
Abstract
Salinity can influence microbial communities and related functional groups in lacustrine sediments, but few studies have examined temporal variability in salinity and associated changes in lacustrine microbial communities and functional groups. To better understand how microbial communities and functional groups respond to salinity, we examined geochemistry and functional gene amplicon sequence data collected from 13 lakes located in Kiritimati, Republic of Kiribati (2° N, 157° W) in July 2014 and June 2019, dates which bracket the very large El Niño event of 2015–2016 and a period of extremely high precipitation rates. Lake water salinity values in 2019 were significantly reduced and covaried with ecological distances between microbial samples. Specifically, phylum- and family-level results indicate that more halophilic microorganisms occurred in 2014 samples, whereas more mesohaline, marine, or halotolerant microorganisms were detected in 2019 samples. Functional Annotation of Prokaryotic Taxa (FAPROTAX) and functional gene results (nifH, nrfA, aprA) suggest that salinity influences the relative abundance of key functional groups (chemoheterotrophs, phototrophs, nitrogen fixers, denitrifiers, sulfate reducers), as well as the microbial diversity within functional groups. Accordingly, we conclude that microbial community and functional gene groups in the lacustrine sediments of Kiritimati show dynamic changes and adaptations to the fluctuations in salinity driven by the El Niño-Southern Oscillation.
Similar content being viewed by others

Introduction
In lacustrine systems, salinity is an important stress selection factor and can influence microbial diversity, community structure, and metabolic activity in surface sediments1,2,3,4,5. Lake salinity can also change abruptly, due to climate variability (e.g., from extreme precipitation events and drought), with substantial impacts to lacustrine ecosystems6,7,8. Notably, previous studies on the effects of salinity on microbial communities have focused on spatial datasets from saline and hypersaline lakes3,9 and estuaries with continuous salinity gradients10,11. Although the temporal response of microorganisms in lacustrine surface sediments to abrupt salinity changes has been documented in the literature, most studies have focused on one lake12,13,14, and few studies have examined temporal shifts in tropical lacustrine sediments.
Microbial functional groups that actively participate in element cycling also vary in response to salinity changes in lacustrine settings. As an example, the relative abundance of nitrogen fixation genes, coding an important pathway for aquatic ecosystem productivity, is often reduced with high salinity in lacustrine sediments, microbial mats, and estuarine environments15,16,17. High salinity, on the other hand, provides favorable conditions for dissimilatory nitrate reduction to ammonium (DNRA) over denitrification in estuarine sediments18,19,20. Supporting this observation, microbial communities that perform these metabolic activities also show significant changes with salinity, partly due to the extreme energetic demands of osmoregulation in high salinity conditions1. Thus, examining temporal changes in community composition and microbial functional groups in response to changes in salinity can improve understanding of functional group roles in element cycling under different salinity conditions in lacustrine ecosystems.
On the island of Kiritimati, Kiribati, in the central tropical Pacific, several hundreds of lakes with salinities ranging from brackish to hypersaline21 are an ideal natural laboratory to investigate salinity influences on microbial communities and functional groups. Here, the interannual El Niño-Southern Oscillation (ENSO) phenomenon results in significant anomalies in atmospheric moisture balance (precipitation minus evaporation)22. Such anomalies greatly affect groundwater, surface water area, and salinity in many of these lakes22,23,24. Thus, Kiritimati is an excellent field site to study the temporal response of microbial communities to lacustrine salinity changes. During 2015–2016, a very strong central-Pacific style El Niño led to abundant rainfall over Kiritimati25, increasing lake surface water area22, and likely leading to significant lake salinity changes. Here we investigate lake water salinity, microbial community and microbial functional groups changes in a set of lake surface (0–5 cm) sediment samples from 2019 and 20149. We used samples from the same lakes taken during these two field seasons to explore the relationship between microbial community and microbial functional groups with changes in lacustrine physiochemical parameters. We hypothesize that between 2014 and 2019, the dominant microbial taxa changed from halophiles to more broadly halotolerant or mesohaline taxa, and that the relative abundance and microbial diversity of functional groups also changed as salinity decreased. We first present the physiochemical data of the lakes from 2014 to 2019 to identify environmental factors that have changed significantly between these two years and discuss the potential causes. We next compare the microbial community of 2014 and 2019 samples to determine the extent of the temporal changes in microbial community and link specific taxa showing significant changes with changes in physiochemical parameters. Finally, we analyze the relationship between salinity and dominant functional groups and the diversity within these groups using taxonomy predictions and functional gene amplicons.
Methods
Field description and sampling
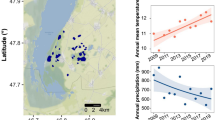
The Kiritimati Atoll (Republic of Kiribati, 1.9° N, 157.4° W), one of the Northern Line Island in the central tropical Pacific, is the largest coral atoll in the world with a surface area of ~ 360 km2. It contains hundreds of brackish to hypersaline carbonate-rich lakes, many of which are connected to a large lagoon (Fig. 1A)21. Nearshore sediment samples were collected from the top 5 cm of the water–sediment/microbial mat interface in the water depth of 20–50 cm from 13 lakes with salinity ranging from freshwater to hypersaline in late July and early August 2014, and June 2019 (Fig. 1A–D).

(A) Map of Kiritimati (Google Earth, 2021) indicating (B–D) locations of sample sites. Samples collected in both 2014 and 2019 are denoted by white circles, and samples only collected in 2019 are denoted by red circles. (E) Total monthly precipitation from the Kiritimati weather station. Markers indicate 2014 and 2019 field seasons. (F) Histogram of lake salinity variations from collected 2014 (red) and 2019 (blue) lake samples.
Temperature (± 0.2 °C), specific conductance relative to 25 °C (± 0.001 mS/cm), pH (± 0.2 units), and dissolved oxygen (DO) content (± 0.2 mg/L; ± 2%) were measured in situ using a YSI ProPlus multiparameter water quality sonde. Salinity was calculated in ppt from these data using the Gibbs Sea Water (GSW) Oceanographic Toolbox26. Alkalinity was measured on-site using a Hach Company alkalinity test kit. For cation and anion analysis, samples were double filtered by 5 μm and 0.22 μm filters and stored refrigerated in the dark in pre-acidified amber plastic bottles until analysis. The major cations concentrations were measured with ICP-OES (Perkin-Elmer, Optima 5300 DV) with a precision of ± 2%27. The concentration of major anions was measured with Ion Chromatography with a precision of ± 5%28. Water samples for water isotope analysis (δ18O and δ2H) were double filtered by 5 μm and 0.2 μm filters, collected in 30 mL brown HPDE vials without headspace and stored in a 4 ℃ freezer before analysis. The water δ18O and δ2H values were measured on a Picarro L2130‐i cavity ringdown isotopic analyzer in the UIUC Department of Geology29. The average precision is ± 0.1‰ for δ18O (VSMOW) and ± 0.8‰ for δ2H (VSMOW).
