
Abstract
Thua Nao is a Thai traditional fermented soybean food and low-cost protein supplement. This study aimed to evaluate the bacterial community in Thua Nao from northern Thailand and assess potentially active short-chain fatty acids (SCFAs)-related bacteria. Sixty-five Thua Nao consisting of 30 wet and 35 dried samples were collected from six provinces: Chiang Rai, Chiang Mai, Mae Hong Son, Lampang, Lamphun, and Phayao. Bacterial diversity was significantly higher in the wet samples than in the dried samples. The dominant phyla were Firmicutes (92.7%), Proteobacteria (6.7%), Actinobacteriota (0.42%), and Bacteroidota (0.26%). The genus Bacillus (67%) was the most represented in all samples. Lactobacillus, Enterococcus, and Globicatella were enriched in the wet samples. Assessment of the SCFA-microbiota relationships revealed that high butyrate and propionate concentrations were associated with an increased Clostridiales abundance, and high acetate concentrations were associated with an increased Weissella abundance. Wet products contained more SCFAs, including acetate (P = 2.8e−08), propionate (P = 0.0044), butyrate (P = 0.0021), and isovalerate (P = 0.017), than the dried products. These results provide insight into SCFA-microbiota associations in Thua Nao, which may enable the development of starter cultures for SCFA-enriched Thua Nao production.
Similar content being viewed by others

Introduction
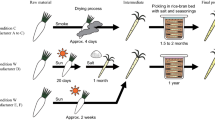
Thua Nao (fermented soybean) is a northern Thai food with high nutritional value and is similar to products such as natto in Japan, chongkukjang in Korea, and kinema in India1. Three main ingredients (i.e., soybeans, water, and natural microbes) are required to produce Thua Nao, which occurs in four major steps: soaking, boiling, fermenting, and post-fermentation. Boiling destroys non-heat-resistant germs and bacteria, thus preserving the heat-resistant bacteria. The fermentation process lasts 3 days under ambient conditions. Freshly prepared or wet Thua Nao can be steamed or roasted before consumption. Under ambient conditions, wet Thua Nao can be stored for approximately 2 days. Therefore, post-fermentation processes have been developed to prolong its shelf-life. For example, cooked Thua Nao is mashed into a circular flat disk and then sun-dried, resulting in the dried form of Thua Nao, which can be stored for several months1.
A previous study demonstrated the value of Thua Nao as a potential resource for bioactive and health-promoting compounds2. Thua Nao contains 57.22–64.78% moisture, 4.70–5.44% ash, 12.92–28.06% crude fiber, 38.94–42.06% crude protein, and 20.37–25.22% fat3. The potential benefits of bioactive compounds, such as phenolic compounds and antioxidant activities, are not only derived from raw materials but also released from bacteria during soybean fermentation3. Using culture-dependent methods, Bacillus species are reported to be predominant microorganisms in Thua Nao4. However, Thua Nao is made from a mixture of natural microflora, which cannot be easily cultured. The fermentation process with mixed microflora allows the microorganisms to help one another produce important substances related to nutrition and health, such as peptides, vitamin B, fatty acids, and short-chain fatty acids (SCFAs). Soybean digestion by the microflora creates the SCFAs, i.e., acetate, propionate, and butyrate, which serve as essential nutrients for colonic mucosal cells, thus stimulating mucosal proliferation and blood flow5 and controlling sugar levels5, metabolism6, and cholesterol levels7. Therefore, Thua Nao is considered a good source of important substances, prebiotics, and bacteria beneficial to health.
Thua Nao is produced in various regions in northern Thailand; however, data on bacterial communities in Thua Nao from different areas is still limited. Studying the bacterial communities may enable a better understanding of the roles and functional properties of fermenting bacteria in Thua Nao. The microbes used for natural fermentation originate from growing soybean. We thus hypothesized that different soybean growing regions or geographical conditions could influence the composition of the microbiota and ultimately the composition of SCFAs. Natto could be a good control compared to other fermented foods (e.g., fermented fish and pickles) because it is also a fermented soybean, but, with the addition of Bacillus as a starter culture. To address this, we first identify the bacterial taxa in Thua Nao from six different areas in northern Thailand using culture-independent approaches (i.e., 16S rRNA gene sequencing). Second, we analyzed the correlations between the bacterial taxa and the types and amounts of SCFAs in Thua Nao using gas–liquid chromatography. The results revealed the beneficial bacteria that may be used as starter cultures for improving Thua Nao production quality.
