
Abstract
Mycoplasma fermentans is a proposed risk factor of several neurological diseases that has been detected in necrotic brain lesions of acquired immunodeficiency syndrome patients, implying brain invasiveness. However, the pathogenic roles of M. fermentans in neuronal cells have not been investigated. In this study, we found that M. fermentans can infect and replicate in human neuronal cells, inducing necrotic cell death. Necrotic neuronal cell death was accompanied by intracellular amyloid-β (1–42) deposition, and targeted depletion of amyloid precursor protein by a short hairpin RNA (shRNA) abolished necrotic neuronal cell death. Differential gene expression analysis by RNA sequencing (RNA-seq) showed that interferon-induced transmembrane protein 3 (IFITM3) was dramatically upregulated by M. fermentans infection, and knockdown of IFITM3 abolished both amyloid-β (1–42) deposition and necrotic cell death. A toll-like receptor 4 antagonist inhibited M. fermentans infection-mediated IFITM3 upregulation. M. fermentans infection also induced necrotic neuronal cell death in the brain organoid. Thus, neuronal cell infection by M. fermentans directly induces necrotic cell death through IFITM3-mediated amyloid-β deposition. Our results suggest that M. fermentans is involved in neurological disease development and progression through necrotic neuronal cell death.
Similar content being viewed by others


Introduction
Mycoplasmas are the smallest and simplest self-replicating prokaryotes. They are found everywhere in nature and are recognized as pathogens and cofactors of several diseases1,2. They belong to the Mollicutes class, and their small size and absence of a cell wall distinguish them from other bacteria. More than 100 species have been identified, and they usually display strict host and tissue specificities. However, despite clinical evidence implicating mycoplasmas in various neurological diseases4,5,6,7, mycoplasma-host cell interactions and pathogenicity remain poorly understood3.
Mycoplasma fermentans has been detected in and isolated from numerous tissues, including blood of human immunodeficiency virus (HIV)-infected patients; hence, it is considered an invasive opportunistic pathogen under immunocompromised conditions4. However, clinical studies also suggest that M. fermentans is a putative risk factor of certain neurological diseases, including chronic fatigue syndrome (CFS)5, Gulf War syndrome (GWS)5, amyotrophic lateral sclerosis (ALS)6, and autism spectrum disorder (ASD)7. Many studies have reported central nervous system (CNS) invasion by mycoplasmas, suggesting that it could negatively affect neuronal cells in various animals8. M. gallisepticum and M. synoviae have been isolated from brains of birds, and M. bovis has been isolated from brain tissue of calves8. Also, M. fermentans can directly disseminate into the CNS since it has been found in brain necrotic lesions of AIDS patients9,10. In addition, in vivo experiments detected M. fermentans DNA in the brain after the bacterium was intraperitoneally injected into nonhuman primates11 and intratracheally injected into hamsters12. M. fermentans was also identified in necrotizing lesions in lymph nodes, spleen, liver, and brain in non-AIDS patients who died of an acute flu-like illness13,14. In a study on M. pneumoniae, bacteria were detected in and isolated from cerebrospinal fluid (CSF) samples of patients presenting various neurological symptoms15, and fatal acute disseminated encephalomyelitis patients showed brain invasion by M. pneumonia16. This evidence suggests that infection with mycoplasmas, including M. fermentans, can result in invasion of the CNS and damage to neuronal cells, promoting pathogenesis in various human neurological diseases.
Even though M. fermentans has been suggested as an etiological factor for several human neurological diseases, interactions between M. fermentans and neuronal cells in neurological diseases remain poorly understood. In this study, cellular and molecular approaches were used to understand the neuropathological changes in patients infected with M. fermentans. We provide evidence for necrotic neuronal cell death caused by M. fermentans in both human neuronal cells and brain organoids. Although we used a brain organoid system instead of a mouse infection model because M. fermentans did not induce toll-like receptor 4 (TLR4)-mediated interferon-induced transmembrane protein 3 (IFITM3) upregulation and necrotic cell death in mouse neuronal cells, our cerebral organoid model system clearly supports M. fermentans infection-mediated necrotic neuronal cell death.
Results
M. fermentans can infect and replicate in SH-SY5Y cells to induce necrotic cell death
To explore the pathogenic role of M. fermentans infection in neuronal cells, we infected human neuroblastoma SH-SY5Y cells and mouse hippocampal neuronal HT-22 cells with M. fermentans. To assess replication in neuronal cells, we used a MOI of 0.01. At 12 days post-infection (dpi), morphological changes, including significant cellular debris and a low cell count, were detected in infected SH-SY5Y cells (Fig. 1a). Although previous studies showed that M. fermentans infection promotes immortalization of Epstein-Barr virus (EBV)-infected human peripheral blood mononuclear cells17 and induces apoptosis in 32D cells18, infected SH-SY5Y cells showed more propidium iodide (PI)-staining and weaker annexin V-staining than uninfected controls (Fig. 1b), indicating that M. fermentans induces necrotic neuronal cell death, not apoptosis. However, HT-22 cells showed no morphological changes (Fig. 1c) or obvious cell death (Fig. 1d). To investigate infection and replication of M. fermentans in SH-SY5Y and HT-22 cells, intracellular and secreted M. fermentans DNA levels were measured by real-time quantitative PCR (qPCR). We detected intracellular and secreted M. fermentans DNA only in infected SH-SY5Y cells, not in HT-22 cells (Supplementary Fig. S1a, b). In addition, we infected another mouse neuroblastoma cell line (neuro2a) with M. fermentans. In contrast to HT-22 cells, we detected intracellular and secreted M. fermentans DNA in infected neuro2a cells (Supplementary Fig. S1c), but no necrotic cell death of mouse neuronal cells (Supplementary Fig. S1d). These results suggest that M. fermentans can directly infect and replicate in neuronal cell lines, but that necrotic cell death may be species-specific.

M. fermentans infection induces necrotic cell death in SH-SY5Y cells. (a) Representative microscopy images of mock- or M. fermentans-infected SH-SY5Y cells at 12 days postinfection (dpi). (b) Representative flow cytometry dot plots from Annexin V/propidium iodide (PI) apoptosis assays for SH-SY5Y cells from (a) and a bar graph of Annexin V-negative PI-positive cells (necrotic cells). (c) Representative microscopy images of HT-22 cells at 12 dpi. (d) Representative flow cytometry dot plots from Annexin V/PI apoptosis assays for HT-22 cells from (c) and a bar graph of Annexin V-negative PI-positive cells. (e, f) Representative flow cytometry dot plots from Annexin V/PI apoptosis assays for SH-SY5Y cells at 12 dpi after inactivation of M. fermentans with 70% EtOH, heating, UV irradiation (e), and mycoplasma antibiotics (f). (g, h) Bar graphs of Annexin V-negative PI-positive cells for data from (e, f). Bar graphs present mean values ± standard deviation (SD). Scale bars = 100 μm for images in (a, c); *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; NS, not significant (unpaired Student’s t-test). Data are averages from three independent experiments.
We also used phorbol 12-myristate 13-acetate (PMA)-induced differentiated SH-SY5Y cells to determine whether infection also affected differentiated neuronal cells (Supplementary Fig. S2a). Differentiated SH-SY5Y cells showed increased microtubule-associated protein 2 (MAP2) gene expression, extensive axon-like processes, and reduced cell growth compared with controls, consistent with previous studies (Supplementary Fig. S2b, c)19. At 19 dpi, M. fermentans-infected differentiated SH-SY5Y cells also exhibited necrotic cell death (Supplementary Fig. S2d–f). To determine the infectiousness of secreted M. fermentans derived from infected differentiated SH-SY5Y cells, we added conditioned medium from infected or noninfected differentiated SH-SY5Y cells into fresh differentiated SH-SY5Y cells at 19 dpi (Supplementary Fig. S2a). Conditioned medium derived from M. fermentans-infected differentiated SH-SY5Y cells induced necrotic cell death in fresh differentiated SH-SY5Y cells (Supplementary Fig. S2g, h). However, M. fermentans inactivated by heating, UV irradiation, and 70% ethanol treatment failed to induce necrotic cell death of SH-SY5Y cells (Fig. 1e–h). In addition, we performed in vitro drug testing by infecting with M. fermentans pretreated with mycoplasma antibiotics, and this drug treatment also failed to induce necrotic cell death. Overall, our results suggest that M. fermentans can infect and replicate in differentiated SH-SY5Y cells to induce necrotic cell death, and secreted M. fermentans then spreads into fresh cells to induce necrotic cell death. Indeed, clinical evidence suggests putative causal links between M. fermentans infection and various neurological diseases including CFS, GWS, ALS, and ASD (Supplementary Table S1)5,6,7,20,21,22,23,24. Therefore, our findings strengthen evidence indicating that M. fermentans may play a pathogenic role in human neurological diseases.