Sediment samples for nucleic acid extraction were sampled from 0 to 5 cm from the sediment/mat-water interface and treated with RNAlater preservative (ThermoFisher Scientific, Waltham, MA), homogenized, and stored on ice. Upon return, they were stored at − 20℃ freezer before DNA extraction and PCR amplification.
DNA extraction and sequencing
DNA extractions for 2019 samples were performed using a modified protocol to extract DNA and RNA from soil using phenol–chloroform methods (see Supplemental Materials S1.1) in Urbana, IL. Details for DNA extractions methods for 2014 samples are given in detail in Schmitt et al. (2019). The quality and quantity of extracted DNA were determined by agarose gel electrophoresis and fluorometry (Qubit 4.0). Primer sets for bacterial- and archaeal 16S rRNA genes, N- and S-cycle genes of interest (narG, nxrB, nirK, nirS, nosZ, nifH, amoA, amoB, nrfA, dsrB, aprA, soxB) were used to generate sequence libraries (Table S1). Specific gene-targeted amplicons for downstream sequencing were generated for all samples using a Fluidigm microarray system through services available in the Functional Genomics Unit of the University of Illinois Urbana-Champaign Carver Biotechnology Center (Fluidigm Array details in Supplemental Materials S1.2). Paired-end sequencing (2 × 250 base pairs) was performed on one lane of a MiSeq Nano (v2) (San Diego, CA, USA) for 2014 samples and one lane of an Illumina NovaSeq 6000 platform (San Diego, CA, USA) for 2019 samples. Raw sequence data were deposited in MG-RAST (2014 samples, project number MGP82583) and NCBI (2019 samples, BioProject ID PRJNA76940).
Microbial community and functional gene analyses
Following sequencing, each primer pair’s amplicon libraries from 2014 and 2019 samples were parallelly processed using Mothur30 and phyloseq31. For 16S rRNA genes, amplicon libraries generated from different years were examined using the exact same pipeline from Mothur and phyloseq. Briefly, paired-end reads were merged for sequences obtained from each primer pair and filtered to the expected amplicon length using Mothur30. For 16S rRNA genes, an OTU file based on 97% sequence similarity that included the number of OTUs found in each sample, and a taxonomy file of the consensus taxonomy for each OTU was generated from the Mothur pipeline, which was later imported into R software using phyloseq packages for downstream analysis. For other functional gene sequences, an OTU file based on 97% sequence similarity that included the number of OTUs found in each sample was generated from the Mothur pipeline and analyzed using phyloseq packages. The recovery reads from different functional genes after quality trimming are listed in Table S5.
Several methods were used to remove the potential batch effects from different primers and sequencing platforms used for 2014 and 2019 samples. First, the merged sequences for 16S rRNA from different years were aligned to the same reference sequence file (SILVA 138 database) and the same taxonomy file (see Supplementary Materials S1.3 for details). Second, for different years, the unique OTUs with the same taxonomy at the genus level were merged using tax_glom in phyloseq to correct for batch effects (this paper referred to as unique populations at the genus level). Genus-level comparisons were then used for downstream analysis to compare relative abundances for 16S rRNA genes in different years, and compared to another method to correct for batch effects (see Supplemental Materials S1.4 for details).
Statistical analysis
The physiochemical parameters of lakes in 2014 and 2019 were compared and visualized using principal component analysis (PCA) to determine which parameters contribute to the variations of environmental data in the two sampling periods. In addition, to determine the parameters that changed significantly from 2014 to 2019, all parameters measured in 2014 and 2019 were assessed with a non-parametric pairwise Wilcoxon rank-sum test, and the p-values were adjusted by the Bonferroni method.
Alpha and beta diversity analyses on the genus level (PERMANOVA, multivariate permutation analysis of variance; mantel test; differential abundance) were conducted in Mothur or the R programming environment using packages phyloseq, vegan, and edgeR30,31,32,33 (Figure S1). Pairwise permutation multivariate analysis of variance (PERMANOVA) was used to determine the significance of microbial community composition differences from 2014 to 2019. In order to test how environmental parameters influence community diversity, nMDS ordination plots for bacterial and archaeal communities based on 16S rRNA gene sequences were overlaid with environmental variables (Spearman coefficients) listed in Table S2 using the function “envfit” from the vegan package32. Environmental variables that were not statistically significant were excluded from the plot and were not discussed. A Mantel test with the vegan package was used to assess the relationship between the microbial community and the measured environmental variables using the Bray–Curtis distance matrix.
The differences in the relative abundance of the most abundant phyla (at least > 1% in one sample) in 2014 versus 2019 lakes were assessed using the pairwise non-parametric Wilcoxon rank-sum test. This test also includes the most abundant subclasses (at least > 1% in one sample of all subclasses) of Proteobacteria due to the diversified nature of this phylum. To further explore differences in microbial communities at a finer level, a pairwise comparison of families that are more abundant in 2014 lakes compared to 2019 lakes was conducted using log twofold changes (logFC) using edgeR33, which expresses the ratio between two quantities. The correlations of relative abundance of most abundant bacterial and archaeal phyla with physiochemical parameters of all sampled lakes were visualized by “aheatmap” from package NMF34.
Functional Annotation of Prokaryotic Taxa (FAPROTAX) was used to make metabolic predictions from the valid 16S rRNA gene sequences obtained from 2014 and 2019 lake sediment samples. The predictions made in correspondence to the OTUs obtained here are based on the characterized strains with putative functional tables in the FAPROTAX database35. In addition, the relative abundances of different metabolic groups across the lakes and their relationship with environmental parameters were visualized by a heatmap.
The functional gene sequences acquired from 2019 lakes were input to R and processed using the same code as the 16S rRNA gene sequences. Mantel tests were used to assess the relationship between functional and taxonomic composition of the bacterial community based on Bray–Curtis distance. For the following analyses, only functional gene compositions correlated significantly with taxonomic composition were used. The alpha diversity (Shannon index) of functional genes of different lakes was calculated using the phyloseq package and correlated with the environmental factors of each lake. Mantel tests were also conducted to determine the correlation between functional genes and environmental factors.
