
Abstract
The nanocomposite coatings composed of carbon nanotubes and various forms of copper were prepared in the two-step process. Firstly, carbon nanotubes were coated on stainless steel substrate using electrophoretic deposition at constant current. Then, the process of electrochemical deposition using copper(II) sulphate solutions was performed under high overpotential conditions. The modification of the copper(II) cations concentration in the solution and the deposition time provided the formation of various forms of crystals. The samples and their cross-sections were observed and examined using scanning electron microscope equipped with electron dispersive spectroscopy system. The analysis of chemical composition revealed that in addition to the pure copper crystals, the crystals characterized by the presence of copper and oxygen were formed. Therefore, Raman spectroscopy was applied to determine the unknown stoichiometry of this copper oxide. The point and in-depth analysis identified copper(I) oxide in the form of different size crystals depending on the concentration of the copper(II) sulphate solution. To confirm these findings, grazing incidence X-ray diffraction measurements were also performed. the combination of the applied methods has provided the detailed description of the preparation of the nanocomposite coatings with the proposed mechanism of copper(I) oxide formation.
Similar content being viewed by others


Introduction
Electrochemical methods are still considered one of the most favorable techniques of production of composite coatings, despite the continuous development of other methods [such as physical vapor deposition (PVD) or chemical vapor deposition (CVD)]. They stand out from the rest because of their high versatility, repeatability and simplicity in combination with precise control over the properties of obtained products1,2. The other advantages are low cost, reduced amount of waste materials and easy scalability of the equipment used in electrochemistry processes. Moreover, by changing the process parameters, it is possible to tailor the crucial properties of coatings such as their thickness, roughness and morphology3,4,5. These advantages, together with variety of received materials and their applications place electrochemistry in the spotlight for researchers from many fields of science. Composite coatings, wherein the particles in the conductive matrix could be metallic, polymeric or ceramic have been used successfully in electronics, surface engineering, aerospace or corrosion protection6,7,8,9,10,11. Electrochemical deposition (ECD) processes have been known since the beginning of the nineteenth century, but research is still ongoing to explain their mechanisms. Although, electrochemical reactions occurring during the electrodeposition process are relatively easy to balance by several redox equations, the individual steps of the process which run according to specific mechanisms are still the subject of research and process modeling12,13. To put it simply, the ECD is based on the modification of the conductive substrate surface with a thin and tight adherent coating of desired material deposited from solution. It takes place at the interface of the two phases: liquid (electrolyte) and solid (electrodes) in the closed electrical circuit. The system could be far from the state of chemical equilibrium, because the applied potentials may differ from the equilibrium values determined by the Nernst equation or the Pourbaix diagrams14. Therefore, by controlling the applied potential and pH, it is possible to obtain various forms (chemical, structural or crystallographic) of material from the same solution15,16,17.
Copper is one of the metals most widely used in the industry and currently most applied material in ECD. This is mainly due to its excellent thermal and electrical conductivity and anti-corrosion properties. In acidic solutions, copper(II) cations (Cu2+) are directly reduced to metallic copper (Cu) according to the following reaction:
Metallic coatings of copper with appropriate thickness are characterized by structure free of porosity and good adhesion18,19. Thin films of electrodeposited copper have been studied extensively in connection to their application in microelectronics, aerospace, gas-sensors, superhydrophobic materials, surface protection or even additive manufacturing20,21,22,23,24,25. However, metallic copper is not always a product of this electrochemical reaction. Due to the water reduction, we can observe local alkalization between the solution and electrode. Under these conditions other forms of copper may be formed. As it is described in the Pourbaix diagram for copper in aqueous system, with use of an appropriate overpotential copper oxides—CuO and Cu2O—could be formed26,27.
Furthermore, by the appropriate control of the deposition time, we are able to obtain crystals of copper and its oxides of various shapes and geometry. It is strictly related to the mass transport (ion diffusion to the electrode) and kinetics of the electrochemical reaction, which are the stages limiting the deposition rate of whole process28,29. It was demonstrated that copper oxides obtained in a simple electrodeposition route by changing only a few parameters (e.g., potential, pH or deposition time) may have nanometric dimensions30.
In the literature, efforts have been made to theoretically understand the mechanisms of copper electrodeposition31,32,33. In turn, in the latest research, articles focusing on potential applications of composite materials containing copper and its oxides obtained electrochemically lead the way34,35,36,37. Therefore, in this work we would like to place special emphasis on the characterization of ECD on the carbon nanotubes coated stainless steel using copper(II) sulphate solutions. Combination of copper and carbon nanotubes in one composite coating obtained by ECD has been the subject of research in recent years in relation to high mechanical, tribological and electrical properties of those materials38,39 and their potential applications, including glucose sensor or anode in Li-on batteries40,41. In this work, we aim to show the influence of deposition parameters and the presence of carbon nanotubes layer on the mechanism of the formation of various structures of copper and copper(I) oxide.
Materials and methods
Preparation of the stainless steel plates
All the samples were prepared using austenitic chromium-nickel steel AISI 304 (1.4301 according to the EN10088-1 standard) in the form of 10 × 20 mm plates as a substrate. Before the deposition of carbon nanotubes (CNT) layer the mechanical and chemical cleaning was performed. It included the following steps: abrasive sandpaper grinding (600 grit), sonication in acetone (≥ 99.5%, Avantor Performance Materials, Poland) and then in ethanol (≥ 96%, Avantor Performance Materials, Poland) for 5 min each using the ultrasonic bath (Sonic-0.5, Polsonic, Poland). Cleaned plates were stored in ethanol and dried just before the preparation of CNT layer.
Chemical oxidation of CNT
Multi-walled CNT (CNT Co. Ltd., South Korea) with a diameter in the range of 5–20 nm and a length of about 10 µm were chemically oxidized in the 3:1 (v/v) mixture of concentrated sulfuric acid (≥ 95%, Avantor Performance Materials, Poland) and concentrated nitric acid (35–38%, Avantor Performance Materials, Poland). For this, they were treated in the solution under a reflux condenser for 4 h at 70 °C and then washed with distilled water and dried. The process was performed to introduce the negatively charged carboxyl (COO−) functional groups into the surface of CNT, as well as carbonyl (C=O) groups and ether (C–O–C) moieties42. Therefore, the resulting negative zeta potential made possible to conduct their electrophoretic deposition (EPD) on the substrate as an anode. Furthermore, the remnants of metallic catalysts from the CNT synthesis could been removed during the process.
Preparation of CNT layers
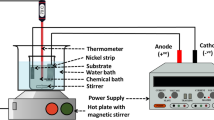
CNT after chemical oxidation were added to the 2:1:1 (v/v/v) mixture of acetone (≥ 99.5%, Avantor Performance Materials, Poland), ethanol (99.8%, Avantor Performance Materials, Poland) and distilled water. The 1 mg/mL solution was sonicated for 5 min using Sonic-0.5 (Polsonic, Poland) ultrasonic bath to improve the CNT dispersion. The process of EPD of CNT on stainless steel plates was performed using the custom-made equipment with PLH250 (Aim-TTi, United Kingdom) DC power supply. Three electrodes arrangement was applied including two counter electrodes with a distance of 15 mm as cathodes and the substrate mounted in a holder symmetrically between them as an anode. The CNT solution was poured into a polypropylene vessel prepared by Fused Deposition Modeling (FDM) 3D printing. For this, 1.75 mm polypropylene filament (Verbatim, Japan) and UBOT 3D S+ (UBOT 3D, Poland) 3D printer were used. The vessel dimensions were designed to fit to the geometry of the applied electrodes arrangement and therefore to reduce the consumption of the solutions. All CNT layers were prepared in the process of EPD using the voltage of 30 V for 60 s. Then, the samples were dried in a room temperature.