Necrotic neuronal cell death caused by M. fermentans is mediated by intracellular Aβ1−42 deposition
Infectious neuro-pathogens can drive amyloidosis and thereby play a protective role in innate immunity in brain25. Therefore, we tested Aβ deposition caused by M. fermentans, and phosphorylated tau (p-tau) that is induced by Aβ deposition, in human neuronal cells26. To measure deposition of p-tau and Aβ1−42, we analyzed these proteins in differentiated SH-SY5Y cells at 7, 12, and 19 dpi using western blotting. The results showed that p-tau (Ser202, Thr205) was significantly increased at 19 dpi, but t-tau was not increased significantly (Fig. 2a– c). We also checked the quality of p-tau bands in the western blot; p-tau bands were confirmed by treating with calf intestinal phosphatase (CIP; Fig. 2d). In addition, intracellular Aβ1−42 levels were increased at 19 dpi (Fig. 2e, f). Increased intracellular Aβ1−42 was also confirmed using flow cytometry analysis (Fig. 2g, h). Immunocytochemical analysis also showed that both intracellular Aβ1−42 and p-tau were increased in differentiated SH-SY5Y cells at 19 dpi, but t-tau was not increased (Fig. 2i, j).

M. fermentans-mediated necrotic cell death induced by intracellular Aβ1–42 deposition. (a) Representative western blotting images from mock- or M. fermentans-infected differentiated SH-SY5Y cells using the indicated antibodies at 1, 7, 12, and 19 dpi. (b, c) Bar graphs of western blotting analysis data from (a) for the ratio of p-tau (b) and t-tau (c) to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) relative to mock controls at each time point. (d) Representative western blotting images from M. fermentans-infected differentiated SH-SY5Y cells using the indicated antibodies at 19 dpi after calf intestinal phosphatase (CIP) treatment. (e) Representative western blotting images from mock- or M. fermentans-infected differentiated SH-SY5Y cells using the indicated antibodies at 1, 7, 12, and 19 dpi. (f) Bar graph of western blotting analysis data from (e) for the ratio of Aβ1–42 to GAPDH relative to mock controls at each time point. (g) Representative flow cytometry histogram of intracellular Aβ1–42 in mock- or M. fermentans-infected differentiated SH-SY5Y cells at 19 dpi. (h) Bar graph for data from (g). (i) Representative immunocytochemistry images from mock- or M. fermentans-infected differentiated SH-SY5Y cells using the indicated antibodies at 19 dpi. (j) Bar graph of mean fluorescence intensity (MFI) for each protein normalized by Hoechst intensity from (i). (k) Bar graph of hAPP gene expression levels normalized against the GAPDH gene of hAPP knockdown SH-SY5Y cells relative to control knockdown SH-SY5Y cells, determined by qPCR. (l) Representative flow cytometry dot plots from Annexin V/PI apoptosis assays for mock- or M. fermentans-infected indicated gene knockdown SH-SY5Y cells at 19 dpi. (m) Bar graph of Annexin V-negative PI-positive cells from (l). Original blots are presented in Supplementary Fig. S9. Con, control knockdown SH-SY5Y cells; APP, hAPP knockdown SH-SY5Y cells. Bar graphs present mean values ± SD. Scale bars = 20 μm for (i); *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; NS, not significant (unpaired Student’s t-test). Data are averages of three or more independent experiments.
To validate necrotic cell death caused by Aβ1–42, we generated stable human amyloid precursor protein (hAPP) knockdown and control knockdown SH-SY5Y cell lines using short hairpin RNA (shRNA) lentivirus, and we observed a decrease in hAPP mRNA level in hAPP knockdown SH-SY5Y cells compared with control cells (Fig. 2k). Necrotic neuronal cell death was dramatically inhibited in hAPP knockdown cells at 19 dpi (Fig. 2l, m). These results indicate that M. fermentans induced intracellular Aβ1–42 deposition, and this caused necrotic cell death.
M. fermentans infection induces Aβ1–42 accumulation and necrotic neuronal cell death in brain organoids
To evaluate the potential pathogenic impact of M. fermentans in a more physiologically relevant system, we utilized human brain organoids instead of a mouse infection model, since previous results showed that mouse neuronal cells are not susceptible to M. fermentans-induced necrotic cell death. We added M. fermentans to human brain organoids at 40 days (Fig. 3a). At 26 dpi, infected brain organoids were decreased in size compared with controls (Fig. 3b, c). To confirm the infection and replication of M. fermentans in brain organoids, intracellular and secreted M. fermentans DNA levels were measured by qPCR. We detected intracellular and secreted M. fermentans DNA only in M. fermentans-infected brain organoids at 26 dpi (Supplementary Fig. S3a–d). We conducted immunohistochemical analysis to confirm necrotic cell death and Aβ1–42 deposition in brain organoids at 26 dpi. After 67 days brain organoids exhibited a ventricular-like structure containing packed SOX2-positive neuronal progenitors, with a beta-tubulin III (TUJ1)-positive neuronal layer at the outer border (Supplementary Fig. S3e)27. Additionally, the infected brain organoids contained more Aβ1–42 in the disrupted TUJ1 and region around the outer border of brain organoids (Fig. 3d), indicating that M. fermentans can induce necrotic cell death through Aβ1–42 deposition in mature neurons of brain organoids. We also confirmed increased p-tau and phosphorylated mixed lineage kinase domain-like (pMLKL) protein in infected brain organoids (Fig. 3e). Collectively, these results suggest that M. fermentans induces necrotic neuronal cell death by inducing Aβ1–42 deposition in brain organoids as well as human neuronal cell lines. Our results also suggest that amyloid pathology can also induce hyperphosphorylated tau as well as necrotic neuronal cell death through pMLKL26.

M. fermentans induces necrotic cell death accompanied by accumulation of Aβ1–42 and p-tau in human induced pluripotent stem cell (hIPSC)-derived brain organoids. (a) Schematic diagram of M. fermentans infection of maturated brain organoids derived from hIPSCs. (b) Representative microscopy images of mock- or M. fermentans-infected brain organoids at 26 dpi. (c) Dot plot of M. fermentans-infected brain organoid sizes relative to mock controls at each time point. (d, e) Representative immunohistochemistry images of mock- or M. fermentans-infected brain organoids using the indicated antibodies at 26 dpi. Scale bars = 1000 μm for (b) and 50 μm for (d, e). Results are expressed as mean ± SD; ***p ≤ 0.001 (unpaired Student’s t-test). Results are averaged from more than three independent experiments.