Results
Characteristics of fermented soybean (Thua Nao) in Thailand
Sixty-five Thua Nao samples, consisting of both wet and dried fermented soybeans, were collected from 37 villages in six provinces of northern Thailand. Commercial natto was also used as a non-traditional fermented soybean sample (control; n = 1), that was derived from a supermarket in Thailand. Figure 1 shows a map of Thailand with the sampling numbers in each province.

Sampling locations and numbers of dried and wet Thua Nao in northern Thailand. The number in the center of the circle represents the number of villages in each province, while the number in the orange and green colors indicates the number of dried and wet samples in each province, respectively. The map was downloaded from Google Data Studio, and additional graph and pointer were drawn using Microsoft Excel v16.66.1, Microsoft PowerPoint v16.66.1, and Inkscape v1.1.
Microbiome statistics
The median number of raw 250-bp paired-end read sequences generated by the Illumina sequencing with the 16S rRNA genes was 66,438 (range 36,740–82,116). After preprocessing, the median number of sequences was reduced to 48,645.5 (range 31,864–60,876). Sequences were then clustered into 652 features based on the amplicon sequence variant (ASV) method with most of the sequence length of 253 bp corresponding to the approximate size of the V4 region of the 16S rRNA genes. Supplementary File S1 shows the taxonomic abundance tables at the phylum and genus levels.
Bacterial taxonomic signatures of Thua Nao
Sixty-six fermented soybean samples (30 wet Thua Nao, 35 dried Thua Nao, and 1 natto) were obtained and analyzed for bacterial communities based on the diversity of 16S rRNA gene sequences. Four phyla were identified in both types of fermented soybeans. Firmicutes was the most common phylum in the dried (95.98% + 11.55%), wet (88.42% + 15.46%), and both (92.66% + 13.71%) types (Fig. 2A). The relative abundances of Proteobacteria, Actinobacteriota, and Bacteroidota were 10.26%, 0.85%, and 0.47% in the wet samples, respectively, and 3.85%, 0.08%, and 0.09% in the dried samples, respectively (Fig. 2B). Twelve genera had ≥ 1% relative abundances (Fig. 2D). Bacillus was the dominant genus in the dried (86.41% + 16.41%), wet (47.19% + 30.63%), and both (66.94% + 32.25%; Fig. 2D) types. Interestingly, Lactobacillus, Enterococcus, and Globicatella were enriched in the wet samples.

Percent relative abundances of the taxonomic compositions were compared between dried (n = 35) and wet (n = 30) fermented soybeans at the phylum (A) and genus (B) levels and among the six provinces at the phylum (C) and genus (D) levels.
Alpha diversities were estimated for all microbiome profiles of the fermented soybeans and compared between dried and wet fermented soybeans based on Shannon’s diversity (Fig. 3A), Simpson’s diversity (Fig. S1B), Pielou’s index (Fig. S1C), and Faith’s phylogenetic diversity (Fig. S1D) using the Wilcoxon rank-sum test. All alpha diversity metrics showed that microbial diversities of the wet fermented soybeans were significantly higher than those of the dried fermented soybeans. Beta diversities were measured between fermented soybean samples (Fig. 3 and S2). Differences between fermented soybean types were evaluated using PERMANOVA tests (Fig. 3B), which indicated that wet and dried fermented soybean and natto had different microbiota patterns (P = 1e−04). Differential abundant taxa between dried and wet fermented soybeans were identified with the cutoff criteria P < 0.05 (Fig. 3C and D).

Comparison of bacterial diversities in dried (red; n = 35) and wet (blue; n = 30) fermented soybeans and natto (green; n = 1). Statistical significance tests for alpha diversity between dried and wet fermented soybeans based on Shannon’s diversity (A). Beta diversity among dried and wet fermented soybeans and natto based on Bray–Curtis dissimilarity and PERMANOVA (B). Differential abundance analysis between dried and wet fermented soybeans at the phylum (C) and genus (D) levels using ANCOM-BC methods; the Q value represents a P-value that has been adjusted for the False Discovery Rate (FDR).