Preparation of the samples
Copper(II) sulfate (≥ 99.99%, Sigma-Aldrich, USA) solutions in distilled water were prepared with the concentration of 1 mM and 100 mM. The process of electrochemical deposition (ECD) on the CNT-coated stainless steel plates was performed using the same equipment as for EPD. As a difference in the arrangement the two counter electrodes performed as anodes whereas the CNT-coated stainless steel plate as a cathode. The samples were prepared using 1 mM or 100 mM CuSO4 solution in the process of ECD with the application of the voltage of 7.0 V for 60 s, 120 s, 180 s, 240 s, 300 s, 450 s or 600 s. Then, they were washed with distilled water and dried in a room temperature. Two series of the samples including A1–A7 and B1–B7, respectively, were obtained (Table 1).
SEM–EDS characterization of the samples
The microstructure and chemical composition of the samples were investigated using Phenom XL (Thermo Fisher Scientific, USA) scanning electron microscope (SEM) equipped with electron dispersive X-ray spectroscopy (EDS) system. The examination included the surfaces and the cross sections. Back-scattered electrons detector and the acceleration voltage of 10 kV (for SEM images) or 15 kV (for EDS analysis) were applied. The characterization of the cross sections required their appropriate preparation. The applied procedure included the following steps: the deposition of a thin gold coating (about 20 nm) to improve the adhesion of an additional nickel layer to the investigated material using EM ACE200 (Leica, Japan) vacuum coater, the application of a protective nickel coating (with the thickness of about 2–3 μm) by electroplating technique, the immersion of the samples in a temperature resistant resin (Struers, Denmark) and finally their polishing according to the standard Struers procedure.
Raman spectroscopy analysis of the samples
The structure of the samples was analyzed by Raman spectroscopy using alpha300 M+ (WITec, Germany) spectrometer equipped with a confocal microscope. All measurements were performed with the application of 488 nm diode laser and 600 grating. The characteristic features observed on the surface of the samples were examined by single point analysis. For this, four accumulations with the integration time of 20 s were collected. The in-depth analysis (Z line scans) was performed to evaluate the inner composition of CNT layers. The 4.0 μm depth profiles with a step of 0.5 μm were characterized by collecting four accumulations with the integration time of 20 s in each step. For the comparison the analysis for pure copper oxides including Cu2O (≥ 99.,99%, Sigma-Aldrich, USA) and CuO (≥ 99.99%, Sigma-Aldrich, USA) were performed. For this, three or six accumulations, respectively, with the integration time of 20 s were collected. The data was processed using Bruker OPUS 7.2 software.
GIXRD measurements of the samples
Phase composition of the samples was characterized by grazing incidence X-ray diffraction (GIXRD) measurements using Empyrean (Malvern Panalytical, United Kingdom) diffractometer with CuKα1 line applied as a radiation source and Ge(111) as a monochromator. The scans were collected applying θ–2θ geometry in 2θ range from 10 to 90° and 0.001° step size with an incident angle of 1.0°. The data was processed using Highscore Plus 3.0 software with PDF-4+ database.
Results and discussion
The scope of the tests
The samples were obtained in the form of CNT layers on the stainless steel plates modified by the process of ECD using CuSO4 solutions. Their examination was focused on the microstructure and composition of the electrodeposited material in relation to the parameters of ECD process. Significantly different concentrations of CuSO4 solutions, i.e. 1 mM and 100 mM, as well as high overpotential conditions were intentionally applied. A general overview of the samples with particular attention to the form of the electrodeposited material on the surface was carried out using SEM. This method was also applied for the cross sections of the selected samples to examine the microstructure of CNT layers. During this analysis, the elemental composition was evaluated by EDS with a special emphasis on copper and oxide. Two samples (A7 and B7) prepared by the longest running process of ECD (600 s) were additionally investigated in detail as the most complex systems in terms of the microstructure. Raman spectroscopy was applied to examine the distinctive forms of the electrodeposited material using the point and in-depth analysis. To confirm the results of the tests the GIXRD measurements were also performed.
Material electrodeposition in terms of the process parameters
The initially applied EPD method enables the preparation of the tight CNT coatings on the stainless steel substrates. In the process of ECD two regions for the material electrodeposition could be distinguished. The first includes the surface of CNT layer whereas the second one is its volume. Therefore, they were analyzed separately in this research. At the beginning, the selected and applied ECD process parameters should be considered. All samples were prepared in high overpotential conditions. It is known43,44,45 that this parameter is critical for the reduction rate of metal cations in similar systems. In order to understand this phenomenon two parts of the solution volume could be distinguished, i.e. a thin interface directly in the vicinity of a substrate and a bulk solution. When a low overpotential is applied Cu2+ cations are relatively slow depleted from the near substrate layer. Consequently, their concentration is easily replenished and maintained by their diffusion from the bulk solution. Crystal formation is limited by the process of Cu2+ cations reduction and driven by the minimization of surface energy. Contrary, when high overpotential is applied the deficiency of Cu2+ cations in the proximity of the substrate is relatively rapidly generated. Therefore, their diffusion rate from the bulk solution is not enough to maintain their concentration in this volume. Crystal growth is limited by the process of mass transport. It could be also mentioned that high overpotential conditions in water solutions promote the evolution of hydrogen on an electrode46,47,48. This effect is important when the aim is to prepare a tight layer of electrodeposited material on a substrate. As a consequence of the production of gas bubbles and their diffusion from the substrate surface to the bulk solution the cavities are formed. It was not observed for the prepared samples, because the duration of ECD process was too short to obtain a high surface coverage by the electrodeposited material. As it is described in the Pourbaix diagram for Cu26,27 the potential which provides the reduction of Cu2+ cations to metal Cu is about hundreds of mV depending on the pH of the solution. Therefore, in this research high overpotential conditions were applied. This results in the domination of the mass transport limited mechanism of the crystals formation.
Characterization of the samples surface by SEM
Two series of the samples were prepared in the process of ECD using the different concentration of CuSO4 solutions, i.e. 1 mM for A1–A7 and 100 mM for B1–B7. Hence, with the application of the same high overpotential the concentration of Cu2+ cations play a major role in the mechanism of the crystals growth. As it is shown in the SEM images of A7 and B7 samples (Figs. 1, 2) a significantly different crystal structures formed on CNT layers. Using 1 mM solutions in the process of ECD the dendritic crystals were obtained (Fig. 3). These structures are typically formed in a local non-equilibrium state49,50,51,52. Hence, in this case the typical mechanism of crystal growth limited by the mass transport may be assumed, as it was explained before. Contrary, using 100 mM CuSO4 solutions the spherical crystals agglomerates were produced (Fig. 4). Well-defined single crystals are formed when the mechanism of their growth is driven entirely by the kinetics of the process of cations reduction. The observation of the agglomerates could indicate the existence of mixed mechanism limited partially by both the mass transport as well as the Cu2+ cations reduction. The effect resulting from the application of high overpotential conditions was partially reduced by a significantly faster diffusion rate of Cu2+ cations from the bulk solution to the interface. This was provided by the significantly higher concentration of Cu2+ cations used for the preparation of B series samples in comparison to A series samples.