Results
Sediment and water properties
The surface water chemistry of Kiritimati lakes varies considerably, both spatially and temporally (Table S2). Salinity in the sampled lakes ranged from 40 ppt (Lake 14, 2019) to 190 ppt (Lake 17, 2014), with a significantly higher median (137.4 ppt vs. 72 ppt) and mean value in 2014 lakes (135.2 ppt vs 74.5 ppt) (Fig. 1F). As expected, the concentrations of the most abundant cations and anions present in seawater, such as Mg2+, Na+, and Sr2+, are highly correlated with salinity (Figures S2, S3). Despite some differences in temperature and pH (Table S2), the median values of these parameters measured from 2014 (temperature: 30.6 °C, pH: 8, respectively) and 2019 lakes (temperature: 30.1 °C, pH: 7.99, respectively) are similar. Alkalinity, K+, and δ18O are significantly higher (p < 0.001 for all variables) in 2014 samples, and Ca2+ is significantly higher (p < 0.001) in 2019 samples (Table S3).
Bacteria and archaea communities
Using 16S rRNA amplicon sequencing, we obtained 267,289 high-quality sequences generated for V4-515f primers from 2014 samples and 2,770,476 sequences for Arc519f-Bac785r from 2019 samples after filtering. There were 1163 unique genera identified for bacteria, which were assigned to 63 bacterial phyla, and 84 unique genera identified for archaea, which were assigned to 12 archaeal phyla (Figure S4). For bacterial phyla, Proteobacteria (33.02 ± 20.44%) and Bacteroidota (16.09 ± 11.16%) are dominant in most of 2014 and 2019 samples. For archaeal phyla, Thermoplasmatota and Halobacterota dominate most of 2014 and 2019 samples and Asgardarchaeota is also predominant in 2014 samples (Table 1). Nanoarchaeota shows high relative abundances in 2019 samples, however it is possible that the different PCR primers used with the 2014 samples would not have amplified sequences from this group. Other bacterial and archaeal phyla that make up at least 1% of the microbial community for at least 1 sample are listed in Table 1 and Figure S4.
Bacteria Shannon diversity index values range from 1.74 to 4.50, while archaeal values range from 0 to 2.8 (Fig. 2). The sample with 0 value from the Shannon diversity index had no Archaea genera detected from one of the 2014 samples. The alpha diversity index has a significant negative correlation with salinity (Bacteria: r = − 0.48, p < 0.01; Archaea: r = − 0.54, p < 0.01) (Table S4). In terms of beta diversity, the pairwise PERMANOVA results indicate that both bacterial and archaeal communities are significantly different from 2014 to 2019 (Bacterial: R2 = 0.37, p = 0.0001, Archaeal: R2 = 0.17, p = 0.0001, respectively). nMDS plots also show that both bacterial and archaeal communities in the 2019 samples plot together and are distinct from the groupings found in the 2014 samples (Fig. 2). When correlating the microbial communities with physiochemical parameters, mantel test results show that the bacterial and archaeal community compositions are significantly positively correlated with salinity, alkalinity, Ca2+, and K+ (p < 0.05), while bacterial community composition is also significantly positively correlated with δ18O (Table S6).

Non-metric multidimensional scaling (nMDS) and Shannon diversity plots of (A, C) bacterial and (B, D) archaeal community composition based on 16S rRNA gene sequencing data. The ordination is based on Bray–Curtis similarities overlaid with significantly correlating environmental variables (p < 0.05) plotted as vectors. Vector length is proportional to the strength of correlation between the variable and community similarity. See Table S4 for environmental fitting statistics. Samples from different years are denoted by different colors. All analyses were performed using R Statistical Software (v4.1.3; R Core Team 2022).
Functional group results
The FAPROTAX prediction analysis identified 35 metabolic functions based on 16S rRNA gene sequence data (relative abundance > 0.01%) in all sediment samples. Comparing samples from 2014 and 2019, aerobic chemoheterotrophy is significantly higher in 2014 samples and photoautotrophy is significantly higher in 2019 samples (Table S7; Fig. 5). Among the predicted microbial functions, many of them show negative correlations with salinity (p < 0.05), including dominant microbial functions (with median value > 1%) such as photoautotrophy and fermentation. In comparison, aerobic chemoheterotrophy is significantly positively correlated with salinity (p < 0.05) (Table S8).
The functional gene amplicon sequence beta diversity, done only with the 2019 samples, showed a significant correlation with the 16S rRNA community composition with the nifH, nrfA, nirS, nosZ, aprA, and soxB genes (Table S9). Salinity significantly correlates with the Shannon index of both nifH (r = − 0.656, p < 0.001, N = 22) and nrfA genes (r = − 0.552, p = 0.008, N = 22) encoding nitrogenase (N2 fixation) and nitrite reductase (NO2− reduction to NH4+), respectively (Figure S7). Besides salinity, DO, pH, alkalinity, Sr2+, δ18O, and δ2H also significantly correlate with the Shannon index of one or more functional genes (Figure S7; Table 2). Salinity strongly correlates with nifH and aprA gene compositions (r = 0.40, 0.54; adjusted p = 0.002, 0.0003, respectively), along with Mg2+, Na+, Sr2+, Cl−, and SO42− (Table 2).
Discussion
El Niño and salinity changes from 2014 to 2019
El Niño years in the central tropical Pacific have much higher precipitation rates, leading to decreased lake salinity23,24. The substantial salinity changes from 2014 to 2019 are likely due to the very strong central Pacific El Niño event in 2015–2016, which caused record high rainfall in the central tropical Pacific (Fig. 1E)25. However, individual lakes may have responded differently to this precipitation anomaly, as lakes in different areas of Kiritimati are more sensitive to precipitation, evaporation, or sea level22. For example, sea level can affect the water budget of lakes that are surficially connected to the main lagoon, or have a stronger subsurface connection to the ocean. Therefore, an increase in sea level during El Niño events can also result in more saline seawater flowing into such lakes, which would contradict the decline in lake salinity caused by freshwater charges from groundwater and precipitation during these periods. When lakes are isolated from the lagoon and the ocean, the major water sources are likely to be precipitation and groundwater fluxes. As a result of high precipitation during El Niño events, such lakes will experience decreased salinity. Evaporation following the termination of an El Niño event will slowly increase lake salinity, but the rates of recovery to pre-event salinity values can vary depending on the rates of the groundwater recharge. Additionally, the permeability of the lake bottoms can also differ, which can impact groundwater flow23. Due to the permeability of the carbonate sediment on Kiritimati atoll, there may be subterranean connections between freshwater lenses and lakes, so proximity of lakes to fresh groundwater can also affect lake salinity21. Moreover, bacterial mat growth provides a sealing effect and can reduce lake bottom permeability, impacting groundwater transportation36. Overall, the observed Kiritimati lake salinity changes are primarily attributed to the extreme El Niño event from 2015 to 2016, with variations in individual lakes due to differences in proximity to freshwater lenses, permeability, and groundwater flow.