Differential gene expression induced by M. fermentans infection
We sought to identify the pathways involved in intracellular Aβ1–42 deposition and necrotic cell death in M. fermentans-infected differentiated SH-SY5Y cells using RNA sequencing (RNA-seq) analysis. To identify genes implicated in necrotic cell death resulting from M. fermentans infection, we compared gene expression profiles between M. fermentans-infected samples collected at 19 dpi, when necrotic cell death was observed, and mock and 1 dpi samples, which did not display necrotic cell death. Analysis of 1dpi samples was conducted to distinguish genes associated with M. fermentans infection from those not related to necrotic cell death. Therefore, overlapping regions in the Venn diagram indicate genes that are specifically upregulated or downregulated in response to M. fermentans infection at 19 dpi and associated with necrotic cell death (Fig. 4a). We identified 1,144 upregulated genes and 543 downregulated genes after induction of necrotic cell death by M. fermentans infection (Fig. 4a), as well as the top five biological process categories and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with either upregulated or downregulated genes (Fig. 4b). The top enriched pathways linked to downregulated genes included cell cycle, cell division, and DNA replication (Fig. 4b), while the top enriched pathways associated with upregulated genes included signal transduction, inflammatory response, immune response, cytokine-cytokine receptor interaction, and tumor necrosis factor (TNF) signaling (Fig. 4b). Notably, inflammatory responses and TNF signaling are central pathways of cognitive decline and Aβ1–42 deposition28. Interestingly, we found that growth differentiation factor 15 (GDF15), a blood marker for myalgic encephalomyelitis (ME)/CFS29 and mitochondria dysfunction30, was upregulated in M. fermentans-infected cells (Fig. 4c). We also identified upregulated genes known to be associated with AD pathogenesis, namely TNF-α, apolipoprotein E (APOE), prion protein (PRNP), IFITM3, and interleukin-1β (IL-1β) (Fig. 4c). These upregulated genes were confirmed by measuring mRNA levels in M. fermentans-infected SH-SY5Y cells by qPCR at 19 dpi (Fig. 4d). TNF and IL-1β are key proinflammatory cytokines in neuroinflammation and degenerative conditions28,31, and APOE interacts with Aβ and promotes the aggregation and pathology of Aβ32. PRNP encodes PrP, a receptor for internalization of oligomeric Aβ, to induce tau hyperphosphorylation and necrotic cell death26,33. IFITM3 in neurons and astrocytes binds to γ-secretase and upregulates its activity, thereby increasing the production of Aβ1–4234. IFITM3 is known to play a role in restricting infection by some viruses and bacteria, such as SARS-CoV-2 and Mycobacterium tuberculosis35,36. To investigate the potential positioning of genes (Fig. 4d) within the signaling pathway involved in M. fermentans-induced Aβ1–42 deposition, we performed experiments to assess the effect of hAPP knockdown on their expression. The results revealed that knockdown of hAPP inhibited hIL-1β and hPRNP expression in M. fermentans-infected SH-SY5Y cells, while hAPOE expression was upregulated (Supplementary Fig. S4). These findings provide further insights into the potential roles of these genes in the signaling pathway, and suggest their potential as upstream or downstream components.

Differential gene expression induced by M. fermentans infection. (a) Venn diagram of the number of upregulated (grey, fold change ≥ 2) or downregulated (blue, fold change ≤ 2) genes of M. fermentans-infected differentiated SH-SY5Y cells at 19 dpi compared to 1 dpi or mock controls, determined by RNA-seq. (b) Gene Ontology (GO) biological process and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway categories related to upregulated (left grey panel) and downregulated (right blue panel) genes. All GO and KEGG groups were identified according to the EASE score (p ≤ 0.05). (c) Scatter plots of RNA-seq analysis data from (a). (d) Bar graph of indicated gene expression levels normalized against the GAPDH gene in M. fermentans-infected SH-SY5Y cells relative to mock-infected SH-SY5Y cells at 19 dpi, determined by qPCR. Bar graphs present mean values ± SD; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001 (unpaired Student’s t-test). Data are averages of three or more independent experiments.
M. fermentans induces IFITM3-mediated Aβ1−42 accumulation, resulting in necrotic neuronal cell death
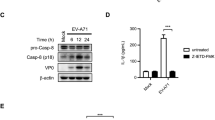
To confirm the function of IFITM3 in Aβ1–42 deposition, we generated stable IFITM3 knockdown SH-SY5Y cells using shRNA lentivirus and infected these with M. fermentans. To evaluate the impact of IFITM3 on mycoplasma infection, we conducted experiments to measure M. fermentans growth in cells with IFITM3 knockdown. The results revealed a significantly higher infection level in cells with IFITM3 knockdown than in control knockdown cells (Supplementary Fig. S5). Moreover, at 12 dpi, M. fermentans-infected IFITM3 knockdown cells showed lower necrotic cell death than the control cells, although M. fermentans levels were higher in IFITM3 knockdown cells (Fig. 5a, b). ELISA demonstrated that M. fermentans increased Aβ1–42 secretion, and that this was reduced by IFITM3 knockdown (Fig. 5c). Furthermore, using western blotting, we confirmed that IFITM3 and intracellular Aβ1–42 protein levels were increased by M. fermentans infection, and that the levels of intracellular Aβ1–42 proteins were reduced in IFITM3 knockdown cells (Fig. 5d).

M. fermentans induces IFITM3-mediated Aβ1-42 accumulation, resulting in necrotic neuronal cell death. (a) Representative flow cytometry dot plots from Annexin V/PI apoptosis assays for mock- or M. fermentans-infected indicated gene knockdown SH-SY5Y cells at 12 dpi. (b) Bar graph of Annexin V-negative PI-positive cells for data from (a). (c) Quantification of secreted Aβ1−42 in the SH-SY5Y culture medium from (a) determined by ELISA. (d) Representative western blotting image from mock- or M. fermentans-infected indicated gene knockdown SH-SY5Y cells using the indicated antibodies at 12 dpi. (e) Representative flow cytometry dot plots from Annexin V/PI apoptosis assays of mock- or M. fermentans-infected SH-SY5Y cells after TLR4 antagonist or the indicated neutralizing antibody (NAb) treatment. (f) Bar graph of Annexin V-negative PI-positive cells from (e). (g) Bar graph of hIFITM3 gene expression levels normalized against the GAPDH gene in infected SH-SY5Y cells from (e). (h) Representative flow cytometry dot plots from Annexin V/PI apoptosis assays of mock- or M. fermentans-infected SH-SY5Y cells after treatment with TLR2 antagonist. (i) Bar graph of Annexin V-negative PI-positive cells from (h). (j, k) Bar graphs of gene expression levels of hTLR4 (j) and hAPP (k) in infected SH-SY5Y cells at 12 dpi, which were determined by qPCR and normalized against the GAPDH gene. Original blots are presented in Supplementary Fig. S9. Con, control knockdown SH-SY5Y cells; TM3, hIFITM3 knockdown SH-SY5Y cells; NAb, neutralizing antibody. Bar graphs present mean values ± SD; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001 (unpaired Student’s t-test). Data are averages of three or more independent experiments.
A previous study showed that TLR2 and TLR4 ligands, and Mycobacterium tuberculosis infection, trigger IFITM3 to restrict mycobacterial growth35. RNA-seq data showed that TLR2 and TLR4 gene expression levels were increased in SH-SY5Y cells by M. fermentans at 19 dpi (Fig. 4c). Because proinflammatory cytokines can be associated with IFITM3 expression35, we explored the roles of IL-1β and TNF-α in IFITM3 upregulation during M. fermentans infection. However, neutralization of these cytokines did not affect IFITM3 upregulation, while the TLR4 antagonist inhibited necrotic cell death (Fig. 5e, f) and IFITM3 upregulation (Fig. 5g). A TLR2 antagonist had no inhibitory effect on necrotic neuronal cell death caused by M. fermentans infection (Fig. 5h, i). Therefore, we further investigated the effect of M. fermentans infection and found a significant upregulation of hTLR4 mRNA levels (Fig. 5j). Interestingly, we did not observe any significant change in the hAPP mRNA expression level, as confirmed by both RNA-seq (Fig. 4c) and qPCR (Fig. 5k). These results suggest that the observed increase in Aβ1–42 production was not due to substrate upregulation, but rather to increased γ-secretase activity. Thus, M. fermentans may induce necrotic cell death via TLR4 signaling to upregulate IFITM3 for Aβ1–42 deposition. In contrast to human neuronal cells, murine IFITM3 gene expression was not upregulated by M. fermentans (Supplementary Fig. S6a). This could be due to sequence differences in the ligand binding sites (560 of 843 amino acids, 66%) between murine and human TLR4 (Supplementary Fig. S6b). These results suggest that M. fermentans infection in human neuronal cells induces TLR4 signaling to increase Aβ1−42 accumulation via an IFITM3-mediated pathway, resulting in necrotic cell death. To determine whether the observed effects are specific to M. fermentans infection or are a general response to TLR4-activating ligands, we performed additional experiments. We added CRX-527, a CD14-independent TLR4 agonist37, and α-enolase, a protein expressed on the cell surface of M. fermentans that can activate the CD14-dependent TLR4 signaling pathway38,39,40, to SH-SY5Y cells. The results showed that neither CRX-527 nor α-enolase alone induced cell death, upregulated hTLR4, hIL-6, hTNF-α, hIL-1β, or hIFITM3 mRNA levels, or increased intracellular Aβ1–42 in SH-SY5Y cells (Supplementary Fig. S7a–e). These results are in line with those of previous research showing no impact of lipopolysaccharide (LPS) on SH-SY5Y cells41,42,43,44,45. Thus, the consequences of M. fermentans infection, such as Aβ1–42 deposition and cell death, would appear to be specifically linked to TLR4 upregulation. We also found no increase in cell death of SH-SY5Y cells after adding the α-enolase to M. fermentans-infected cells (Supplementary Fig. S7f) while an TLR4 antagonist blocked IFITM3 upregulation and necrotic cell death. These findings suggest that M. fermentans is sufficient for the activation of TLR-4 because α-enolase did not increase necrotic cell death.