The comparison of SEM images of A7 sample (ECD deposition time of 600 s, CuSO4 concentration of 1 mM) (a) and B7 sample (ECD deposition time of 600 s, CuSO4 concentration of 100 mM) (b) at the magnification of ×1000.

The comparison of SEM images of A7 sample (ECD deposition time of 600 s, CuSO4 concentration of 1 mM) (a) and B7 sample (ECD deposition time of 600 s, CuSO4 concentration of 100 mM) (b) at the magnification of ×2000.

SEM images of the dendritic crystals formed on CNT layer in A6 sample (ECD deposition time of 450 s, CuSO4 concentration of 1 mM) at the magnification of ×5000 (a) and ×10,000 (b).

SEM images of the crystals agglomerate formed on CNT layer in B7 sample (ECD deposition time of 600 s, CuSO4 concentration of 100 mM) at the magnification of ×15,000 (a) and ×30,000 (b).
To examine the crystal growth two series of the samples were prepared. The SEM images (Figs. S1–S7 and Figs. S8–S14, respectively, in supplementary data) reveal their gradual formation. For A series samples the first well-defined dendrites were observed after 120 s of the electrodeposition (Fig. S2). With the increasing duration of the process, they progressive growth was revealed and the surface coverage was higher. The examination of B series samples shows a significantly different picture. The formation of the spherical agglomerates is already noticed after 60 s of the electrodeposition (Fig. S8). They are growing bigger with time. The other effect is the formation of crystals within CNT layer. At the beginning they are small and could be noticed as its local inhomogeneity (Fig. S8), but with the increasing duration of the process they are also growing bigger. Finally, some of these crystals get a spherical shape, but the other obtain a well-defined octahedral shape (Figs. 5, 6). It was also found that after 600 s of the electrodeposition the octahedral crystals grow to the surface of CNT layer.

SEM images of the crystals which formed within CNT layer in B6 sample (ECD deposition time of 450 s, CuSO4 concentration of 100 mM) at the magnification of ×5000 (a) and ×10,000 (b).