Bacterial compositions among the fermented soybeans showed that Lampang had the most bacterial genera (14 genera) with ≥ 1% relative abundances, followed by Chiang Mai, Chiang Rai, Phayao, Mae Hong Son, and Lamphun (Fig. 2D). Interestingly, Proteobacteria had lower abundances in Chiang Rai and Phayao than in the other provinces (Fig. 2C).
Alpha diversities in terms of Shannon’s diversity, Simpson’s diversity, Pielou’s index, and Faith’s phylogenetic diversity were compared among the six provinces using Kruskal–Wallis tests (Fig. S3). The statistical results indicated that microbial diversities among the six provinces differed significantly in the wet condition based on Faith’s phylogenetic diversity (P = 0.0011; Fig. 4A). However, alpha diversities were not significantly different among the six provinces in the dried condition (P = 0.06; Fig. 4B). Alpha diversities in Chiang Rai, Chiang Mai, and Lampang were higher than those in Lamphun, Mae Hong Son, and Phayao. Differences between any two samples were measured based on Bray–Curtis dissimilarity, Jaccard distance, weighted UniFrac distance, and unweighted UniFrac distance (Fig. S4). Beta diversities based on four metrics were analyzed and visualized using principal coordinate analysis (PCoA) plots (Fig. S4). PERMANOVA indicated significant differences in the bacterial profiles from the six provinces (P = 0.001; Figs. 4C and S3).

Comparison of bacterial phylogenetic diversities and dissimilarity in fermented soybeans among provinces. Statistical significance tests for alpha diversity among the provinces based on Faith’s phylogenetic diversity in wet (n = 30) (A) and dried (n = 35) (B) conditions using Kruskal–Wallis tests. Beta diversity among dried and wet fermented soybeans from six provinces and Japanese natto (n = 1) based on Bray–Curtis dissimilarity was analyzed via PERMANOVA (C).
Natural SCFA levels of Thua Nao
The fermented soybeans were measured for their SCFA levels. Acetate (P = 2.8e−08), propionate (P = 0.0044), butyrate (P = 0.0021), and isovalerate (P = 0.017) were greater in wet fermented soybeans (n = 30) than in dried fermented soybeans (n = 35) based on Wilcoxon rank-sum tests (Fig. 5A). The dried and wet forms of the fermented soybeans did not differ significantly for isobutyrate, valerate, hexanoate, or total SCFAs (Fig. 5A). The fermentation concentrations in the dried and wet forms of the fermented soybeans were higher for acetic acid than for the other SCFAs (Fig. 5A). Acetate and isovalerate were the most abundant in Chiang Rai and Lampang; isobutyrate and total SCFAs were dominant in Chiang Rai (Fig. 5B). Lampang and Chiang Mai contained the highest amounts of propionate and butyrate, respectively (Fig. 5B). Chiang Mai had the greatest amount of hexanoate (Fig. 5B). All six provinces had the highest concentrations of acetate fermentation in Thua Nao (Fig. 5B).

Comparison of SCFA concentrations in the fermented soybeans. Dried (n = 35) and wet (n = 30) fermented soybeans were compared using Wilcoxon’s rank-sum test (A) and assessed among six provinces using the Kruskal–Wallis test with a cutoff of P < 0.05 (B).