Discussion
M. fermentans infection is associated with various neurological diseases such as CFS, GWS, ALS, and ASD5,6,7,20,21,22,23,24. This suggests that M. fermentans infection could have pathological relevance for neurological changes associated with shared symptoms in CFS, and GWS, ALS, and ASD, such as cognitive and neurological problems46. Indeed, mycoplasmas have been isolated from the brains of various types of animals, suggesting they can directly invade the CNS8,9,11,15,16,47,48,49. Additionally, M. fermentans have been detected in numerous tissues including the brains of AIDS patients9,10, and was also detected in a case study of non-AIDS patients with acute fatal disease13,14. Thus, M. fermentans may be an underestimated pathogen in neurological diseases. We therefore investigated pathological changes in neuronal cells infected with M. fermentans. Surprisingly, M. fermentans could infect human neuronal cells, in which it replicated and induced necrotic cell death, but it could not induce necrotic cell death in mouse neuronal cells.
IFITM3, a member of the interferon-induced transmembrane protein family, is a restriction factor that prevents enveloped viral particles from entering host cells by blocking membrane fusion during endocytosis50,51. It also blocks mycobacterial growth35, plays a critical role in intrinsic antiviral immunity of human cells, and restricts cell entry of diverse viruses including influenza virus, West Nile virus, dengue virus, Ebola virus, and coronavirus52. IFITMs are potent antiviral effectors known for their ability to inhibit fusion between viral and cellular membranes, and they are widely studied for their potential as antiviral therapies51,52. Interestingly, a study revealed that IFITM3 in neurons can bind to γ-secretase to upregulate its activity, thereby increasing the production of Aβ1–4234. In our current study, we also found that M. fermentans could upregulate IFITM3, which led to deposition of Aβ1–42. Eventually, this triggered necrotic cell death in infected human neuronal cells. Thus, our results suggest that M. fermentans may contribute to deposition of Aβ in the CNS of M. fermentans-infected patients. Analysis of the mechanism showed that a TLR4 antagonist reduced IFITM3 gene expression and necrotic cell death in M. fermentans-infected SH-SY5Y cells, suggesting that TLR4 is involved in IFITM3 upregulation in M. fermentans infection. Although mycoplasmas do not possess LPS, M. fermentans cells express α-enolase, which can activate the TLR4 signaling pathway on their cell surfaces38,39,40. However, we observed that neither α-enolase nor the TLR4 agonist CRX-527 alone induced cell death or activated the TLR4 signaling pathway in SH-SY5Y cells. This finding is consistent with previous studies showing that direct application of LPS, a TLR4 agonist, has no effect on SH-SY5Y cell death43,44,45. Unlike other cells, SH-SY5Y cells do not exhibit cytokine transcription or release in response to LPS, possibly due to their low levels of TLR4 expression41,42. Thus, our results suggest that M. fermentans infection-mediated additional processes are required for necrotic cell death. In addition, addition of α-enolase did not enhance the cell death effect beyond that observed with M. fermentans infection alone, whereas the TLR4 antagonist blocked cell death. This also suggests that M. fermentans infection is sufficient for TLR4 activation and induction of cell death.
In our experiments, we tested two mouse neuronal cell lines, neuro2a and HT-22, but neither of these cell lines exhibited IFITM3 upregulation or necrotic cell death after M. fermentans infection. However, we observed necrotic cell death of the human SH-SY5Y neuronal cell line. In addition, this effect by M. fermentans infection was observed in organoid cultures, minimizing the concern about extrapolating from a pure cell line approach. Thus, our results may indicate that M. fermentans-induced immunological responses to pathogen infection can differ depending on the species, as shown in a previous study53. Overall, our findings suggest that intracellular Aβ1–42 deposition by TLR4-mediated IFITM3 upregulation is the main inducer of necrotic cell death.
The amphipathic helix of IFITM3 is essential for its antiviral activity by inducing membrane deformation in virus-infected cells54. However, a previous study revealed that IFITM3 also plays a modulatory role in γ-secretase activity by binding to PS1 near the active site, resulting in an increase in γ-secretase activity and Aβ production34. Thus, it is essential to consider the potential antiviral effects of any therapeutic targeting of IFITM3 since the determinants involved in its antiviral activity may also play a role in its interaction with γ-secretase and regulation of Aβ production. It is worth noting that oligomerization of IFITM3 is necessary for its antiviral activity through the induction of membrane stiffening55, whereas IFITM3 binds to PS1-NTF as a monomer or dimer34. This suggests that the antiviral mechanism of IFITM3 and its role in modulating γ-secretase activity may involve distinct determinants. Therefore, further investigations are necessary to fully comprehend the determinants of IFITM3 involved in this complex and their impact on γ-secretase activity, which could have significant implications for the development of targeted therapies that balance its antiviral and modulatory effects.
In our study, we observed a significant downregulation of IL-1β and PRNP gene expression in M. fermentans-infected SH-SY5Y cells following hAPP knockdown. This suggests that these genes may function downstream of the signaling pathway leading to Aβ1–42 deposition in our experimental system, which is consistent with previous studies implicating IL-1β and PRNP in Aβ-induced neuroinflammation and neurotoxicity26,28,31,33. Interestingly, we also observed upregulation of APOE expression following hAPP knockdown in M. fermentans-infected SH-SY5Y cells. APOE has been implicated in AD pathology, where it plays a complex role in the clearance and aggregation of Aβ in an isoform-specific manner32. Therefore, this finding may reflect a compensatory mechanism that counteracts Aβ-induced neurotoxicity. However, the exact role of the Aβ pathway in human PRNP or APOE remains unclear. Therefore, it is important to note that the observed effects of hAPP knockdown on gene expression may be indirect, and further studies are needed to confirm their precise positioning within the signaling pathway leading to Aβ deposition. Nonetheless, our findings provide important insights into the potential roles of IL-1β, PRNP, and APOE in this process, and suggest new avenues for future research.
M. fermentans has been found in various tissues of AIDS patients, leading to its designation as an opportunistic pathogen4. Interestingly, several studies showed that M. fermentans is present in saliva in about half of the population (110 of 201, 54.7%; 49 of 110, 44%), suggesting that the organism colonizes the human mouth and is transmitted easily to others56,57. However, blood infection did not occur frequently in healthy individuals5,6,7,20,21,22,23,24. It is possible that the organism invades blood and various tissues including the brain in immunocompromised individuals, altering physiological conditions and causing M. fermentans-related neurological diseases. Thus, our results suggest the possibility that opportunistic CNS infection by M. fermentans may occur in immune-altered individuals, resulting in other types of neurological diseases.
In conclusion, in vitro and ex vivo analyses revealed that M. fermentans can function in a pathogenic capacity in human neuronal cells. Cellular and molecular analyses of M. fermentans pathogenicity revealed that M. fermentans directly induces necrotic neuronal cell death by IFITM3-mediataed Aβ1–42 deposition. Thus, our results suggest that M. fermentans is involved in the pathogenicity of neurological diseases. Furthermore, our findings could help expand our understanding of neurological diseases caused by mycoplasmal infections.