SEM images of the crystals which grew to the surface of CNT layer in B7 sample (ECD deposition time of 600 s, CuSO4 concentration of 100 mM) at the magnification of ×10,000 (a) and ×20,000 (b).
Analysis of the crystals structure by Raman spectroscopy
Raman spectroscopy was used to examine the formation of crystals on CNT layer (Fig. 7). For this, the single point measurements were performed for B7 sample in the characteristic points chosen using confocal microscopy (Fig. 8). The first one is positioned on pure CNT layer (point A) whereas the others correspond to the different crystal structures, including the spherical agglomerate which partially grew to the surface (point B) and the octahedral crystal on the surface (point C). In this microphotography the spherical Cu agglomerates are also noticed. Copper, like every metal, is not active in Raman spectroscopy, so the appropriate spectra are not obtained. The first spectrum (for point A) is typical for CNT layer on the stainless steel substrate as we reported previously53,54. Two the most intense bands are located at 1357 cm−1 (so called D band) and at 1587 cm−1 (so called G band). The first one is related to the presence of disorder in sp2 carbon structure (A1g mode) whereas the second one is associated with the C–C vibrations, both along the CNT axis and circumferential (E2g mode). The other weak band observed at about 2710 cm−1 is a second-order harmonic of the D mode (so called 2D band) which could be attributed to the three dimensional long range order of CNT55,56. The third spectrum (for point C) was obtained for the octahedral crystal on the surface. A set of bands typical for Cu2O at the range from 150 to 650 cm−1 are found. This could be confirmed by the comparison (Table 2) with the spectra obtained for pure copper oxides (Fig. S15, in supplementary data). The most intense band at 647 cm−1 is associated with the T1u(LO) mode. The other ones at 151 cm−1 and 219 cm−1 are attributed to the T1u(TO) and 2Eu modes, respectively. Finally, the weak bands at 417 cm−1 and 501 cm−1 could be related to the four-phonon 3Eu + T1u(TO) mode and to the T2g mode, accordingly. Therefore, this assignment proves that the observed octahedral crystals are composed of Cu2O57,58. In case of the measurement for point B, the spectrum is a result of the probing depth characteristic of Raman spectroscopy. The bands at 1366 cm−1, 1585 cm−1 and 2708 cm−1 are associated with CNT which form the layer. They are analogous to those observed in the spectrum obtained at point A. Additional weak bands between 150 and 650 cm−1 indicate the presence of Cu2O in the crystal structures that are formed within the CNT layer. Their positions are similar to the positions of the most intense and characteristic bands as it was noted in the spectrum obtained at point C. Thus, it can be confirmed that the probing depth made it possible to determine the presence of both CNT and Cu2O. No bands characteristic of CuO were observed in the spectrum at any of the points studied59.

Confocal microscope image of B7 sample (ECD deposition time of 600 s, CuSO4 concentration of 100 mM) for the single point Raman analysis.

Raman spectra of the B7 sample (ECD deposition time of 600 s, CuSO4 concentration of 100 mM) in points A–C.
Raman spectroscopy was also used to investigate the inner composition of CNT layers after the process of ECD. The in-depth analysis was performed for A7 and B7 samples from the surface to the depth of 4.0 µm. In the both sets of spectra (Figs. 9, 10) the bands associated with the presence of CNT at about 1360 cm−1 (D band), 1585 cm−1 (G band) and 2710 cm−1 (2D band) are visible, as it is expected. Apart from them, the bands characteristic for Cu2O at about 150 cm−1 (T1u(TO) mode), 220 cm−1 (2Eu mode) and 650 cm−1 (T1u(LO) mode) are found in each of the spectra. This indicates the formation of its crystals within CNT layer during the process of ECD using 1 mM or 100 mM CuSO4 solutions. Based on the intensities of the bands observed in the spectra obtained at different depths, it could be concluded that Cu2O do not develop any distinctive clusters within the investigated part of the coatings.

Raman in-depth analysis of the A7 sample (ECD deposition time of 600 s, CuSO4 concentration of 1 mM) from the surface to the depth of 4.0 µm.

Raman in-depth analysis of the B7 sample (ECD deposition time of 600 s, CuSO4 concentration of 100 mM) from the surface to the depth of 4.0 µm.
Examination of the CNT layer microstructure by SEM–EDS
The results of Raman analysis indicated the formation of copper oxides within CNT layers after the process of ECD. Therefore, the examination of their microstructure was also performed. For this, the cross sections of the samples were prepared and analyzed by SEM and EDS.
Figure 11 shows the exemplary SEM images for the cross sections of A7 sample. CNT layer with the dendritic Cu crystals on the surface could be identified (Fig. 11a). In its entire volume the nanometric crystals are visible. They are also present in the fragments without Cu deposits on the surface (Fig. 11b). The distributions of the chosen elements obtained by EDS for the cross section of A7 sample are presented in Fig. 12. Chromium and iron are the main components of the stainless steel substrate (Fig. 12d,e) whereas nickel and gold were introduced as the additional layers in preparation for the analysis (Fig. 12h,i). The distribution of copper and oxygen (Fig. 12c,f) within CNT layer confirms the presence of copper oxides and the previously described Raman measurements indicated that this is Cu2O. Some small share of the oxygen content could be also associated with the functional groups of CNT introduced during their initial modification.

SEM images of the cross sections of A7 sample (ECD deposition time of 600 s, CuSO4 concentration of 1 mM) at the magnification of ×10,000.

