplate reader (Synergy HT, Biotek, Winooski, Vermont) at 520 nm/480 nm and correlated to DNA standard prepared according to CyQuant manufacture brochure.
The DNA content in cell-seeded collagen scaffolds was recalculated to cell number according to cell number calibration curve made from the cells seeded usually on a plate.
Data processing and statistical analysis
Data were analyzed using R statistical software14. R packages ‘beeswarm’15 and ‘vioplot’16 were used for data visualization.
The accuracies were compared using: (i) bias from the theoretical values, indicating how much the average estimates from the StereoCount and DNA content estimators differ from the Bürker, reference method. Next, we used two measures reflecting dispersion, ignoring the potential bias and thus enabling meaningful comparison of the Bürker vs. other methods. These are (ii) deviation from [group-specific] mean and (iii) squared deviation from mean. Finally, two other measures indicate overall accuracy of the methods and reflect both the bias, as well as the dispersion: (iv) deviation from theoretical value and (v) squared deviation from the theoretical value (Fig. 3).
All these five measures of accuracy were originally compared using generalized least square (GLS), with variance function allowing estimator-specific variance, using ‘nlme’ package17. As models of squared deviations exhibited non-normal distribution of standardized residuals (checked visually using histograms and Q-Q plots and via Shapiro–Wilk test), they were re-analyzed using generalized linear models with Gamma distribution and log-link (Gamma GLM).
In each model, one of the five above-mentioned measures represented the outcome (response variable) whereas the type of the method represented a predictor (explaining variable). P-value (two-sided) was based on permutational t-test (bias and absolute deviations) or permutational Gamma GLM (squared deviations), according to the script shown previously18 using 5000 Monte-Carlo randomizations. The permutation approach was chosen because it estimates statistical significances reliably even if sample sizes are small and assumptions of the fully parametric methods (including normal distribution of residuals) are not met19. P-values were not corrected for multiple comparisons because we concentrated particularly on the single comparison (StereoCount vs. DNA content) and 95% confidence intervals for effect size were derived from the GLS and Gamma GLM models.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
Change history
21 June 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41598-023-37265-z
References
Bott, K. et al. The effect of matrix characteristics on fibroblast proliferation in 3D gels. Biomaterials https://doi.org/10.1016/j.biomaterials.2010.07.046 (2010).
Wang, H., Pieper, J., Péters, F., Van Blitterswijk, C. A. & Lamme, E. N. Synthetic scaffold morphology controls human dermal connective tissue formation. J. Biomed. Mater. Res. Part A https://doi.org/10.1002/jbm.a.30232 (2005).
Schindelin, J. et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Ghiasi, M., Kalhor, N., Tabatabaei Qomi, R. & Sheykhhasan, M. The effects of synthetic and natural scaffolds on viability and proliferation of adipose-derived stem cells. Front. Life Sci. https://doi.org/10.1080/21553769.2015.1077477 (2016).
Rothan, H. A. et al. Three-dimensional culture environment increases the efficacy of platelet rich plasma releasate in prompting skin fibroblast differentiation and extracellular matrix formation. Int. J. Med. Sci. 11, 1029–1038 (2014).
Guo, J., Chen, H., Wang, Y., Cao, C. B. & Guan, G. Q. A novel porcine acellular dermal matrix scaffold used in periodontal regeneration. Int. J. Oral Sci. 5, 37–43 (2013).
Dong, C. & Lv, Y. Application of collagen scaffold in tissue engineering: Recent advances and new perspectives. Polymers https://doi.org/10.3390/polym8020042 (2016).
Chan, E. C. et al. Three dimensional collagen scaffold promotes intrinsic vascularisation for tissue engineering applications. PLoS ONE 11, e0149799 (2016).
Adan, A., Kiraz, Y. & Baran, Y. Cell proliferation and cytotoxicity assays. Curr. Pharm. Biotechnol. 17, 1213–1221 (2016).
Salinas, M., Rosas, J., Iborra, J., Manero, H. & Pascual, E. Comparison of manual and automated cell counts in EDTA preserved synovial fluids: Storage has little influence on the results. Ann. Rheum. Dis. https://doi.org/10.1136/ard.56.10.622 (1997).
Camacho-Fernández, C., Hervás, D., Rivas-Sendra, A., Marín, M. P. & Seguí-Simarro, J. M. Comparison of six different methods to calculate cell densities. Plant Methods https://doi.org/10.1186/s13007-018-0297-4 (2018).
Mouton, P. R. Principles and Practices of Unbiased Stereology: An Introduction for Bioscientists (Johns Hopkins University Press, 2002).
Arzt, M. et al. LABKIT: Labeling and segmentation toolkit for big image data. Front. Comput. Sci. https://doi.org/10.3389/fcomp.2022.777728 (2022).
R Development Core Team. R: A Language and Environment for Statistical Computing. (2020).
Eklund, A. beeswarm: The Bee Swarm Plot, an Alternative to Stripchart. (2016).
Adler, D. & Kelly, S. T. vioplot: Violin plot. (2020).
Pinheiro, J., Bates, D., DebRoy, S., Sarkar, D. & R Core Team. nlme: Linear and Nonlinear Mixed Effects Models. (2018).
Tichanek, F. et al. Hippocampal mitochondrial dysfunction and psychiatric-relevant behavioral deficits in spinocerebellar ataxia 1 mouse model. Sci. Rep. 10, 1–14 (2020).
Good, P. I. Permutation, Parametric and Bootstrap Tests of Hypotheses (Springer, 2005). https://doi.org/10.1007/b138696.
Acknowledgements
The study was supported by Project No. CZ.02.1.01/0.0/0.0/16_019/0000787 “Fighting INfectious Diseases”, awarded by the MEYS CR, financed from EFRR, by the Cooperatio Program, research area DIAG and research area MED/DIAG, by the profiBONE project (TO01000309) benefitting from a € (1.433.000) grant from Iceland, Liechtenstein and Norway through the EEA Grants and the Technology Agency of the Czech Republic and by a Grant (#1926990) to PRM and SRC Biosciences from the National Science Foundation (U.S. Public Health Service). The authors acknowledge the invaluable assistance provided by Iveta Paurova via her support in terms of the provision of laboratory services.
Author information
Authors and Affiliations
Contributions
Conceptualization, A.Z. and L.V., Methodology, A.Z., L.V., T.B., F. T. and P.M., Data Analysis A.Z., L.V., T.B., D. B. and F.T., Writing, A.Z., F.T., T.B., L.V. and P.M., Creating images, A.Z. and F.T.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained errors in the Materials and methods section as well as in the Supplementary Information. Full information regarding the corrections made can be found in the correction for this article.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zavadakova, A., Vistejnova, L., Belinova, T. et al. Novel stereological method for estimation of cell counts in 3D collagen scaffolds. Sci Rep 13, 7959 (2023). https://doi.org/10.1038/s41598-023-35162-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-35162-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