Identification of beneficial microbes associated with SCFA production
The correlations (R) between bacterial abundances and SCFA concentrations based on Spearman’s correlation coefficient were low and not significant (P > 0.05) (Fig. 6 and Table S4). The greatest R values were found between increasing Weissella (R = 0.45), Lactobacillus (R = 0.47), or Clostridium_sensu_stricto-18 (R = 0.46) abundances and acetate concentrations (Table S4). Using a cutoff of 1% for relative abundance, Weissella was found in Chiang Rai, Lampang, and Phayao; Lactobacillus was found in Chiang Rai and Lampang, and Clostridium_sensu_stricto-18 was found only in Lampang (Table S4). To increase propionate levels, Globicatella (R = 0.40), Ignatzschineria (R = 0.5), Clostridium_sensu_stricto-15 (R = 0.44), Clostridium_sensu_stricto-18 (R = 0.66), Paenalcaligenes (R = 0.40), or Staphylococcus (R = 0.46) abundances were the greatest bacterial members in fermented soybeans (Table S4). Globicatella and Clostridium_sensu_stricto-15 were found in Lampang; Ignatzschineria was found in Lampang, Chiang Mai, and Chiang Rai; Paenalcaligenes was found in Lampang and Chiang Mai, and Staphylococcus was found in Chiang Mai and Phayao, with a ≥ 1% relative abundance (Table S3). Compared with other bacterial abundances, Globicatella (R = 0.44), Ignatzschineria (R = 0.56), Corynebacterium (R = 0.49), Sphingobacterium (R = 0.49), Clostridium_sensu_stricto-15 (R = 0.50), Clostridium_sensu_stricto-18 (R = 0.59), Vagococcus (R = 0.41), Paenalcaligenes (R = 0.40), Staphylococcus (R = 0.44), and Comamonas (R = 0.40) were the most impacts on increased butyrate concentrations (Table S4). In addition, increasing total SCFAs were based on increased Weissella (R = 0.44) and Lactobacillus (R = 0.46) abundances (Table S4). Corynebacterium was found in Chiang Mai; Sphingobacterium was found in Lampang; Vagococcus was found in Lampang and Chiang Mai, and Comamonas was found in Mae Hong Son, all with a ≥ 1% relative abundance (Table S3).

Pairwise correlations between SCFAs and dominant bacterial genera with the highest total abundances across samples of > 0.1% using Spearman’s correlation coefficient.
Discussion
In this study, we characterized the microbiota of Thua Nao from different areas of northern Thailand and identified the beneficial bacteria potentially synthesizing SCFAs. We hypothesized that raw materials used in Thua Nao production (i.e., soybean and the natural flora of each area) would yield diverse bacterial classification results. We also assessed two types of Thua Nao, wet and dried, using 16S rRNA gene sequencing, a culture-independent method. At the phylum level, Firmicutes was the most abundant and common in both wet and dried fermented soybean products (Fig. 2A). Members of Firmicutes mainly include beneficial bacteria that play significant roles in the relationship between gut bacteria and human health. Some Firmicutes members can ferment dietary fiber from legumes to metabolite end-products, including SCFAs8. We further analyzed the predominant genera from Firmicutes with ≥ 1% abundances. The present results suggested that Bacillus was the most abundant in both wet and dried samples, which was consistent with previously reported microbial compositions2,4,9. As expected, Bacillus was also the predominant bacterial genus in the natto sample, which was the control in this work. However, we had only one sample for natto, which did not allow us to perform any statistical test between natto and Thua Nao.
Considering the differences in bacterial diversity between wet and dried Thua Nao, differential abundance analyses showed high abundances of lactic acid-producing bacteria (LAB). LAB inoculants are used to produce various animal and vegetable products and for developing flavors, aromas, and textures in fermented foods10. The different proportions of LAB in the wet and dried samples could be factors in the viability of LAB in preserved Thua Nao. Two LAB genera, Lactobacillus and Enterococcus, highly abundant in wet samples, have been reported to be able to hydrolyze soy proteins11,12. Lactobacillus spp. are commonly used as probiotics for human and animal supplements, and mixed cultures of Lactobacillus are used to ferment soybean meal13. Although some enterococci are considered pathogenic determinants, these bacteria also have several positive features. Many effective enterococci strains have been described as potential starters in various fermented products. The use of enterococci as starter cultures or co-cultures has increased considerably. However, the mixed natural factors of soybean substrates and other predominant bacteria (such as Bacillus, LAB, and other bacterial members) as well as other microbes (such as yeasts and moulds) might create unique flavors, aromas, textures, and nutritive values in wet Thua Nao.
Thua Nao is a popular fermented food in northern Thailand because it can be used as an ingredient to increase the flavor in cooked foods and can be eaten directly with glutinous rice or rice1. Various factors in Thua Nao manufacturing, including the types and amount of fibers in raw soybeans, the natural microbiota, environment, and fermentation duration vary among the provinces, resulting in distinct microbiome profiles and different tastes of Thua Nao. Our results indicate that Thua Nao samples from Lampang, Chiang Mai, and Chiang Rai had higher microbial diversity than those from Lamphun, Mae Hong Son, and Phayao in the wet condition (Fig. 4A).