SEM images of A7 sample (ECD deposition time of 600 s, CuSO4 concentration of 1 mM): cross section (a), cross section with elements distribution (b), single element distribution (c–i) including copper (c), iron (d), chromium (e), oxygen (f), carbon (g), nickel (h) and gold (i).
The exemplary SEM images for the cross sections of B7 sample are shown in Fig. 13. In the entire volume of CNT layer, the crystals with dimensions of hundreds of nanometers are observed (Fig. 13a). Therefore, they are significantly bigger than the ones noticed in A7 samples. Some large agglomerates within CNT layer which partially grow to its surface are also identified (Fig. 13b). The others could be formed on the surface or entirely in the volume of CNT layer (Fig. 14a). The distributions of the chosen elements obtained by EDS for the cross section of B7 sample are presented in Fig. 14. The origin of chromium and iron (Fig. 14d,e) as well as nickel and gold (Fig. 14h,i) is the same as for A7 sample. The distribution of copper and oxygen (Fig. 14c,f) within CNT layer also confirms the presence of copper oxides in form of the larger nanometric crystals. Based on the previously described Raman measurements it is known that this is Cu2O. Careful analysis of the oxygen distribution also reveals that the large agglomerates are composed only of Cu, but the one on the surface additionally shows the traces of surface oxidation. This result is in accordance with the confocal microscope image (Fig. 7) and the Raman analysis in point B (Fig. 8) which in the similar situation indicates the presence of some amount of Cu2O. In the SEM image a local inhomogeneity of CNT layer is observed and explained as related to the crystals growing in its volume. The octahedral Cu2O and spherical Cu agglomerates are formed and could grow to the surface. It is known41,60 that the thermodynamic deposition potential of metal cations on the same metal substrate is lower in comparison to the one for the substrate composed of the other materials. This is due to the formation of the same continuous crystal lattice by the electrodeposited substance. This effect could be the explanation for the observed Cu agglomerate which grows on the other one partially located within CNT layer (Fig. 14a). When Cu agglomerate grows to the surface it provides a preferred site for the Cu deposition in comparison with pure CNT layer.

SEM images of the cross sections of B7 sample (ECD deposition time of 600 s, CuSO4 concentration of 100 mM) at the magnification of ×10,000.

SEM images of B7 sample (ECD deposition time of 600 s, CuSO4 concentration of 100 mM): cross section (a), cross section with elements distribution (b), single element distribution (c–i) including copper (c), iron (d), chromium (e), oxygen (f), carbon (g), nickel (h) and gold (i).
Examination of the CNT layer composition be GIXRD
To support the findings of the SEM–EDS and Raman analysis the GIXRD measurements of the A7 and B7 samples were performed (Fig. 15). This method allows to assess the surface composition of the layers because of the limited penetration depth of X-rays. The diffraction peaks assigned to Cu (COD 4105681) and CNT (COD 1566075) could be identified in both diffractograms, but the ones attributed to Cu2O (COD 1000063) may be observed only for B7 sample (Table 3). This is consistent with the previous results and confirms the presence of the octahedral Cu2O crystals which grow to the surface of CNT layer during the process of ECD using 100 mM CuSO4 solution.