Community composition changes from 2014 to 2019
We found significant differences in the sediment bacterial and archaeal communities between 2014 and 2019, with these differences associated with changes in the physiochemical parameters of salinity (Tables S3 and S4; Fig. 2). Among the physiochemical parameters that changed substantially from 2014 to 2019, salinity is likely the major driver of the observed changes in microbial communities. Salinity is often considered a primary control on the composition of microbial communities in lakes, in both the water column and the underlying sediments3,4,37,38,39. High salinity can decrease the taxonomic diversity of sediment microbial communities, since high osmotic pressure during high salinity requires specific strategies for adaptation40,41. In addition, low salt conditions can also be deleterious for halophiles that use a “salt-in” strategy (accumulating KCl equal to NaCl within their cells), since many of the halophilic proteins of such halophiles will become unstable or denature in low salinity42. Therefore, the decrease in salinity and other solutes from 2014 to 2019 likely impacted the diversity of the sediment microbial communities.
Considering relative abundance changes of specific phyla, classes, and families from 2014 to 2019, Gammaproteobacteria (phylum Proteobacteria) is significantly more abundant in 2014 samples (Table 1), and its abundance is positively correlated with salinity (Fig. 3A). Gammaproteobacteria include families that contain halophilic or marine bacteria that can survive in saline environments, such as Alteromonadaceae, Pseudoalteromonas, Idiomarina, and Halomonadaceae43,44. These families are significantly more abundant in 2014 lakes with overall higher salinities (Fig. 4). The more abundant phyla in 2019 lake sediments exhibit a significant negative correlation with salinity (Fig. 3A) and include Cyanobacteria, Desulfobacterota (formerly Deltaproteobacteria), Chloroflexi, Gemmatimonadetes, and Actinobacteria. These phyla are also more abundant in brackish lakes from the 2014 spatial survey9. Cyanobacteria, Chloroflexi, and Actinobacteria are generally more abundant in low-salinity settings, since salinity is an important abiotic stress for these phyla2,45,46. For the class Alphaproteobacteria, the families Hyphomonadaceae, Kiloniellales, Rhizobiales, Rhodobacteraceae, and Geminicoccaceae are more abundant in 2019 samples. Among them, Hyphomonadaceae and Kiloniellales are predominantly mesophilic, marine bacteria47,48, and Rhodobacteraceae is one of the most widely distributed bacterial lineages in marine habitats49. Their normal presence in marine habitats can possibly explain their high abundance in 2019 samples with lower salinity.

Heatmap of correlations between environmental factors and (A) bacterial phyla and (B) top 20 unique genus groups. Corresponding phylum of each genus group is also indicated in (B). For rows (microbial communities) and columns (environmental factors), dendrograms from hierarchical clustering using the distance and clustering methods distfun and hclustfun are made and shown in the figure. All analyses were performed using R Statistical Software (v4.1.3; R Core Team 2022, https://www.R-project.org/).

LogFC pairwise comparison between (A) bacterial and (B) archaeal families within 2014 and 2019 samples with significant differences (p < 0.05). Positive logFC values on the x-axis indicate families that are more abundant in 2019 lake sediment samples, and negative logFC values on the x-axis indicate families that are more abundant in 2014 lake sediment samples. Colors of circles denote the phyla for each listed family, shown in the legend. All analyses were performed using R Statistical Software (v4.1.3; R Core Team 2022, https://www.R-project.org/).
Additionally, some substantial changes can occur at the taxonomic level of families even if the corresponding phylum (class) shows no overall significant change from 2014 to 2019. For example, the family Cyclobacteriaceae from phylum Bacteroidota, which contains species isolated from foreshore soils and saline lakes50,51, has a higher relative abundance in 2014. Similarly, the families Bacillaceae and Halanaerobiaceae from phylum Firmicutes are relatively more abundant in 2014 lakes compared to 2019 lakes (Fig. 4). The Halanaerobiaceae and Bacillaceae families can either be halotolerant (family Halanaerobiaceae) or capable of forming endospores, which can help them survive in hypersaline environments52,53. In addition, Bacillaceae is one of the most abundant families in 2014 and shows a significant correlation with salinity (Fig. 3B). Lastly, even though class Gammaproteobacteria is more abundant in 2014 samples, the Chromatiaceae family (also known as phototrophic purple sulfur bacteria) from this class is more predominant in most of 2019 samples.
The only archaeal phylum that differs significantly between 2014 and 2019 lakes is Nanoarchaeota (Table 1), which is not present in any of the 2014 samples. As the universal primer pair used in 2014 samples54 shows poorer coverage of archaeal communities than the primer set used in 2019 samples55, we cannot exclude the possibility that Nanoarchaeota may have been present in 2014 samples but were not detected due to methodological limitations. The logFC plot of the comparison of changes in archaeal community families indicates that, surprisingly, many family members from phylum Halobacterota have significantly higher relative abundances in 2019 lake sediments (Fig. 4B). Despite this, since the detections of archaeal communities in 2014 primers were poor, comparisons of archaeal communities from 2014 to 2019 samples may not be statistically valid. When examining the 2019 spatial samples, many of the detected genera from phylum Halobacterota are only present in a small set of samples (< 5 samples) (Table S10). When looking at the 5 genera that are found in at least half of the samples (> 11 samples), 2 of them (unclassified Halomicrobiacea and uncultured Haloferacaceae) show significant positive correlation with salinity (r = 0.471, 0.584; p = 0.03, 0.005, respectively), and are among the dominant genera of the Halobacterota phylum (Table S10). To be noticed, there are dominant genera from the Halobacterota phylum that show no significant correlations with salinity. Although members from phylum Halobacterota mainly inhabit high salinity environments56, 16S rRNA gene results show that members are also found from low- to moderate-salinity systems57,58,59. Recent studies have shown that Halobacteria can adapt to different salinity conditions through changing membrane permeability to different solutes60, and can rapidly repopulate sediments when salinity drops, such as after rainfall61,62. Specifically, one dominant genera (Halomarina) that occurs in 15 lake samples can grow in a wide range of salt concentrations, and survive at low salt concentrations and can recover after prolonged exposure to distilled water63. Therefore, many halophilic archaea in Kiritimati lake systems may be able to adapt to abrupt changes in salinity and may not be restricted to hypersaline environments. Future isolation and characterization of those Halobacterota strains can be used to test our hypothesis.