Methods
Cell lines
SH-SY5Y cells from the Korean Cell Line Bank (KCLB, Seoul, Republic of Korea, Cat. No. 22266), HT-22 cells (gifted from Dr. Inhee Mook-Jung, Seoul National University), and neuro2a cells from the American Type Culture Collection (ATCC, Manassas, VA, USA, Cat. No. CCL-131) were cultured in 10% fetal bovine serum (FBS, Hyclone, Logan, UT, USA, Cat. No. SH3008403), 100 U/ml penicillin, and 100 μg/ml streptomycin-supplemented Dulbecco’s modified Eagle’s medium (DMEM, Hyclone, Cat. No. SH30243.01) at 37 °C with 5% CO2. Human induced pluripotent stem cells (IPSCs) CMC-hiPSC-003 and CMC-hiPSC-009 were provided by the National Stem Cell Bank of Korea (Korea National Institute of Health), originally provided from Catholic University. IPSCs were cultured using an mTeSR 1 Complete Kit (StemCell Technologies, Vancouver, BC, Canada, Cat. No. 85850). Control-, hAPP-, and hIFITM3-knockdown SH-SY5Y cells were established via infection of control shRNA lentivirus particle-A (Santa Cruz Biotechnology, Dallas, TX, USA, Cat. No. sc-108080), hAPP shRNA lentivirus particles (Santa Cruz Biotechnology, Cat. No. sc-29677-V), and hIFITM3 shRNA lentivirus particles (Santa Cruz Biotechnology, Cat. No. sc-97053-V). Knockdown SH-SY5Y cells were maintained under 5 μg/ml puromycin dihydrochloride (Sigma-Aldrich, St. Louis, MO, USA, Cat. No. P8833-10 mg).
Preparation and infection of M. fermentans
M. fermentans strain PG-18 (ATCC, Cat. No. ATCC 19989) was used in this study. M. fermentans strains were grown in ATCC mycoplasma medium (ATCC medium 243) under anaerobic conditions using an anaerobic jar and indicator. We followed the instructions suggested for culturing Mollicutes from ATCC. The number of viable mycoplasma cells was determined by plating and was calculated as the number of colony-forming units (CFU) per milliliter under anaerobic conditions. For infection of M. fermentans, SH-SY5Y cells were seeded into 3 ml culture medium in a 6-well cell culture plate at a density of 5 × 105 cells per well, while HT-22 cells were seeded at 0.5 × 105 cells per well, and neuro2a cells were seeded at 1 × 105 cells per well. We infected these cells with M. fermentans at MOI = 0.01–1 and confirmed that necrotic cell death of SH-SY5Y cells occurred at earlier time points at higher MOI. Therefore, we used MOI = 0.01 throughout for the in vitro infection system. Before M. fermentans infection, we centrifuged M. fermentans cells at 26,000 g for 30 min at 4 °C and replaced mycoplasma medium with 1 ml of cell culture medium, and repeated this twice. After 24 h of infection, we replaced the cell culture medium. In the case of brain organoids, Matrigel (Corning, Corning, NY, USA, Cat. No. 354277) around organoids could inhibit the infection, so we increased the MOI to 0.05. We estimated the number of cells in one brain organoid to be ~ 1 × 105 cells, and we replaced the medium after 72 h of infection. Inactivated M. fermentans particles were prepared by preincubation with 70% EtOH for 10 min, heating at 56 °C for 30 min, and exposure to UV irradiation (253.7 nm) for 10 min. For mycoplasma antibiotic drug testing, we used M. fermentans pretreated with 1 mg/ml Primocin (InvivoGen, San Diego, CA, USA, Cat. No. ant-pm-05) and 125 μg/ml Myco-Guard Mycoplasma Elimination Reagent (Biomaxinc, Seoul, Republic of Korea, Cat. No. SMD022) for 3 h at 37 °C, then diluted 1:10 in culture medium before infection.
Annexin V/PI apoptosis assay
To confirm the mechanism of cell death, we performed annexin V/PI apoptosis assays using an Annexin V Apoptosis Detection Kit APC (eBioscience, San Diego, CA, USA, Cat. No. 88-8007-74) according to the manufacturer’s instructions, and analyzed the results on a Guava EasyCyte HT instrument (Merck Millipore, Burlington, MA, USA) or a BD FACSCanto II (Becton, Dickinson and Company, Franklin Lakes, NJ, USA), then analyzed with FlowJo v10.7.1 (Becton, Dickinson and Company) data analysis software.
Differentiation of SH-SY5Y cells and preparation of conditioned medium
To induce differentiated SH-SY5Y cells, they were seeded into 3 ml of medium at a density of 2 × 105 cells per a well in a 6-well cell culture plate. After 24 h, cells were treated with 80 mM PMA (Sigma-Aldrich, Cat. No. P1585-1MG) for 7 days. Infection was performed at an MOI of 0.01, and medium was replaced with fresh medium after 24 h. At 7, 12, and 19 dpi, cells were used for analysis. At 19 dpi, the cell culture medium was replaced with fresh medium, and after 2 days of incubation, the conditioned medium was harvested and centrifuged at 900 × g at 4 °C for 30 min, and supernatants were passed through a 0.45 μm pore size membrane filter. The conditioned medium was then diluted 1:3 in fresh culture medium and used to treat fresh differentiated SH-SY5Y cells.
Quantification of M. fermentans DNA levels
For quantification of intracellular or secreted M. fermentans DNA levels, total DNA was extracted using a G-spin Total DNA Extraction Mini Kit (iNtRON Biotechnology, Gyeonggi-do, Republic of Korea, Cat. No. TCS0803). qPCR was then performed using SYBR Green qPCR 2 × Premix (Enzynomics) on a CFX Connect Real-Time PCR Detection System (Bio-Rad). Intracellular M. fermentans DNA levels were normalized against internal control DNA levels. qPCR primer sequences were as follows: M. fermentans-F, 5′-GGACTATTGTCTAAACAATTTCCC-3′; M. fermentans-R, 5′-GGTTATTCGATTTCTAAATCGCCT-3′; mouse internal control-F, 5′-CTGTGGATGGCCCCTCTG-3′; mouse internal control-R, 5′-CACGACGGACACATTGGG-3′; human internal control-F, 5′- GGCTGTTGTCATACTTCTCATG-3′; human internal control-R, 5′- GGACCAAGATCCCTCCAAAAT-3′.
Quantification of gene expression levels
To quantify gene expression, total cellular RNA was isolated using an RNeasy Mini Kit (Qiagen, Hilden, Germany, Cat. No. 74104), and cDNA was prepared using TOPscript RT Drymix (Enzynomics, Daejeon, Republic of Korea, Cat. No. RT100). qRT-PCR was then performed using SYBR Green qPCR 2 × Premix (Enzynomics, Cat. No. RT500-M) on a CFX Connect Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Expression levels of each gene were normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene expression levels. qRT-PCR primer sequences were as follows: hGAPDH-F, 5′-GGAGCGAGATCCCTCCAAAAT-3′; hGAPDH-R, 5′-GGCTGTTGTCATACTTCTCATG-3′; hAPP-F, 5′-GCTGGTGGAGACACACATG-3′; hAPP-R, 5′-GGATCTGAGCGGCTTTCTTG-3′; hMAP2-F, 5′-TGCGCCCAGATTTTATTGATC-3′; hMAP2-R, 5′-GTTCGTTGTGTCGTGTTCTCA-3′; hAPOE-F, 5′-GGGTCGCTTTTGGGATTACCTG-3′; hAPOE-R, 5′-CAACTCCTTCATGGTCTCGTCC-3′; hIL-1β-F, 5′-CCACAGACCTTCCAGGAGAATG-3′; hIL-1β-R, 5′-GTGCAGTTCAGTGATCGTACAGG-3′; hTNF-α-F, 5′-ACGCTCTTCTGCCTGCTG-3′; hTNF-α-R, 5′-GCTTGAGGGTTTGCTACAACA-3′; hPRNP-F, 5′-CTGCTGGATGCTGGTTCTCT-3′; hPRNP-R, 5′-GTGTTCCATCCTCCAGGCTT-3′; hIFITM3-F, 5′-GGTCTTCGCTGGACACCAT-3′, hIFITM3-R, 5′-TGTCCCTAGACTTCACGGAGTA-3′; mGAPDH-F, 5′-GCACAGTCAAGGCCGAGAAT-3′; mGAPDH-R, 5′-GCCTTCTCCATGGTGGTGAA-3′; hTLR4-F, 5′-GACTTGCGGGTTCTACATCA-3′; hTLR4-R, 5′-CATAGGGTTCAGGGACAGGT-3′. hIL-6-F, 5′-TGTGAAAGCAGCAAAGAGGC-3′; hIL-6-R, 5′-CCAGGCAAGTCTCCTCATTG-3′.