GIXRD patterns for A7 sample (ECD deposition time of 600 s, CuSO4 concentration of 1 mM) and B7 sample (ECD deposition time of 600 s, CuSO4 concentration of 100 mM).
The mechanism of copper(I) oxide formation within CNT layers
Raman and SEM–EDS analysis revealed the formation of Cu2O within CNT layer during the process of ECD. For the samples prepared in the process of ECD using lower concentration of CuSO4 solution, i.e. 1 mM (A series), the nanometric crystals were formed whereas using higher concentration of the solution, i.e. 100 mM (B series), the crystals with dimensions of hundreds of nanometers were grown. To explain this effect, we propose the mechanism based on the reaction:
It consists of two stages:
CNT in the layer are functionalized by the initial introduction of the negatively charged functional groups (mainly carboxyl COO− groups). Some of Cu2+ cations which diffuse from the bulk solution to their proximity are electrically attracted by them. This bonding enables only the partial reduction of Cu+ cations instead of the total reduction to metal Cu. These cations remain bonded to the functionalized CNT. If their local concentration reaches a specific threshold and all the functional groups are occupied, then the crystallization of Cu2O occurs. In case of the lower concentration of CuSO4 solution, i.e. 1 mM, the diffusion rate significantly decrease. Therefore, the nucleation provides the formation of crystals of smaller size. Contrary, for the higher concentration of the solution, i.e. 100 mM, the increased diffusion rate allows the growth of bigger crystals.
Conclusions
The high overpotential conditions and the significantly different concentrations of CuSO4 solutions applied in the ECD process enabled the formation of various forms of crystals on the surface and within the volume of CNT layer. This indicates that by changing these parameters, the microstructure and chemical composition of the electrodeposited material could be easily controlled. SEM–EDS and Raman spectroscopy, with the addition of GIXRD to confirm the results obtained, were shown as a complete set of methods providing detailed characterization of the samples prepared in this way. Specific attention was given to the formation of crystals within the CNT layer. This required in-depth Raman spectroscopy measurements and SEM–EDS analysis for the cross sections. The study showed that in the high overpotential conditions and using CuSO4 solutions with a concentration of 1 mM the formation of dendritic crystals is favored. However, at their concentration of 100 mM, well-defined single crystals are formed. Such differences could be explained by various mechanisms involving the diffusion and reduction during the ECD process. At the lower concentrations of the solutions crystal growth is limited by mass transport, and at the higher concentrations—by the kinetics of the process of cations reduction. A noteworthy phenomenon that was observed for the samples prepared was the formation of Cu2O crystals. A mechanism for this process was proposed based on the electrostatic bonding of Cu2+ cations by the negatively charged functional groups of CNT, which results only in their partial reduction to Cu+ cations. When their local concentration reaches a certain threshold, the crystallization of Cu2O occurs. The results obtained may be supportive in the preparation of composite materials based on Cu and CNT with the designed microstructure easily controlled by the ECD process parameters.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information files. Additional data related to this study are available from the corresponding author on reasonable request.
References
Kadekar, V., Fang, W. & Liou, F. Deposition technologies for micromanufacturing: A review. J. Manuf. Sci. Eng. 126(4), 787–795. https://doi.org/10.1115/1.1811118 (2004).
Liu, P. S. & Liang, K. M. Review functional materials of porous metals made by P/M, electroplating and some other techniques. J. Mater. Sci. 36(21), 5059–5072. https://doi.org/10.1023/A:1012483920628 (2001).
Kozlovskiy, A. L. et al. Influence of electrodeposition parameters on structural and morphological features of Ni nanotubes. Phys. Met. Metallogr. 118(2), 164–169. https://doi.org/10.1134/S0031918X17020065 (2017).
Raghavendra, C. R., Basavarajappa, S., Sogalad, I., Kallappa, V. & Saunshi, V. Study on surface roughness parameters of nano composite coatings prepared by electrodeposition process. Mater. Today: Proc. 38(5), 3110–3115. https://doi.org/10.1016/j.matpr.2020.09.493 (2020).
Mureşan, A. C., Buruiană, D. L., Istrate, G. G. & Pintilie, S. C. Effect of electrodeposition parameters on the morphology, topography and corrosion resistance of epoxy resin/zinc hybrid coatings. Materials 14(8), 1991. https://doi.org/10.3390/ma14081991 (2021).
Walsh, F. C., Wang, S. & Zhou, N. The electrodeposition of composite coatings: diversity, applications and challenges. Curr. Opin. Electrochem. 20, 8–19. https://doi.org/10.1016/j.coelec.2020.01.011 (2020).
Ünal, E., Yaşar, A. & Karahan, I. H. A review of electrodeposited composite coatings with Ni–B alloy matrix. Mater. Res. Express. 6, 092004 (2019).
Karimzadeh, A., Aliofkhazraei, M. & Walsh, F. C. A review of electrodeposited Ni–Co alloy and composite coatings: Microstructure, properties and applications. Surf. Coat. Technol. 372, 463–498. https://doi.org/10.1016/j.surfcoat.2019.04.079 (2019).
Waware, U. S., Hamouda, A. M. S., Bajaj, B., Borkar, T. & Pradhan, A. K. Synthesis and characterization of electrodeposited Ni-B-Tl2O3 composite coatings. J. Alloys Compd. 769, 353–359. https://doi.org/10.1016/j.jallcom.2018.08.017 (2018).
Jabbar, A. et al. Electrochemical deposition of nickel graphene composite coatings: effect of deposition temperature on its surface morphology and corrosion resistance. RSC Adv. 7(49), 31100–31109. https://doi.org/10.1039/C6RA28755G (2017).
Raghupathy, Y., Kamboj, A., Rekha, M. Y., Narasimha Rao, N. P. & Srivastava, C. Copper-graphene oxide composite coatings for corrosion protection of mild steel in 3.5% NaCl. Thin Solid Films 636, 107–115. https://doi.org/10.1016/j.tsf.2017.05.042 (2017).