Overall, bacterial communities show more halophilic taxa in 2014 samples and more halotolerant or marine taxa in 2019 samples, suggesting salinity was a major driver of the observed bacterial community changes. However, the diversity and changes of archaeal groups in Kiritimati lake sediments need to be further evaluated due to the limitations of the universal primer chosen in 2014 and their overall unstudied nature.
Functional groups change with salinity
As salinity influences the microbial community composition, the relative distribution of microbial metabolisms that directly impact lake ecosystem functions can also vary2. Accordingly, we modeled the relative abundances of different metabolic pathways for each sample in 2014 and 2019 using FAPROTAX, a program that uses 16S rRNA gene phylogeny (Fig. 5). The results suggest that chemoheterotrophy (48.4 ± 9.76%) is dominant in 2014 samples, while chemoheterotrophy (19.21 ± 6.89%) and phototrophy (14.12 ± 4.68%) are both important metabolisms in 2019 samples (Table S7). Furthermore, we find significant positive correlations between aerobic chemoheterotrophy and salinity (r = 0.49, p = 0.00, N = 31), and negative correlations between photoautotrophy and salinity (r = − 0.41, p = 0.02, N = 31). Salinity can alter microbial community composition from primarily oxygenic phototrophs to primarily heterotrophs in sabkha and ocean ecosystems64,65. Furthermore, heterotrophs in the sediments can also utilize the organic matter substrates produced by phototrophs. For example, Gammaproteobacteria, a dominant class of heterotrophs in 2014 samples (median 53.7%), can play important roles in organic matter degradation in sediments by assimilating intermediate products (acetate) or initializing the decomposition of algal-derived organic matter66,67. In applying FAPROTAX to our data, the principal limitation is that FAPROTAX implicitly assumes that if all cultured members of a taxon (genus or species) can perform a particular function, then all members of the taxon (cultured and uncultured) can perform that function35,68. As more organisms are cultured in the future, it might lead to false generalizations. Therefore, we need to take precautions when interpreting the metabolic results obtained from FAPROTAX. Previous validation of FAPROTAX shows that some functional groups show good agreement in FAPROTAX and metagenomic results, including aerobic chemoheterotrophs and photoautothrophs35, which are also major functional groups shifts observed from 2014 to 2019 samples (Table S7; Fig. 5). Accordingly, FAPROTAX results may still be valid when it comes to predicting and comparing changes in aerobic chemoheterotrophy and photoautotrophy between 2014 and 2019 (Table S7; Fig. 5). In addition, previous biomarker results at Lake 30 support that microbial functional groups change with salinity over long timescales, since the absence of phytol (produced by photosynthetic organisms) and low δ2H values of C16:0 and C18:0 (indicating chemoautotrophic organisms) coincide with periods of high salinity over the last millennium69.

Heatmap showing relative abundances of metabolisms retrieved from Functional Annotation of Prokaryotic Taxa (FAPROTAX) for each sample. For each metabolism (rows), the relative abundances of each sample are centered, standardized and scaled to [− 1, 1] interval. For rows (metabolisms) and columns (lake samples), dendrograms from hierarchical clustering using the distance and clustering methods distfun and hclustfun are made and shown in the figure. All analyses were performed using R Statistical Software (v4.1.3; R Core Team 2022, https://www.R-project.org/).
Considering the limitations of the metabolic results predicted by FAPROTAX, we also checked the functional gene results to further evaluate how salinity may impact the functional potentials. The functional gene results acquired from the 2019 spatial survey also suggest that salinity may affect microbial diversity within functional groups. As an example, the richness and diversity of nrfA and nifH genes show a significant negative correlation with salinity (Figure S7), indicating that the diversity of these genes may decrease with salinity. Additionally, salinity significantly correlated with nifH and aprA in mantel tests, showing that a change in these genes would follow salinity (Table 2). Salinity is known to increase the activity of nitrogenase (NifH) genes and decrease the abundance of Cyanobacteria in intertidal microbial mats15. As salinity changes, we may see changes in dominant taxa harboring nifH genes. According to the nifH phylogenetic tree results, the dominant nifH OTUs of hypersaline lake site 17 (113 ppt) cluster near references Desulfovibrio marinus and Halothece (Fig. 6), which are both salt-tolerant halophilic taxa70,71. In contrast, the predominant nifH OTUs of brackish Lake 30 (1.7 ppt, Fig. 6) are from taxa Desulfobacter that are common in marine and non-halophilic aquatic environments72,73. Similarly, a study conducted in Tirez Lagoon suggests that aprA genes can be used as indicators of salinity74. The relative abundances of aprA OTUs in lake site 17 also differ significantly from those in Lake 30 on Kiritimati (Figure S8).

Distribition of nifH OTUs in the Kiritimati sediments under investigation and the phylogenetic tree for the nifH genes. The phylotypes under consideration (blue) comprised of the top 5 abundant OTUs identified in all the samples. The relative abundances of OTUs are presenting using a log2 transformation. The reference nifH genes (pink) were identified via BLAST against the NCBI-non redundant database and the gene sequences for the cultivated species were preferentially selected. The phylogenetic tree is generated using iTOL (v6, https://itol.embl.de/).
Metagenomic data from a specific hypersaline lake (Lake 1) sampled in 201775 provide additional information regarding how functional genes may change with salinity. The annotated nifH genes from metagenomic data (metagenome-assembly genomes and assembled sequences) of Lake 1 from 2017 (salinity: 44.7 ppt) cluster with nifH OTUs from amplicon sequences from 2019 (salinity: 71 ppt) (Figure S9), suggesting that those nifH genes from different years have close phylogenetic affiliations. Many of the annotated nifH genes from 2017 and 2019 belong to a clade of sulfate-reducing bacteria with known references that live at lower salinity conditions, including marine or slightly halophilic environments (e.g., Desulfovibrio marinus strain CS1, Desulfosarcina widdelii PP31, and Chloroherpeton thalassium)70,76,77, which is consistent with the relatively low salinity of the lakes in those two sampling years. Some of the annotated nifH genes from both 2017 and 2019 samples clustered with known references of halophilic Cyanobacteria (e.g., Halothece sp. PCC 7418). Even though halophilic Cyanobacteria from the clade “Halothece” often have optimal growth in hypersaline conditions, they may still grow suboptimally in conditions of relatively lower salinity71. Considering this, their presence in sediments with lower salinity suggests these Cyanobacteria can survive a wide range of salinity fluctuations in responses to precipitation anomalies on Kiritimati.