Western blotting analysis
Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 12–15% gels and transferred to a polyvinylidene difluoride membrane. Membranes were then incubated with the following primary antibodies: β-Amyloid (1–42 Specific, D9A3A, Cell Signaling Technology, Danvers, MA, USA, Cat. No. 14974S), Tau (HT7, Thermo Fisher Scientific, Waltham, MA, USA, Cat. No. MN1000), Phospho-Tau (Ser202, Thr205, AT8, Thermo Fisher Scientific, Cat. No. MN1020), GAPDH (H-12, Santa Cruz Biotechnology, Cat. No. sc-166574), and IFITM3 (F-41, Santa Cruz Biotechnology, Cat. No. sc-100768). Peroxidase-conjugated anti-rabbit and anti-mouse secondary antibodies (Sigma-Aldrich, Cat. No. A0545 and A2554) for ECL substrate (GE Healthcare, Barrington, IL, USA) were then employed. To check the quality of p-tau bands, we used CIP at 10 and 40 units (New England Biolabs, Ipswich, MA, USA, Cat. No. M0290) for 60 min at 37 °C. It is important to note that the original blots were cut before hybridization with antibodies. For this reason, full-length blots cannot be provided. The images in Supplementary Fig. S9 depict the blots after cropping.
Immunocytochemistry and immunohistochemistry analysis
For immunocytochemical staining, cells were fixed with 2% paraformaldehyde (PFA) for 20 min and permeabilized with 0.3% Triton X-100 in phosphate-buffered saline (PBS) for 20 min, and then blocked with 0.5% bovine serum albumin (BSA) in PBS for 1 h at room temperature (RT). Cells were stained with primary antibodies in 0.5% BSA in PBS for 2 h at RT, and then stained with Alexa Fluor 488-conjugated anti-mouse IgG antibody and 594-conjugated anti-rabbit IgG antibody (Thermo Fisher Scientific, Cat. No. A-11029 and A-11037) in 0.5% BSA in PBS for 45 min at RT. Images were captured with an FV1000 confocal microscope (Olympus, Tokyo, Japan). Quantification analyses were performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA; https://imagej.nih.gov/ij/features.html).
For immunohistochemical staining, brain organoids were embedded in optimal cutting temperature (OCT) compound after fixation with 4% PFA for 24 h at 4 °C and submerged in 30% sucrose. After slicing the OCT-embedded frozen block into 12 μm sections, brain organoids were permeabilized with 0.3% Triton X-100 in PBS for 20 min and blocked with 1% BSA in PBS for 1 h, and tissues were stained with primary antibodies in 0.5% BSA in PBS for 24 h at 4 °C, followed by Alexa Fluor 488-conjugated anti-mouse IgG antibody and 594-conjugated anti-rabbit IgG antibody (Thermo Fisher Scientific). Images were captured with a Leica TCS SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany). Analyses were performed using Leica Application Suite X software (Leica Microsystems). The following primary antibodies were used: SOX2 (D6D9, Cell Signaling Technology, Cat. No. C3579S), Tubulin β 3 (TUBB3, BioLegend, Cat. No. 801201), β-Amyloid (1–42 Specific, D9A3A, Cell Signaling Technology, Cat. No. 14974S), Phospho-Tau (Ser202, Thr205, AT8, Thermo Fisher Scientific, Cat. No. MN1020), Phospho-MLKL (Ser358, Cell Signaling Technology, Cat. No. D6H3V), and β-actin (ABclonal, Woburn, MA, USA, Cat. No. AC026). Nuclei were visualized by staining with 20 μg/ml of Hoechst 33342 (Sigma-Aldrich, Cat. No. B2261-25MG) in PBS.
Generation of brain organoids
To generate brain organoids, we used a STEMdiff Cerebral Organoid Kit (StemCell Technologies, Cat. No. 08570) and a Cerebral Organoid Maturation Kit (StemCell Technologies, Cat. No. 08571) according to the manufacturer’s protocols. Briefly, to stimulate embryoid body (EB) formation, iPSCs were dissociated with ReLeSR (StemCell Technologies, Cat. No. 05872) and seeded in 100 μl of EB seeding medium containing 10 μM Y-27632 (StemCell Technologies, Cat. No. 72302) at 9,000 cells per well in a 96-well round-bottom ultralow attachment plate. On days 2 and 4, 100 μl of EB formation medium was added. On day 5, EBs of 400–600 μm in size with round and smooth edges were used for induction. Briefly, one or two EBs were incubated in 500 μl of induction medium per well in a 24-well ultralow attachment plate. On day 7, EBs were embedded in Matrigel (Corning), and 12–16 embedded EBs were incubated in 3 ml of expansion medium per well in a 6-well ultralow adherent plate. On day 10, brain organoids were matured in maturation medium on an orbital shaker at 65 rpm, and maturation medium was changed every 3 days.
RNA-seq analysis
For RNA-seq, libraries were prepared using a TruSeq Stranded Total RNA LT Sample Prep Kit (Illumina, San Diego, CA, USA) according to the TruSeq stranded total RNA samples prep guide. Sequencing was performed with a NovaSeq 6000 system (Illumina). Genes with a fold change ≥ 2 and transcripts per million (TPM) ≥ 1 were defined as differentially expressed genes and displayed in a scatter plot. Gene functional classification was performed using the DAVID (release 6.8; https://david.ncifcrf.gov). EASE scores, a modified Fisher Exact p-value, (p ≤ 0.05) were used to identify gene enrichment of functional pathways by estimating the number of genes belonging to functional pathways involving the selected target proteins.
Enzyme-linked immunosorbent assay (ELISA)
For quantification of secreted Aβ1–42 in culture medium, we used a commercial ELISA Kit for Amyloid Beta Peptide 1–42 (Wuhan USCN Business Co., Ltd., Wuhan, China, Cat. No. CEA946Hu) according to the manufacturer’s protocols.
Treatments with neutralizing antibody, antagonist, and ligands
We used 50 μM human TLR2/1 and TLR2/6 and murine TLR2/1 signaling inhibitor (InvivoGen, Cat. No. inh-c29) as a TLR2 antagonist, and 10 μg/ml of ultrapure lipopolysaccharide from Rhodobacter sphaeroides (InvivoGen, Cat. No. tlrl-prslps) as a TLR4 antagonist. For IL-1β neutralization, we used 1 μg/ml of human IL-1β neutralizing antibody (4H5, InvivoGen, Cat. No. mabg-hil1b-3), and 100 ng/ml TNF-α neutralizing antibody (SinoBiological Inc. Beijing, China, Cat. No. 10602-MM0N1) was used for TNF-α neutralization. CRX-527 (InvivoGen, Cat. No. tlrl-crx527) at 1 μg/ml was used as an CD14-independent TLR4 agonist37. Additionally, 10 μg/ml of active α-enolase protein (Mybiosource, Vancouver, Canada, Cat. No. MBS203689) was added as an activator of the CD14-dependent TLR4 signaling pathway40. SH-SY5Y cells were pretreated with these inhibitors for 1 h before M. fermentans infection at 0.1 MOI. After 48 h, we replaced the medium with fresh medium containing inhibitors. At 3 dpi we analyzed the M. fermentans-infected cells.