Lacitignola, D., Bozzini, B., Peipmann, R. & Sgura, I. Cross-diffusion effects on a morphochemical model for electrodeposition. Appl. Math. Model. 57, 492–513. https://doi.org/10.1016/j.apm.2018.01.005 (2018).
Wang, K., Xiao, Y., Pei, P., Liu, X. & Wang, Y. A phase-field model of dendrite growth of electrodeposited zinc. J. Electrochem. Soc. 166(10), D389–D394. https://doi.org/10.1149/2.0541910jes (2019).
Gao, J., Tao, H. & Liu, B. Progress of nonprecious-metal-based electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 33, 2003786. https://doi.org/10.1002/adma.202003786 (2021).
Manivannan, R. & Victoria, S. N. Preparation of chalcogenide thin films using electrodeposition method for solar cell applications—a review. Sol. Energy 173, 1144–1157. https://doi.org/10.1016/j.solener.2018.08.057 (2018).
Hoang, T. T. H., Ma, S., Gold, J. I., Kenis, P. J. A. & Gewirth, A. A. Nanoporous copper films by additive-controlled electrodeposition: CO2 reduction catalysis. ACS Catal. 7(5), 3313–3321. https://doi.org/10.1021/acscatal.6b03613 (2017).
Agasti, A., Mallick, S. & Bhargava, P. Electrolyte pH dependent controlled growth of co-electrodeposited CZT films for application in CZTS based thin film solar cells. J. Mater. Sci. Mater. Electron. 29(5), 4065–4074. https://doi.org/10.1007/s10854-017-8350-z (2018).
Okamoto, N., Wang, F. & Watanabe, T. Adhesion of electrodeposited copper, nickel and silver films on copper, nickel and silver substrates. Mater. Trans. 45(12), 3330–3333. https://doi.org/10.2320/matertrans.45.3330 (2004).
Kim, S. & Duquette, D. J. Effect of chemical composition on adhesion of directly electrodeposited copper film on TiN. J. Electrochem. Soc. 153(6), C417. https://doi.org/10.1149/1.2189971 (2006).
Frost, G. & Ladani, L. Development of high-temperature-resistant seed layer for electrodeposition of copper for microelectronic applications. J. Electron. Mater. 49, 1387–1395. https://doi.org/10.1007/s11664-019-07826-y (2019).
Kuang, Y. et al. One-step electrodeposition of superhydrophobic copper coating from ionic liquid. Mater. Lett. 303, 130579. https://doi.org/10.1021/am501539b (2021).
Ge, M., Gondosiswanto, R. & Zhao, C. Electrodeposited copper nanoparticles in ionic liquid microchannels electrode for carbon dioxide sensor. Inorg. Chem. Commun. 107(3), 107458. https://doi.org/10.1016/j.inoche.2019.107458 (2019).
Jain, R. & Pitchumani, R. Facile fabrication of durable copper-based superhydrophobic surfaces via electrodeposition. Langmuir 34(10), 3159–3169. https://doi.org/10.1021/acs.langmuir.7b02227 (2017).
Chang, T., Leygraf, C., Wallinder, I. O. & Jin, Y. Understanding the barrier layer formed via adding BTAH in copper film electrodeposition. J. Electrochem. Soc. 166(2), D10–D20. https://doi.org/10.1149/2.0041902jes (2019).
Kim, M. J. et al. One-step electrodeposition of copper on conductive 3D printed objects. Addit. Manuf. 27, 318–326. https://doi.org/10.1016/j.addma.2019.03.016 (2019).
Beverskog, B. Revised pourbaix diagrams for copper at 25 to 300°C. J. Electrochem. Soc. 144(10), 3476–3483. https://doi.org/10.1149/1.1838036 (1997).
Brandt, I. S., Tumelero, M. A., Pelegrini, S., Zangari, G. & Pasa, A. A. Electrodeposition of Cu2O: growth, properties, and applications. J. Solid State Electrochem. 21(7), 1999–2020. https://doi.org/10.1007/s10008-017-3660-x (2017).
Radisic, A., Ross, F. M. & Searson, P. C. In situ study of the growth kinetics of individual island electrodeposition of copper. J. Phys. Chem. 110(15), 7862–7868. https://doi.org/10.1021/jp057549a (2006).
Radisic, A., Vereecken, P. M., Searson, P. C. & Ross, F. M. The morphology and nucleation kinetics of copper islands during electrodeposition. Surf. Sci. 600(9), 1817–1826. https://doi.org/10.1016/j.susc.2006.02.025 (2006).
Khattar, H. K., Juda, A. M. & Al-Saady, F. A. A. Preparation of Cu2O nanoparticles as a catalyst in photocatalyst activity using a simple electrodeposition route. Nano Biomed. Eng. 10(4), 406–416. https://doi.org/10.5101/nbe.v10i4.p406-416 (2018).
Diao, S., Wang, Y. & Jin, H. Electronucleation mechanism of copper in wastewater by controlled electrodeposition analysis. RSC Adv. 10, 38683–38694. https://doi.org/10.1039/D0RA07380F (2020).
Riveros, G. et al. Study of the nucleation and growth mechanisms of copper electrodeposition on bare and nitrogen-doped reduced graphene oxide modified SnO2:F/glass substrates. J. Electrochem. Soc. 167, 122508. https://doi.org/10.1149/1945-7111/abb281 (2020).
Luo, Z. et al. Electrodeposition of copper nanopowder with controllable morphology: influence of pH on the nucleation/growth mechanism. J. Solid State Electrochem. 25, 1611–1621. https://doi.org/10.1007/s10008-021-04913-1 (2021).
Islam, M., Arya, N., Weidler, P. G., Korvink, J. G. & Badilita, V. Electrodeposition of chitosan enables synthesis of copper/carbon composites for H2O2 sensing. Mater. Today Chem. 17, 100338. https://doi.org/10.1016/j.mtchem.2020.100338 (2020).
Hagio, T. et al. Electrodeposition of nano-diamond/copper composite platings: Improved interfacial adhesion between diamond and copper via formation of silicon carbide on diamond surface. Surf. Coat. Technol. 403, 126322. https://doi.org/10.1016/j.surfcoat.2020.126322 (2020).
Cai, B., Gui, Z., Guo, T. & Kang, Z. Fabrication of self-recovering superhydrophobic Cu-CNTs composite coatings via co-electrodeposition: Wettability transition is due to spontaneous adsorption of airborne hydrocarbons. Colloids Interface Sci. Commun. 46, 100524. https://doi.org/10.1016/j.colcom.2021.100524 (2022).
Li, Z. et al. Binder free Cu2O/CuO/Cu/carbon-polymer composite fibers derived from metal/organic hybrid materials through electrodeposition method as high performance anode materials for lithium-ion batteries. J. Alloys Compd. 864, 158585. https://doi.org/10.1016/j.jallcom.2020.158585 (2021).
Wang, Z., Cai, X., Yang, C. & Zhou, L. Improving strength and high electrical conductivity of multi-walled carbon nanotubes/copper composites fabricated by electrodeposition and powder metallurgy. J. Alloys Compd. 735, 905–913. https://doi.org/10.1016/j.jallcom.2017.11.200 (2018).
Zhou, M. et al. Electrodeposition of CNTs/copper composite coatings with enhanced tribological performance from a low concentration CNTs colloidal solution. Mater. Res. Bull. 97, 537–543. https://doi.org/10.1016/j.materresbull.2017.09.062 (2018).