In conclusion, FAPROTAX, functional gene amplicon sequences, and previous metagenomic data altogether suggest that functional group relative abundance, diversity, and affiliated dominant taxa likely change in response to abrupt salinity changes. Also, some functional groups (e.g. Cyanobacteria) can survive under unfavorable conditions and flourish when the environment becomes more ideal in relation to ENSO events. Future metagenomic, metatranscriptomic, and isolation studies from different lakes in different years can provide additional details of how functional groups can change with salinity in these tropical sediments.
Conclusion
In a suite of lakes with varying salinities on Kiritimati, lake salinity decreased between 2014 and 2019 due to anomalously high precipitation rates during the 2015–2016 El Niño event. Both alpha and beta diversity metrics suggest that microbial community also changed significantly from 2014 to 2019, and that these changes were correlated with salinity. Phylum- and family-level results indicate that halophilic microorganisms are more abundant in 2014 samples, whereas halo-tolerant or mesohaline microorganisms are more abundant in 2019 samples. The FAPROTAX and functional gene amplicon sequencing results suggest that the salinity-induced microbial community changes altered the relative abundance of functional groups (chemoheterotrophs, phototrophs, nitrogen fixation, denitrification, sulfate reduction) as well as microbial diversity (alpha diversity and dominant taxa) within these groups. Although some differences in primer sets and sequencing techniques could potentially account for some of these variations, we conducted thorough adjustments to account for batch effects, and our results demonstrate that these effects had negligible or no impact on our conclusions. Based on the results of our analysis and observations, we conclude that the significant decrease in salinity altered microbial communities in Kiritimati lake sediments, with a change from a dominance of halophilic microbes to mesosaline and salinity-sensitive microbes. In addition, the functional groups also changed from aerobic chemoheterotroph dominance to photoautotroph dominance in response to salinity changes. Additionally, we discovered microbial communities and functional groups living at salinity levels that were outside of their optimal growth ranges, suggesting that microbial communities on Kiritimati Island may be able to adapt to large salinity fluctuations associated with interannual precipitation anomalies. The study demonstrates how abrupt changes in salinity, induced by climate variability, can impact microbial community metabolism in tropical near-marine lacustrine environments, offering insights on the factors influencing community diversity and stability in extreme ecosystems. Moreover, this study sheds light on microbial responses to future potential climatic and anthropogenic salinity fluctuations in lacustrine environments, as previous studies showed that artificial and anthropogenic salinization and desalinization processes can substantially shape microbial communities and functions in lake systems39,78,79,80.
Data availability
Raw sequence data from this study has been deposited in MG-RAST (2014 samples, project number MGP82583) and NCBI (2019 samples, BioProject ID PRJNA76940). Raw sequence data for 2019 samples will be made publicly available to download upon publication. Reviewers can use the link listed below to get access to the submission of 2019 samples. https://dataview.ncbi.nlm.nih.gov/object/PRJNA769403?reviewer=jc7aunebjhi97a77ajnc7c730f.
References
Oren, A. Thermodynamic limits to microbial life at high salt concentrations. Environ. Microbiol. 13, 1908–1923 (2011).
Oren, A. Microbial life at high salt concentrations: Phylogenetic and metabolic diversity. Saline Syst. 4, 1–13 (2008).
Yang, J., Ma, L., Jiang, H., Wu, G. & Dong, H. Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai–Tibetan Lakes. Sci. Rep. 6, 1–6 (2016).
Jiang, H. et al. Microbial response to salinity change in Lake Chaka, a hypersaline lake on Tibetan plateau. Environ. Microbiol. 9, 2603–2621 (2007).
Yang, J., Jiang, H., Wu, G. & Liu, W. Phylum-level archaeal distributions in the sediments of Chinese lakes with a large range of salinity. Geomicrobiol. J. 35, 404–410 (2018).
Jeppesen, E., Beklioğlu, M., Özkan, K. & Akyürek, Z. Salinization increase due to climate change will have substantial negative effects on inland waters: A call for multifaceted research at the local and global scale. Innovation 1, 66 (2020).
Jeppesen, E. et al. Ecological impacts of global warming and water abstraction on lakes and reservoirs due to changes in water level and related changes in salinity. Hydrobiologia 750, 201–227 (2015).
Han, L. et al. Relationship between lake salinity and the climatic gradient in northeastern China and its implications for studying climate change. Sci. Total Environ. 805, 150403 (2022).
Schmitt, S. et al. Salinity, microbe and carbonate mineral relationships in brackish and hypersaline lake sediments: A case study from the tropical Pacific coral atoll of Kiritimati. Depos. Rec. 5, 212–229 (2019).
Herlemann, D. P. et al. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5, 1571–1579 (2011).
Bouvier, T. C. & Del Giorgio, P. A. Compositional changes in free-living bacterial communities along a salinity gradient in two temperate estuaries. Limnol. Oceanogr. 47, 453–470 (2002).
Almeida-Dalmet, S., Sikaroodi, M., Gillevet, P. M., Litchfield, C. D. & Baxter, B. K. Temporal study of the microbial diversity of the North Arm of Great Salt Lake, Utah, U.S.. Microorganisms 3, 310–326 (2015).
Zhang, L. et al. Temporal and spatial variations in the bacterial community composition in Lake Bosten, a large, brackish lake in China. Sci. Rep. 10, 1–10 (2020).
Ghori, N. U. H., Wise, M. J. & Whiteley, A. S. Temporal microbial community dynamics within a unique acid saline lake. Front. Microbiol. 12, 1739 (2021).
Severin, I., Confurius-Guns, V. & Stal, L. J. Effect of salinity on nitrogenase activity and composition of the active diazotrophic community in intertidal microbial mats. Arch. Microbiol. 194, 483–491 (2012).
Herbst, D. B. Potential salinity limitations on nitrogen fixation in sediments from Mono Lake, California. Int. J. Salt Lake Res. 7, 261–274 (1998).
Hou, L., Wang, R., Yin, G., Liu, M. & Zheng, Y. Nitrogen fixation in the intertidal sediments of the Yangtze estuary: Occurrence and environmental implications. J. Geophys. Res. Biogeosci. 123, 936–944 (2018).
Giblin, A. E., Weston, N. B., Banta, G. T., Tucker, J. & Hopkinson, C. S. The effects of salinity on nitrogen losses from an oligohaline estuarine sediment. Estuaries Coasts 33, 1054–1068 (2010).