Thua Nao contains high concentrations of non-digestible carbohydrates, which cannot be digested by endogenous enzymes in the stomach and small intestines but are a potential fermentation substrate for beneficial microbes in the gut and are considered prebiotics14,15,16. Fermentation using natural beneficial microbes in fermented food production involves hydrolyzing non-digestible carbohydrates to sugars, which are subsequently fermented to produce primarily SCFAs as well as H2 and CO216,17. Several studies reported that the types and amounts of SCFAs depend greatly on bacterial digestibility and the fermentability and viscosity of fiber16,17. The three main SCFAs, acetic acid (C2), propionic acid (C3), and butyric acid (C4), help to release energy for gut microbial growth and physiological effects in humans16. SCFAs are volatile and can be produced by anaerobic fermentation of dietary fibers6,18. These metabolites were significantly higher in wet Thua Nao than in dried Thua Nao (Fig. 5A), possibly owing to their volatility and destroyed anaerobic enzymatic activities during the drying process15,18,19.
Acetic acid was the most abundant SCFA in both wet and dried Thua Nao samples. Previous studies revealed that the main acetic acid producers in the solid fermentation included anaerobic Clostridium, Ignatzschineria, LAB and aerobic acetic acid bacteria (AAB)20,21,22. In this study, Clostridium sensu stricto, Ignatzschineria, LAB (i.e., Lactobacillus, Pediococcus, Streptococcus, Enterococcus, Globicatella, Vagococcus, and Weissella), and AAB (i.e., Acetobacter) in Thua Nao might be associated with acetic acid production during fermentation. The two highest concentrations of acetic acid found in Chiang Rai and Lampang corresponded to the predominance of LAB, AAB, Ignatzschineria, and Clostridium spp.
The three major propionate formation pathways are the succinate, acrylate, and propanediol pathways23. Propionibacterium and Clostridium, which are anaerobes, have been explored as potential propionate producers via the succinate pathway20,24. Correlation analysis showed that the propionate concentrations were closely related to Globicatella, Ignatzschineria, Clostridium sensu stricto, Paenalcaligenes, and Staphylococcus abundances. Clostridium, Ignatzschineria, and Paenalcaligenes are reported to be potential propionic acid producers25,26. Although Ignatzschineria and Paenalcaligenes belong to the phylum Proteobacteria, implicated in human infections27, these genera were found in very low amounts (< 1%) in dried Thua Nao (Table S2). The highest amount of propionic acid was observed in Lampang, followed by Chiang Mai, and corresponded to the highest relative abundances of Globicatella, Clostridium sensu stricto, and Staphylococcus.
Correlation analysis showed that the butyrate concentrations were closely related to the abundances of Globicatella, Ignatzschineria, Corynebacterium, Sphingobacterium, Clostridium sensu stricto, Vagococcus, Paenalcaligenes, Staphylococcus, and Comamonas. Previous studies showed that butyrate can be generated from the fermentation of LAB such as Globicatella, Vagococcus, Staphylococcus; Bacillus spp.; Corynebacterium, Comamonas, Sphingobacterium, Ignatzschineria; and Clostridium sensu stricto28,29,30. The highest amount of butyric acid was observed in Chiang Mai, followed by Lampang, and corresponded to the highest relative abundances of Corynebacterium in Chiang Mai, Sphingobacterium and Vagococcus in Chiang Mai and Lampang, Globicatella and Clostridium sensu stricto in Lampang, and Comamonas in Mae Hong Son. Correlation analysis showed that the total SCFA concentrations were closely related to the relative abundances of Weissella and Lactobacillus. The highest amounts of total SCFAs observed in Chiang Rai corresponded to the highest relative abundances of Weissella and Lactobacillus.
Our results indicate that both wet and dried Thua Nao from the northern provinces of Thailand are potential sources of SCFAs. Notably, we used an amplicon-based microbiome approach to observe the bacterial communities in Thua Nao, and the results were based on genus-level bacteria. This study provides insight into SCFA-microbiota associations in Thua Nao, which could be used to develop starter culture for SCFA-enriched fermented soybean production as a supplementary food to modulate lifelong health. The effect of the microbiome in Thua Nao on SCFA fermentation end-products should be further investigated using other approaches, including shotgun metagenomics, genome sequencing, and metabolomics.