Statistical analysis
The significance of differences between two groups was analyzed using unpaired Student’s t-tests using Origin2021 software (OriginLab Corporation, Northampton, MA, USA). Quantification of protein expression levels in western blotting was performed using ImageJ software, GAPDH served as an internal control for normalization, and Hoechst was used for normalization of immunocytochemical data. All results are expressed as the mean ± standard deviation (SD). For all statistical tests, p-values ≤ 0.05 were considered significant.
Data availability
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author. All RNA-seq data generated in this study are available through the NCBI Sequence Read Archive (SRA) through accession numbers SRR22252649-SRR22252651 under BioProject PRJNA900032.
References
Benedetti, F., Curreli, S. & Zella, D. Mycoplasmas-host interaction: Mechanisms of inflammation and association with cellular transformation. Microorganisms 8, 1351. https://doi.org/10.3390/microorganisms8091351 (2020).
Young, L., Sung, J., Stacey, G. & Masters, J. R. Detection of mycoplasma in cell cultures. Nat. Protoc. 5, 929–934. https://doi.org/10.1038/nprot.2010.43 (2010).
Razin, S., Yogev, D. & Naot, Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol. Mol. Biol. Rev. 62, 1094–1156. https://doi.org/10.1128/MMBR.62.4.1094-1156.1998 (1998).
Montagnier, L. & Blanchard, A. Mycoplasmas as cofactors in infection due to the human immunodeficiency virus. Clin. Infect. Dis. 17(Suppl 1), S309-315 (1993).
Vojdani, A. & Franco, A. R. Multiplex PCR for the detection of Mycoplasma fermentans, M. hominis, and M. penetrans in patients with chronic fatigue syndrome, fibromyalgia, rheumatoid arthritis, and gulf war syndrome. J. Chron. Fatigue Syndr. 5, 187–197. https://doi.org/10.1300/J092v05n03_16 (1999).
Gil, C. et al. Detection of mycoplasmas in patients with amyotrophic lateral sclerosis. Adv. Microbiol. 4, 712–719 (2014).
Nicolson, G. L., Gan, R., Nicolson, N. L. & Haier, J. Evidence for Mycoplasma ssp., Chlamydia pneunomiae, and human herpes virus-6 coinfections in the blood of patients with autistic spectrum disorders. J. Neurosci. Res. 85, 1143–1148. https://doi.org/10.1002/jnr.21203 (2007).
Rosales, R. S., Puleio, R., Loria, G. R., Catania, S. & Nicholas, R. A. J. Mycoplasmas: Brain invaders?. Res. Vet. Sci. 113, 56–61. https://doi.org/10.1016/j.rvsc.2017.09.006 (2017).
Blanchard, A. & Montagnier, L. AIDS-associated mycoplasmas. Annu. Rev. Microbiol. 48, 687–712. https://doi.org/10.1146/annurev.mi.48.100194.003351 (1994).
Lo, S. C. et al. Identification of Mycoplasma incognitus infection in patients with AIDS: An immunohistochemical, in situ hybridization and ultrastructural study. Am. J. Trop. Med. Hyg. 41, 601–616. https://doi.org/10.4269/ajtmh.1989.41.601 (1989).
Lo, S. C. et al. Fatal systemic infections of nonhuman primates by Mycoplasma fermentans (incognitus strain). Clin. Infect. Dis. 17(Suppl 1), S283-288. https://doi.org/10.1093/clinids/17.supplement_1.s283 (1993).
Yanez, A. et al. Animal model of Mycoplasma fermentans respiratory infection. BMC Res Notes 6, 9. https://doi.org/10.1186/1756-0500-6-9 (2013).
Lo, S. C. et al. Association of the virus-like infectious agent originally reported in patients with AIDS with acute fatal disease in previously healthy non-AIDS patients. Am. J. Trop. Med. Hyg. 41, 364–376 (1989).
Sasaki, T. et al. Evidence that Lo’s mycoplasma (Mycoplasma fermentans incognitus) is not a unique strain among Mycoplasma fermentans strains. J. Clin. Microbiol. 30, 2435–2440. https://doi.org/10.1128/jcm.30.9.2435-2440.1992 (1992).
Socan, M. et al. Neurological symptoms in patients whose cerebrospinal fluid is culture- and/or polymerase chain reaction-positive for Mycoplasma pneumoniae. Clin. Infect. Dis. 32, E31-35. https://doi.org/10.1086/318446 (2001).
Stamm, B. et al. Neuroinvasion by Mycoplasma pneumoniae in acute disseminated encephalomyelitis. Emerg. Infect. Dis. 14, 641–643. https://doi.org/10.3201/eid1404.061366 (2008).
Zhang, S. et al. Mycoplasma fermentans infection promotes immortalization of human peripheral blood mononuclear cells in culture. Blood 104, 4252–4259. https://doi.org/10.1182/blood-2004-04-1245 (2004).
Liu, W. & Shou, C. Mycoplasma hyorhinis and Mycoplasma fermentans induce cell apoptosis and changes in gene expression profiles of 32D cells. Biol. Res. 44, 383–391 (2011).
Zogovic, N. et al. Coordinated activation of AMP-activated protein kinase, extracellular signal-regulated kinase, and autophagy regulates phorbol myristate acetate-induced differentiation of SH-SY5Y neuroblastoma cells. J. Neurochem. 133, 223–232. https://doi.org/10.1111/jnc.12980 (2015).
Choppa, P. C., Vojdani, A., Tagle, C., Andrin, R. & Magtoto, L. Multiplex PCR for the detection of Mycoplasma fermentans, M. hominis and M. penetrans in cell cultures and blood samples of patients with chronic fatigue syndrome. Mol. Cell. Probes 12, 301–308. https://doi.org/10.1006/mcpr.1998.0186 (1998).
Nijs, J., Nicolson, G. L., De-Becker, P., Coomans, D. & De Meirleir, K. High prevalence of Mycoplasma infections among European chronic fatigue syndrome patients. Examination of four Mycoplasma species in blood of chronic fatigue syndrome patients. FEMS Immunol. Med. Microbiol. 34, 209–214. https://doi.org/10.1111/j.1574-695X.2002.tb00626.x (2002).
Nasralla, M., Haier, J. & Nicolson, G. L. Multiple mycoplasmal infections detected in blood of patients with chronic fatigue syndrome and/or fibromyalgia syndrome. Eur. J. Clin. Microbiol. Infect. Dis. 18, 859–865. https://doi.org/10.1007/s100960050420 (1999).
Nicolson, G. L., Nasralla, M. Y., Nicolson, N. L. & Haier, J. High prevalence of mycoplasma infections in symptomatic (chronic fatigue syndrome) family members of mycoplasma-positive gulf war illness patients. J. Chron. Fatigue Syndrome 11, 21–36. https://doi.org/10.1300/J092v11n02_03 (2003).
Nicolson, G. L., Nasralla, M. Y., Haier, J. & Pomfret, J. High frequency of systemic mycoplasmal infections in Gulf War veterans and civilians with Amyotrophic Lateral Sclerosis (ALS). J. Clin. Neurosci. 9, 525–529. https://doi.org/10.1054/jocn.2001.1075 (2002).
Bourgade, K. et al. Protective effect of amyloid-beta peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J. Alzheim. Dis. 50, 1227–1241. https://doi.org/10.3233/JAD-150652 (2016).
Larson, M. et al. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer’s disease. J. Neurosci. 32, 16857–16871a. https://doi.org/10.1523/JNEUROSCI.1858-12.2012 (2012).
Zhao, J. et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat. Commun. 11, 5540. https://doi.org/10.1038/s41467-020-19264-0 (2020).
Kinney, J. W. et al. Inflammation as a central mechanism in Alzheimer’s disease. Alzheim. Dement. (N. Y.) 4, 575–590. https://doi.org/10.1016/j.trci.2018.06.014 (2018).
Melvin, A., Lacerda, E., Dockrell, H. M., O’Rahilly, S. & Nacul, L. Circulating levels of GDF15 in patients with myalgic encephalomyelitis/chronic fatigue syndrome. J. Transl. Med. 17, 409. https://doi.org/10.1186/s12967-019-02153-6 (2019).