Marchant, H. K., Lavik, G., Holtappels, M. & Kuypers, M. M. M. The fate of nitrate in intertidal permeable sediments. PLoS ONE 9, e104517 (2014).
Mobberley, J. M. et al. Organismal and spatial partitioning of energy and macronutrient transformations within a hypersaline mat. FEMS Microbiol. Ecol. 93, 1–13 (2017).
Saenger, C., Miller, M., Smittenberg, R. H. & Sachs, J. P. A physico-chemical survey of inland lakes and saline ponds: Christmas Island (Kiritimati) and Washington (Teraina) Islands, Republic of Kiribati. Saline Syst. 2, 1–15 (2006).
Higley, M. C. & Conroy, J. L. The hydrological response of surface water to recent climate variability: A remote sensing case study from the central tropical Pacific. Hydrol. Process. 33, 2227–2239 (2019).
Falkland, A. C. & Woodroffe, C. D. Geology and hydrogeology of Tarawa and Christmas Island, Kiribati. Dev. Sedimentol. 54, 577–610 (2004).
Schoonmaker, J., Tribble, G., Smith, S. & MacKenzie, F. Geochemistry of saline ponds, Kiritimati. In Proceedings of the Fifth International Coral Reef Congress, Tahiti vol. 3 439–444 (1985).
Gasparin, F. & Roemmich, D. The strong freshwater anomaly during the onset of the 2015/2016 El Niño. Geophys. Res. Lett. 43, 6452–6460 (2016).
McDougall, T. & Barker, P. M. Getting started with TEOS-10 and the Gibbs Seawater (GSW)Oceanographic Toolbox. SCOR/IAPSO WG 127, 1–28 (2011).
Olesik, J. W. Elemental analysis using icp-oes and icp/ms. Anal. Chem. 63, 12A-21A (1991).
Weiss, J. Handbook of Ion Chromatography, 3 Volume Set. vol. 1 (Wiley, 2016).
Berden, G., Peeters, R. & Meijer, G. Cavity ring-down spectroscopy: Experimental schemes and applications. Int. Rev. Phys. Chem. 19, 565–607 (2000).
Schloss, P. D. et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537 (2009).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Oksanen, J. et al. The vegan package. Community Ecol. Packag. (2008).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139 (2010).
Gaujoux, R. & Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinf. 11, 1–9 (2010).
Louca, S., Parfrey, L. W. & Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277 (2016).
Davis, J. S. Biological communities of a nutrient enriched salina. Aquat. Bot. 4, 23–42 (1978).
Jiang, H. et al. Microbial diversity in water and sediment of Lake Chaka, an athalassohaline lake in northwestern China. Appl. Environ. Microbiol. 72, 3832–3845 (2006).
Wu, Q. L., Zwart, G., Schauer, M., Kamst-Van Agterveld, M. P. & Hahn, M. W. Bacterioplankton community composition along a salinity gradient of sixteen high-mountain lakes located on the Tibetan Plateau, China. Appl. Environ. Microbiol. 72, 5478–5485 (2006).
Huang, J. et al. Microbial responses to simulated salinization and desalinization in the sediments of the Qinghai-Tibetan lakes. Front. Microbiol. 66, 1772 (2020).
Oren, A. The bioenergetic basis for the decrease in metabolic diversity at increasing salt concentrations: implications for the functioning of salt lake ecosystems. Hydrobiology 466, 61–72 (2001).
Oren, A. Molecular ecology of extremely halophilic Archaea and bacteria. FEMS Microbiol. Ecol. 39, 1–7 (2002).
Siglioccolo, A., Paiardini, A., Piscitelli, M. & Pascarella, S. Structural adaptation of extreme halophilic proteins through decrease of conserved hydrophobic contact surface. BMC Struct. Biol. 11, 1–12 (2011).
de la Haba, R. R., Arahal, D. R., Sánchez-Porro, C. & Ventosa, A. The family Halomonadaceae. in The Prokaryotes: Gammaproteobacteria vol. 9783642389 325–360 (Springer, 2014).
Ivanova, E. P. & Mikhailov, V. V. A new family, Alteromonadaceae fam. nov., including marine proteobacteria of the genera Alteromonas, Pseudoalteromonas, Idiomarina, and Colwellia. Microbiology 70, 10–17 (2001).
Dupont, C. L. et al. Functional tradeoffs underpin salinity-driven divergence in microbial community composition. PLoS ONE 9, e89549 (2014).
Canfora, L. et al. Salinity and bacterial diversity: To what extent does the concentration of salt affect the bacterial community in a saline soil?. PLoS ONE 9, e106662 (2014).
Abraham, W.-R. & Rohde, M. The family Hyphomonadaceae. The Prokaryotes: Alphaproteobacteria and Betaproteobacteria vol. 9783642301 (Springer, 2014).
Imhoff, J. F. & Wiese, J. The order Kiloniellales. in The Prokaryotes: Alphaproteobacteria and Betaproteobacteria vol. 9783642301 301–306 (Springer, 2014).
Pujalte, M. J., Lucena, T., Ruvira, M. A., Arahal, D. R. & Macián, M. C. The Family Rhodobacteraceae. in The Prokaryotes: Alphaproteobacteria and Betaproteobacteria vol. 9783642301 439–512 (Springer, 2014).
Han, M.-X. et al. Lunatibacter salilacus gen. nov., sp. nov., a member of the family Cyclobacteriaceae, isolated from a saline and alkaline lake sediment. Int. J. Syst. Evol. Microbiol. 71, 004621 (2021).
Irshad, A., Ahmad, I. & Kim, S. B. Culturable diversity of halophilic bacteria in foreshore soils. Brazilian J. Microbiol. 45, 563 (2014).
Oren, A. The order Halanaerobiales, and the families Halanaerobiaceae and Halobacteroidaceae. in The Prokaryotes: Firmicutes and Tenericutes 153–177 (Springer, 2014). https://doi.org/10.1007/978-3-642-30120-9_218.
Echigo, A. et al. Endospores of halophilic bacteria of the family Bacillaceae isolated from non-saline Japanese soil may be transported by Kosa event (Asian dust storm). Saline Syst. 1, 1–13 (2005).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 108, 4516–4522 (2011).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1–e1 (2013).
Oren, A. The ecology of the extremely halophilic archaea. FEMS Microbiol. Rev. 13, 415–439 (1994).
Purdy, K. J. et al. Isolation of haloarchaea that grow at low salinities. Environ. Microbiol. 6, 591–595 (2004).