Methods
Sample collection
Sixty-five Thua Nao products (naturally fermented soybean samples) consisting of 30 wet and 35 dried samples were collected from 36 villages in six northern provinces of Thailand: Chiang Rai (14 dried, 5 wet), Chiang Mai (5 dried, 5 wet), Mae Hong Son (5 dried, 5 wet), Lampang (5 dried, 8 wet), Lamphun (5 dried, 5 wet), and Phayao (1 dried, 2 wet). We also used natto, a soybean from Japan fermented using a leavening agent as the control. In 27 of 36 villages, we collected both wet and dried fermented soybean samples. Moisture content was analyzed for weight determination.
DNA extraction and 16S rRNA sequencing
Total genomic DNA was extracted from 67 samples using the DNeasy® Power Food® Microbial Kit (Qiagen, MD, USA). The DNA concentration was determined using a Nanodrop instrument (Thermo Scientific, USA). DNA integrity was monitored on 1% agarose gel. The extracted DNA samples were shipped to Genome Quebec Innovation Center (McGill University, Montreal, QC, Canada) for PCR amplification, library preparation, and sequencing. PCR was performed to produce V4 regions of the 16S rDNA gene using the conserved primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). A FastStart® High Fidelity PCR System (Roche, Mannheim, Germany) was used for 26 amplification cycles. A blank sample without a DNA template was used as a negative control. The library was sequenced on an Illumina MiSeq platform per the company's protocol, and 250-bp paired-end reads were generated. A mock community (Zymo Research, USA), including 10 bacterial species, was used as a positive control.
Determination of SCFAs in Thua Nao
SCFA analysis was performed using gas chromatography per the methods of31,32 with slight modifications. One gram of ground Thua Nao samples was mixed with 3 mL of internal standard solution (heptanoic acid, 5.04 µM/g sample). Samples were vortexed and centrifuged at 2,500 × g at 4 °C for 10 min, then 10 µL of 1 M phosphoric acid was added to 300 µL of the supernatant. The supernatant was filtered through a nylon filter with 0.45-µm pore size, and 0.2 μL of the filtrate was injected into a gas chromatograph equipped with a flame ionization detector (GC-FID, model 7890A; Agilent Technologies, Santa Clara, CA, USA) and capillary column (DB-FFAP, 30 m × 0.53 mm × 0.5 µm). The initial oven temperature was held at 90 °C for 1 min, then increased at 20 °C/min to 190 °C and held for 2.5 min. The injector and detector temperatures were set at 210 °C. Helium was used as a carrier gas with the gas flow and septum purge rates at 7.7 and 3.0 mL/min, respectively. A standard SCFA mixture containing acetate, propionate, butyrate, isobutyrate, valerate, isovalerate, and hexanoate as well as phenol and p-cresol was used for the calculation. The SCFA concentrations were calculated in µM/g. Quantification was performed based on the relative peak area of each SCFA standard, adjusting the quantity based on the internal standard.
Microbiome data analysis
Sequences were 250-bp paired-end reads in the FASTQ format. Sequences were imported into QIIME2-2022.233. Adapters and primers were trimmed from the front of both forward and reverse reads using q2-cutadapt. Sequences were truncated at the forward and reverse positions; paired-end reads were then merged with overlapping regions into sequences representative of V4 16S rRNA, and chimeric sequences were discarded using q2-dada2. Preprocessed sequences were clustered into features based on the amplicon sequence variant method, and the feature counts in the samples were arranged in an abundance table34. Features were taxonomically annotated based on SILVA v13835. Rarefying normalization was applied to make all samples comparable to the equal number of reads. The taxonomic abundances of all samples were calculated using q2-classify-sklearn.
Diversity analysis
Taxonomic abundances at the genus level were used to calculate diversity metrics. Alpha diversity based on Shannon’s diversity, Simpson’s diversity, Pielou’s evenness, and Faith’s phylogenetic distance were estimated for all samples. Alpha diversity was compared between soybean types using Wilcoxon’s rank-sum tests and evaluated among provinces using Kruskal–Wallis tests. Pairwise comparisons between provinces were conducted using Wilcoxon’s rank-sum tests with multiple hypothesis corrections. Beta diversity in terms of Bray–Curtis dissimilarity, Jaccard index, UniFrac phylogenetic distance, and weighted UniFrac phylogenetic distance was calculated for all samples. PCoA was performed using Phyloseq v1.36.036.