Fujita, Y., Ito, M. & Ohsawa, I. Mitochondrial stress and GDF15 in the pathophysiology of sepsis. Arch. Biochem. Biophys. 696, 108668. https://doi.org/10.1016/j.abb.2020.108668 (2020).
Shaftel, S. S., Griffin, W. S. & O’Banion, M. K. The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J. Neuroinflamm. 5, 7. https://doi.org/10.1186/1742-2094-5-7 (2008).
Wisniewski, T. & Drummond, E. APOE-amyloid interaction: Therapeutic targets. Neurobiol. Dis. 138, 104784. https://doi.org/10.1016/j.nbd.2020.104784 (2020).
Foley, A. R. et al. Evidence for aggregation-independent, PrP(C)-mediated Abeta cellular internalization. Proc. Natl. Acad. Sci. U. S. A. 117, 28625–28631. https://doi.org/10.1073/pnas.2009238117 (2020).
Hur, J. Y. et al. The innate immunity protein IFITM3 modulates gamma-secretase in Alzheimer’s disease. Nature 586, 735–740. https://doi.org/10.1038/s41586-020-2681-2 (2020).
Ranjbar, S., Haridas, V., Jasenosky, L. D., Falvo, J. V. & Goldfeld, A. E. A role for IFITM proteins in restriction of mycobacterium tuberculosis infection. Cell Rep. 13, 874–883. https://doi.org/10.1016/j.celrep.2015.09.048 (2015).
Prelli Bozzo, C. et al. IFITM proteins promote SARS-CoV-2 infection and are targets for virus inhibition in vitro. Nat. Commun. 12, 4584. https://doi.org/10.1038/s41467-021-24817-y (2021).
Legat, A. et al. CD14-independent responses induced by a synthetic lipid A mimetic. Eur. J. Immunol. 40, 797–802. https://doi.org/10.1002/eji.200939992 (2010).
Yiwen, C., Yueyue, W., Lianmei, Q., Cuiming, Z. & Xiaoxing, Y. Infection strategies of mycoplasmas: Unraveling the panoply of virulence factors. Virulence 12, 788–817. https://doi.org/10.1080/21505594.2021.1889813 (2021).
Yavlovich, A., Rechnitzer, H. & Rottem, S. Alpha-enolase resides on the cell surface of Mycoplasma fermentans and binds plasminogen. Infect. Immun. 75, 5716–5719. https://doi.org/10.1128/IAI.01049-07 (2007).
Guillou, C. et al. Soluble alpha-enolase activates monocytes by CD14-dependent TLR4 signalling pathway and exhibits a dual function. Sci. Rep. 6, 23796. https://doi.org/10.1038/srep23796 (2016).
Klegeris, A. & McGeer, P. L. Inflammatory cytokine levels are influenced by interactions between THP-1 monocytic, U-373 MG astrocytic, and SH-SY5Y neuronal cell lines of human origin. Neurosci. Lett. 313, 41–44. https://doi.org/10.1016/s0304-3940(01)02251-0 (2001).
Lawrimore, C. J. & Crews, F. T. Ethanol, TLR3, and TLR4 agonists have unique innate immune responses in neuron-like SH-SY5Y and microglia-like BV2. Alcohol. Clin. Exp. Res. 41, 939–954. https://doi.org/10.1111/acer.13368 (2017).
Monaghan, T. K., Pou, C., MacKenzie, C. J., Plevin, R. & Lutz, E. M. Neurotrophic actions of PACAP-38 and LIF on human neuroblastoma SH-SY5Y cells. J. Mol. Neurosci. 36, 45–56. https://doi.org/10.1007/s12031-008-9082-6 (2008).
Liu, Y. et al. Butein attenuates the cytotoxic effects of LPS-stimulated microglia on the SH-SY5Y neuronal cell line. Eur. J. Pharmacol. 868, 172858. https://doi.org/10.1016/j.ejphar.2019.172858 (2020).
Zhu, L. et al. Luteolin inhibits SH-SY5Y cell apoptosis through suppression of the nuclear transcription factor-kappaB, mitogen-activated protein kinase and protein kinase B pathways in lipopolysaccharide-stimulated cocultured BV2 cells. Exp. Ther. Med. 7, 1065–1070. https://doi.org/10.3892/etm.2014.1564 (2014).
Golomb, B. Oxidative stress and mitochondrial injury in chronic multisymptom conditions: From gulf war illness to autism spectrum disorder. Nature Prec. https://doi.org/10.1038/npre.2012.6847.1 (2012).
Much, P., Winner, F., Stipkovits, L., Rosengarten, R. & Citti, C. Mycoplasma gallisepticum: Influence of cell invasiveness on the outcome of experimental infection in chickens. FEMS Immunol. Med. Microbiol. 34, 181–186. https://doi.org/10.1111/j.1574-695X.2002.tb00622.x (2002).
Chin, R. P., Meteyer, C. U., Yamamoto, R., Shivaprasad, H. L. & Klein, P. N. Isolation of Mycoplasma synoviae from the brains of commercial meat turkeys with meningeal vasculitis. Avian Dis. 35, 631–637 (1991).
Ayling, R. et al. Mycoplasma bovis isolated from brain tissue of calves. Vet. Rec. 156, 391–392. https://doi.org/10.1136/vr.156.12.391-b (2005).
McMichael, T. M., Chemudupati, M. & Yount, J. S. A balancing act between IFITM3 and IRF3. Cell. Mol. Immunol. 15, 873–874. https://doi.org/10.1038/cmi.2017.18 (2018).
Shi, G., Schwartz, O. & Compton, A. A. More than meets the I: The diverse antiviral and cellular functions of interferon-induced transmembrane proteins. Retrovirology 14, 53. https://doi.org/10.1186/s12977-017-0377-y (2017).
Majdoul, S. & Compton, A. A. Lessons in self-defence: Inhibition of virus entry by intrinsic immunity. Nat. Rev. Immunol. 22, 339–352. https://doi.org/10.1038/s41577-021-00626-8 (2022).
Boys, I. N. et al. RTP4 is a potent IFN-inducible anti-flavivirus effector engaged in a host-virus arms race in bats and other mammals. Cell Host Microbe 28, 712–723. https://doi.org/10.1016/j.chom.2020.09.014 (2020).
Chesarino, N. M. et al. IFITM3 requires an amphipathic helix for antiviral activity. EMBO Rep. 18, 1740–1751. https://doi.org/10.15252/embr.201744100 (2017).
Rahman, K. et al. Homology-guided identification of a conserved motif linking the antiviral functions of IFITM3 to its oligomeric state. Elife 9, 159. https://doi.org/10.7554/eLife.58537 (2020).
Shibata, K. et al. Detection of Mycoplasma fermentans in saliva sampled from infants, preschool and school children, adolescents and adults by a polymerase chain reaction-based assay. Microbiol. Immunol. 43, 521–525. https://doi.org/10.1111/j.1348-0421.1999.tb02437.x (1999).
Chingbingyong, M. I. & Hughes, C. V. Detection of Mycoplasma fermentans in human saliva with a polymerase chain reaction-based assay. Arch. Oral Biol. 41, 311–314. https://doi.org/10.1016/0003-9969(96)84556-0 (1996).
Acknowledgements
We thank W. Sun (Korea University) for technical comments on brain organoid experiments. This research was supported by the Ministry of Health & Welfare, Republic of Korea (grant number: HI22C0828) and by the National Research Foundation of Korea (NRF-2021R1A2C3011211, NRF-2022M3A9I2017587).
Author information
Authors and Affiliations
Contributions
Conceptualization: K.Y.S., S.G.P.; data curation: K.Y.S., Y.B., S.E.B., T.Y., C.R.L.; formal analysis: K.Y.S., Y.B., S.G.P.; writing - original draft: K.Y.S.; writing - review and editing: S.G.P. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sim, KY., Byeon, Y., Bae, SE. et al. Mycoplasma fermentans infection induces human necrotic neuronal cell death via IFITM3-mediated amyloid-β (1–42) deposition. Sci Rep 13, 6864 (2023). https://doi.org/10.1038/s41598-023-34105-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-34105-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