Vauclare, P., Natali, F., Kleman, J. P., Zaccai, G. & Franzetti, B. Surviving salt fluctuations: stress and recovery in Halobacterium salinarum, an extreme halophilic Archaeon. Sci. Rep. 10, 1–10 (2020).
Takai, K. & Horikoshi, K. Rapid detection and quantification of members of the archaeal community by quantitative PCR using fluorogenic probes. Appl. Environ. Microbiol. 66, 5066 (2000).
Dawson, K. S., Freeman, K. H. & Macalady, J. L. Molecular characterization of core lipids from halophilic archaea grown under different salinity conditions. Org. Geochem. 48, 1–8 (2012).
Genderjahn, S., Alawi, M., Mangelsdorf, K., Horn, F. & Wagner, D. Desiccation-and saline-tolerant bacteria and archaea in kalahari pan sediments. Front. Microbiol. 9, 2082 (2018).
Kulp, T. R. et al. Effects of imposed salinity gradients on dissimilatory arsenate reduction, sulfate reduction, and other microbial processes in sediments from two California soda lakes. Appl. Environ. Microbiol. 73, 5130–5137 (2007).
Inoue, K., Itoh, T., Ohkuma, M. & Kogure, K. Halomarina oriensis gen. nov., sp. nov., a halophilic archaeon isolated from a seawater aquarium. Int. J. Syst. Evol. Microbiol. 61, 942–946 (2011).
Diloreto, Z. A., Garg, S., Bontognali, T. R. R. & Dittrich, M. Modern dolomite formation caused by seasonal cycling of oxygenic phototrophs and anoxygenic phototrophs in a hypersaline sabkha. Sci. Rep. 11, 1–13 (2021).
Berner, C., Bertos-Fortis, M., Pinhassi, J. & Legrand, C. Response of microbial communities to changing climate conditions during summer cyanobacterial blooms in the Baltic Sea. Front. Microbiol. 66, 1562 (2018).
Teeling, H. et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611 (2012).
Dyksma, S., Lenk, S., Sawicka, J. E. & Mußmann, M. Uncultured Gammaproteobacteria and Desulfobacteraceae account for major acetate assimilation in a coastal marine sediment. Front. Microbiol. 9, 3124 (2018).
Kumar, A., Ng, D. H. P., Wu, Y. & Cao, B. Microbial community composition and putative biogeochemical functions in the sediment and water of tropical granite quarry lakes. Microb. Ecol. 77, 1–11 (2019).
Wyman, D. A., Conroy, J. L., Osburn, M. R. & Atwood, A. R. Coeval drying across the central tropical Pacific over the last millennium. Paleoceanogr. Paleoclimatology 36, e2021PA004311 (2021).
Thabet, O. B. D. et al. Desulfovibrio marinus sp. nov., a moderately halophilic sulfate-reducing bacterium isolated from marine sediments in Tunisia. Int. J. Syst. Evol. Microbiol. 57, 2167–2170 (2007).
Garcia-Pichel, F., Nübel, U. & Muyzer, G. The phylogeny of unicellular, extremely halotolerant cyanobacteria. Arch. Microbiol. 169, 469–482 (1998).
Li, Z. & Brand, J. Leptolyngbya nodulosa sp. nov. (Oscillatoriaceae), a subtropical marine cyanobacterium that produces a unique multicellular structure. Phycologia 46, 396–401 (2007).
Trüper, H. G. Ectothiorhodospira mobilis Pelsh, a photosynthetic sulfur bacterium depositing sulfur outside the cells. J. Bacteriol. 95, 1910–1920 (1968).
Montoya, L., Lozada-Chávez, I., Amils, R., Rodriguez, N. & Marín, I. The sulfate-rich and extreme saline sediment of the ephemeral Tirez Lagoon: A biotope for acetoclastic sulfate-reducing bacteria and hydrogenotrophic methanogenic archaea. Int. J. Microbiol. 2011, 753758 (2011).
Chen, M., Conroy, J. L., Sanford, R. A., Chee-Sanford, J. C. & Connor, L. M. Interpreting lacustrine bulk sediment δ15N values using metagenomics in a tropical hypersaline lake system. J. Paleolimnol. 65, 151–168 (2020).
Imhoff, J. F. & Mikrobiologie, M. The Family Chlorobiaceae. in The Prokaryotes: Other Major Lineages of Bacteria and The Archaea vol. 9783642389 501–514 (Springer, 2014).
Watanabe, M., Higashioka, Y., Kojima, H. & Fukui, M. Desulfosarcina widdelii sp. Nov. and Desulfosarcina alkanivorans sp. nov., hydrocarbon-degrading sulfate-reducing bacteria isolated from marine sediment and emended description of the genus Desulfosarcina. Int. J. Syst. Evol. Microbiol. 67, 2994–2997 (2017).
Mo, Y. et al. Low shifts in salinity determined assembly processes and network stability of microeukaryotic plankton communities in a subtropical urban reservoir. Microbiome 9, 128 (2021).
Corsi, S. R., Graczyk, D. J., Geis, S. W., Booth, N. L. & Richards, K. D. A fresh look at road salt: Aquatic toxicity and water-quality impacts on local, regional, and national scales. Environ. Sci. Technol. 44, 7376–7382 (2010).
Selak, L., Marković, T., Pjevac, P. & Orlić, S. Microbial marker for seawater intrusion in a coastal Mediterranean shallow Lake, Lake Vrana, Croatia. Sci. Total Environ. 849, 157859 (2022).
Acknowledgements
This research was funded by ACS-PRF 57417-DNI2 and NSF-EAR 1602590 to JLC. The authors thank the Environmental Ministry of the Republic of Kiribati for the research permit to complete this work. We thank Melinda Higley, Christina Karamperidou, Nicole Murray, and Nooreen Meghani for field assistance, Mark Band and Ted Flynn for the technical support, and Bruce Fouke, Thomas Johnson, and Chris Field for valuable comments and advice. We thank the UIUC Roy J. Carver Biotechnology Center, specifically the High-Throughput Sequencing and Genotyping Unit, for DNA sample analysis.
Author information
Authors and Affiliations
Contributions
M.C. and J.L.C. designed the study, M.C., D.A.W–F., and R.A.S. performed data analysis. M.C. and J.L.C. wrote the first draft of the manuscript. All authors contributed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, M., Conroy, J.L., Sanford, R.A. et al. Tropical lacustrine sediment microbial community response to an extreme El Niño event. Sci Rep 13, 6868 (2023). https://doi.org/10.1038/s41598-023-33280-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-33280-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