Differential abundance analysis
ANCOM-BC v1.2.237,38,39 was performed to identify differentially abundant microbes between wet and dried fermented soybeans with the criteria P < 0.05.
Association analysis
Taxa with total relative abundances < 0.1% were filtered out from the taxonomic table. Pairwise correlations between taxonomic profiles and SCFA concentrations were calculated using Spearman’s correlation coefficient and visualized using ggplot2 v3.3.5.
Statistical tests
Wilcoxon rank-sum tests and Kruskal–Wallis tests were conducted in R, v4.1.0. Boxplots and PCoA plots were created using ggplot2 v3.3.5.
Data availability
Sequencing data were submitted to NCBI SRA with accession numbers SRR20217885– SRR20217951 under the BioProject accession number PRJNA852866.
References
Chukeatirote, E. Thua nao: Thai fermented soybean. J. Ethn. Foods 2, 115–118 (2015).
Inatsu, Y. et al. Characterization of Bacillus subtilis strains in Thua nao, a traditional fermented soybean food in northern Thailand. Lett. Appl. Microbiol. 43, 237–242 (2006).
Dajanta, K., Chukeatirote, E. & Apichartsrangkoon, A. Nutritional and physicochemical qualities of Thua nao (Thai traditional fermented soybean). Chiang Mai J. Sci. 39, 562–574 (2012).
Petchkongkaew, A., Taillandier, P., Gasaluck, P. & Lebrihi, A. Isolation of Bacillus spp. from Thai fermented soybean (Thua-nao): Screening for aflatoxin B1 and ochratoxin A detoxification. J. Appl. Microbiol. 104, 1495–1502 (2008).
Henningsson, Å., Björck, I. & Nyman, M. Short-chain fatty acid formation at fermentation of indigestible carbohydrates. Näringsforskning 45, 165–168 (2001).
Demigne, C. et al. Effect of propionate on fatty acid and cholesterol synthesis and on acetate metabolism in isolated rat hepatocytes. Br. J. Nutr. 74, 209–219 (1995).
Roshanravan, N. et al. Effect of butyrate and inulin supplementation on glycemic status, lipid profile and glucagon-like peptide 1 level in patients with type 2 diabetes: A randomized double-blind, placebo-controlled trial. Horm. Metab. Res. 49, 886–891 (2017).
Calatayud, M. et al. Comparative effect of 22 dietary sources of fiber on gut microbiota of healthy humans in vitro. Front. Nutr. 8, 700571 (2021).
Leejeerajumnean, A. Thua nao: Alkali fermented soybean from Bacillus subtilis. Silpakorn Univ. Int. J. 3, 277–292 (2007).
Pescuma, M., Hebert, E. M., Mozzi, F. & de Valdez, G. F. Functional fermented whey-based beverage using lactic acid bacteria. Int. J. Food. Microbiol. 141, 73–81 (2010).
Shirotani, N. et al. Proteolytic activity of selected commercial Lactobacillus helveticus strains on soy protein isolates. Food Chem. 340, 128152 (2021).
Biscola, V. et al. Proteolytic activity of Enterococcus faecalis VB63F for reduction of allergenicity of bovine milk proteins. J. Dairy Sci. 99, 5144–5154 (2016).
Xia, Y. et al. Effects of dietary Lactobacillus rhamnosus JCM1136 and Lactococcus lactis subsp. lactis JCM5805 on the growth, intestinal microbiota, morphology, immune response and disease resistance of juvenile Nile tilapia, Oreochromis niloticus. Fish Shellfish Immunol. 76, 368–379 (2018).
Alexander, C., Swanson, K. S., Fahey, G. C. & Garleb, K. A. Perspective: physiologic importance of short-chain fatty acids from nondigestible carbohydrate fermentation. Adv. Nutr. 10, 576–589 (2019).
De Baere, S. et al. Development of a HPLC-UV method for the quantitative determination of four short-chain fatty acids and lactic acid produced by intestinal bacteria during in vitro fermentation. J. Pharm. Biomed. Anal. 80, 107–115 (2013).
Pylkas, A. M., Juneja, L. R. & Slavin, J. L. Comparison of different fibers for in vitro production of short chain fatty acids by intestinal microflora. J. Med. Food 8, 113–116 (2005).