Wu, L., Lu, Z. & Ye, J. Enzyme-free glucose sensor based on layer-by-layer electrodeposition of multilayer films of multi-walled carbon nanotubes and Cu-based metal framework modified glassy carbon electrode. Biosens. Bioelectron. 135, 45–49. https://doi.org/10.1016/j.bios.2019.03.064 (2019).
Yehezkel, S., Auinat, M., Sezin, N., Starosvetsky, D. & Ein-Eli, Y. Distinct copper electrodeposited carbon nanotubes (CNT) tissues as anode current collectors in Li-ion battery. Electrochim. Acta 229, 404–414. https://doi.org/10.1016/j.electacta.2017.01.175 (2017).
Stobinski, L. et al. Multiwall carbon nanotubes purification and oxidation by nitric acid studied by the FTIR and electron spectroscopy methods. J. Alloys Compd. 501(1), 77–84. https://doi.org/10.1016/j.jallcom.2010.04.032 (2010).
Chen, L., Wang, G., He, Y., Wang, Y. & Zhang, M. The influence of overpotential on electrodeposited aluminum coating onto zincated ZM5 magnesium alloy in ionic liquid. Mat. Lett. 258, 126814. https://doi.org/10.1016/j.matlet.2019.126814 (2020).
Foroozan, T. et al. Non-dendritic Zn electrodeposition enabled by zincophilic graphene substrates. ACS Appl. Mater. Interfaces 11(47), 44077–44089. https://doi.org/10.1021/acsami.9b13174 (2019).
Motobayashi, K., Shibamura, Y. & Ikeda, K. Origin of a high overpotential of co electrodeposition in a room-temperature ionic liquid. J. Phys. Chem. Lett. 11(20), 8697–8702. https://doi.org/10.1021/acs.jpclett.0c02605 (2020).
Nikolić, N. D., Popov, K. I., Pavlović, L. J. & Pavlović, M. G. The effect of hydrogen codeposition on the morphology of copper electrodeposits. I. The concept of effective overpotential. J. Electroanal. Chem. 588(1), 88–98. https://doi.org/10.1016/j.jelechem.2005.12.006 (2006).
Eiler, K., Suriñach, S., Sort, J. & Pellicer, E. Mesoporous Ni-rich Ni–Pt thin films: electrodeposition, characterization and performance toward hydrogen evolution reaction in acidic media. Appl. Catal. B. 265, 118597. https://doi.org/10.1016/j.apcatb.2020.118597 (2020).
Nikitin, V. S., Ostanina, T. N., Rudoi, V. M., Kuloshvili, T. S. & Darintseva, A. B. Features of hydrogen evolution during Electrodeposition of loose deposits of copper, nickel and zinc. J. Electroanal. Chem. 870, 114230. https://doi.org/10.1016/j.jelechem.2020.114230 (2020).
Nanda, B. & Mallik, M. Production of copper powder by electrodeposition with different equilibrium crystal shape. Trans. Indian Inst. Met. 73(8), 2113–2119. https://doi.org/10.1007/s12666-020-02015-6 (2020).
Han, Y. J., Zhang, X. & Leach, G. W. Shape control of electrodeposited copper films and nanostructures through additive effects. Langmuir 30(12), 3589–3598. https://doi.org/10.1021/la500001j (2014).
Wahyudi, S., Soepriyanto, S., Mubarok, M. Z. & Sutarno. Effect of pulse parameters on the particle size of copper powder electrodeposition. IOP Conf. Ser. Mater. Sci. Eng. 547, 012020. https://doi.org/10.1088/1757-899X/547/1/012020 (2019).
Santos, N. M. & Santos, D. M. F. A fractal dimension minimum in electrodeposited copper dendritic patterns. Chaos Solitons Fractals 116, 381–385. https://doi.org/10.1016/j.chaos.2018.09.042 (2018).
Marchewka, J., Jeleń, P., Długoń, E., Sitarz, M. & Błażewicz, M. Spectroscopic investigation of the carbon nanotubes and polysiloxane coatings on titanium surface. J. Mol. Struct. 1212, 128176. https://doi.org/10.1016/j.molstruc.2020.128176 (2020).
Gawęda, M. et al. Polysiloxane-multiwalled carbon nanotube layers on steel substrate: microstructural, structural and electrochemical studies. J. Electrochem. Soc. 166(13), D707–D717. https://doi.org/10.1149/2.1261913jes (2019).
Lehman, J. H., Terrones, M., Mansfield, E., Hurst, K. E. & Meunier, V. Evaluating the characteristics of multiwall carbon nanotubes. Carbon 49(8), 2581–2602. https://doi.org/10.1016/j.carbon.2011.03.028 (2011).
Jeleń, P. et al. Free carbon phase in SiOC glasses derived from ladder-like silsesquioxanes. J. Mol. Struct. 1126, 172–176. https://doi.org/10.1016/j.molstruc.2016.03.096 (2016).
Carbó-Argibay, E. et al. Up-scaling the synthesis of Cu2O submicron particles with controlled morphologies for solar H2 evolution from water. J. Colloid Interface Sci. 456, 219–227. https://doi.org/10.1016/j.jcis.2015.06.014 (2015).
Sander, T. et al. Correlation of intrinsic point defects and the Raman modes of cuprous oxide. Phys. Rev. B. 90, 045203. https://doi.org/10.1103/PhysRevB.90.045203 (2014).
Debbichi, L., Marco de Lucas, M. C., Pierson, J. F. & Krüger, P. Vibrational properties of CuO and Cu4O3 from first-principles calculations, and Raman and infrared spectroscopy. J. Phys. Chem. C. 116(18), 10232–10237. https://doi.org/10.1021/jp303096m (2012).
Nikolić, N. D., Popov, K. I., Pavlović, L. J. & Pavlović, M. G. Morphologies of copper deposits obtained by the electrodeposition at high overpotentials. Surf. Coat. Technol. 201(3–4), 560–566. https://doi.org/10.1016/j.surfcoat.2005.12.004 (2006).
Acknowledgements
The authors would like to thank Piotr Jeleń, PhD for his support in the Raman measurements as well as Maciej Bik, PhD for the preparation of the cross sections samples. This research was funded by the National Science Centre, Poland, Grant number 2017/25/B/ST8/02602. Research project supported by program “Excellence initiative-research university” for the AGH University of Science and Technology.
Author information
Authors and Affiliations
Contributions
J.M.—conceptualization, J.M., E.K. and P.B.—methodology, J.M., E.K. and P.B.—validation, J.M., E.K. and P.B.—formal analysis, J.M., E.K. and P.B.—investigation, J.M. and P.B.—writing—original draft preparation, J.M. and P.B.—writing—review and editing, J.M.—visualization, M.S.—supervision, M.S.—project administration, M.S.—funding acquisition. All authors have read, reviewed and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marchewka, J., Kołodziejczyk, E., Bezkosty, P. et al. Characterization of electrochemical deposition of copper and copper(I) oxide on the carbon nanotubes coated stainless steel substrates. Sci Rep 13, 6786 (2023). https://doi.org/10.1038/s41598-023-33963-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-33963-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