Hye Jeong, Y., Sunmin, P., Valeriy, P., Kyung Rhan, C. & Dae Young, K. Fermented soybean products and their bioactive compounds, in Soybean and Health. (ed. E.-S. Hany) Ch. 2 (IntechOpen, Rijeka; 2011).
den Besten, G. et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 54, 2325–2340 (2013).
Hoffmann, S. & Justesen, T. Effect of temperature, humidity and exposure to oxygen on the survival of anaerobic bacteria. J. Med. Microbiol. 13, 609–612 (1980).
Hu, C. et al. A comprehensive analysis of the colonic flora diversity, short chain fatty acid metabolism, transcripts, and biochemical indexes in heat-stressed pigs. Front. Immunol. 12, 717723 (2021).
Fusco, V. et al. The genus Weissella: Taxonomy, ecology and biotechnological potential. Front. Microbiol. 6, 155 (2015).
Gonzalez-Garcia, R. A. et al. Microbial propionic acid production. Fermentation 3, 21 (2017).
Reichardt, N. et al. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 8, 1323–1335 (2014).
Flint, H. J., Duncan, S. H., Scott, K. P. & Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 74, 13–22 (2015).
Chen, L. et al. Effect of lactic acid bacteria and propionic acid on conservation characteristics, aerobic stability and in vitro gas production kinetics and digestibility of whole-crop corn based total mixed ration silage. J. Integr. Agric. 16, 1592–1600 (2017).
Jia, T., Yun, Y. & Yu, Z. Propionic acid and sodium benzoate affected biogenic amine formation, microbial community, and quality of oat silage. Front. Microbiol. 12, 750920 (2021).
Deslandes, V., Haney, C., Bernard, K. & Desjardins, M. Ignatzschineria indica bacteremia in a patient with a maggot-infested heel ulcer: A case report and literature review. Access Microbiol. 2, acmi000078 (2020).
Chen, Y. et al. Directional production of even carbon volatile fatty acids from peanut meal: Effects of initial pH and hydraulic residence time. Environ. Eng. Res. 27, 210190–210190 (2022).
Njalam’mano, J. B. J. & Chirwa, E. M. N. Indigenous butyric acid-degrading bacteria as surrogate pit latrine odour control: Isolation, biodegradability performance and growth kinetics. Ann. Microbiol. 69, 107–122 (2019).
Jung, J. Y., Lee, S. H. & Jeon, C. O. Microbial community dynamics during fermentation of doenjang-meju, traditional Korean fermented soybean. Int. J. Food Microbiol. 185, 112–120 (2014).
Charoensiddhi, S., Conlon, M. A., Vuaran, M. S., Franco, C. M. M. & Zhang, W. Impact of extraction processes on prebiotic potential of the brown seaweed Ecklonia radiata by in vitro human gut bacteria fermentation. J. Funct. Foods 24, 221–230 (2016).
McOrist, A. L., Abell, G. C. J., Cooke, C. & Nyland, K. Bacterial population dynamics and faecal short-chain fatty acid (SCFA) concentrations in healthy humans. Br. J. Nutr. 100, 138–146 (2008).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Callahan, B. J., McMurdie, P. J. & Holmes, S. P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643 (2017).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 41, D590-596 (2013).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Lin, H. & Peddada, S. D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 11, 3514 (2020).
Kaul, A., Mandal, S., Davidov, O. & Peddada, S. D. Analysis of microbiome data in the presence of excess zeros. Front. Microbiol. 8, 2114 (2017).
Mandal, S. et al. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 26, 27663 (2015).
Acknowledgements
We thank Traci Raley, MS, ELS, from Edanz (www.edanz.com/ac) for editing a draft of this manuscript.
Funding
PJI was supported by BRAND’s Health Research Award 2019 and the MU-KMUTT Biomedical Engineering and Biomaterials Consortium Grant 2021.
Author information
Authors and Affiliations
Contributions
All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wongsurawat, T., Sutheeworapong, S., Jenjaroenpun, P. et al. Microbiome analysis of thai traditional fermented soybeans reveals short-chain fatty acid-associated bacterial taxa. Sci Rep 13, 7573 (2023). https://doi.org/10.1038/s41598-023-34818-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-34818-0
This article is cited by
-
Comprehensive bacterial-metabolite profiles of Hawaijar, Bekang, and Akhone: a comparative study on traditional fermented soybeans of north-east India
World Journal of Microbiology and Biotechnology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
