
Abstract
This work reports a thermo-kinetic study on unimolecular thermal decomposition of some ethoxyquinolines and ethoxyisoquinolines derivatives (1-ethoxyisoquinoline (1-EisoQ), 2-ethoxyquinoline (2-EQ), 3-ethoxyquinoline (3-EQ), 3-ethoxyisoquinoline (3-EisoQ), 4-ethoxyquinoline (4-EQ), 4-ethoxyisoquinoline (4-EisoQ), 5-ethoxyquinoline (5-EQ), 5-ethoxyisoquinoline (5-EisoQ), 8-ethoxyquinoline (8-EQ) and 8-ethoxyisoquinoline (8-EisoQ)) using density functional theory DFT (BMK, MPW1B95, M06-2X) and ab initio complete basis set-quadratic Becke3 (CBS-QB3) calculations. In the course of the decomposition of the investigated systems, ethylene is eliminated with the production of either keto or enol tautomer. The six-membered transition state structure encountered in the path of keto formation is much lower in energy than the four-membered transition state required to give enol form. Rate constants and activation energies for the decomposition of 1-EisoQ, 2-EQ, 3-EQ, 3-EisoQ, 4-EQ, 4-EisoQ, 5-EQ, 5-EisoQ, 8-EQ, and 8-EisoQ have been estimated at different temperatures and pressures using conventional transition state theory combined with Eckart tunneling and the unimolecular statistical Rice–Ramsperger–Kassel–Marcus theories. The tunneling correction is significant at temperatures up to 1000 K. Rate constants results reveal that ethylene elimination and keto production are favored kinetically and thermodynamically over the whole temperature range of 400–1200 K and the rates of the processes under study increase with the rising of pressure up to 1 atm.
Similar content being viewed by others

Introduction
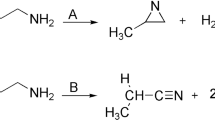
Quinoline and its derivatives, as important naturally occurring compounds, are found in coal tar as well as bone oil and have biological and pharmaceutical effects1,2,3,4,5,6,7,8,9, including antimalarial, antineoplastic, anticonvulsant, antibacterial, antifungal, anticancer, anti-inflammatory, and analgesic activity1,2,3,4,5,6. For the last two decades, nitrogen- and oxygen-containing heterocyclic compounds have been attractive in biology due to their pharmaceutical action, mainly attributable to their ability to make hydrogen bonds. Quinoline research is now one of the most prominent areas in organic, inorganic, pharmaceutical, and theoretical chemistry, as well as dye manufacturing. Tautomerism is a fundamental notion in organic chemistry and an intriguing phenomenon because it is linked to numerous essential chemical and biological processes10.
The study of how tautomerism affects the chemical, biological, and pharmacological properties of heterocyclic compounds is of great interest to many researchers, particularly medicinal chemists, as it may be related to the pharmacological properties of these compounds. Experimentally and theoretically, the tautomeric equilibrium of heterocyclic compounds has been investigated11,12,13, and a detailed analysis of the changes in structural, geometric, and energetic parameters caused by the transfer of H atoms can help us understand the different properties of tautomers. Understanding the relative stabilities of tautomeric forms of heterocycles and how they convert from one form to another is important in the field of structural chemistry. Quinolines and isoquinolines have been extensively investigated because they are relevant to physics7, chemistry8, and medicine9. The thermal decomposition of these materials is essential to understand their behavior and stability in different environments14,15,16,17,18,19,20,21,22. Similar to esters23,24,25,26, it was reported that thermolysis of alkoxy benzene and heteroaromatics produces olefins and the corresponding keto or enol form with activation energies depend on the structure of the reactants14,15,16,17,18,19,20,21,22. These gas phase degradation reactions are unimolecular, homogeneous, and pass over six-membered ring transition states14,15,16,17,18,19,20,21,22. In the course of these pyrolytic reactions, different tautomers can be formed. However, the formation of the keto tautomer needs less energy than that required for enol because the former passes over a six-membered ring transition state whereas the latter is formed through a four-membered transition state. Therefore, the keto form appears as a dominant product. If the energy barrier to producing enol from its keto tautomer is low, a state of equilibrium between the two forms might be established.
Experimental14,16 and theoretical22 studies have been conducted on the formation of hydroxyquinoline and quinolone by the removal of ethylene from ethoxyquinoline and ethoxyisoquinoline. Al-Awadi and colleagues14 looked at the rates of thermal ethylene removal from substances such as 2-ethoxyquinoline, 1- and 3-ethoxyisoquinoline, and 1-ethoxythiazole. They also investigated the rates of gas-phase pyrolytic reactions of 2-pyridine and 8-quinoline sulfonic acid esters16. Each of the pyridine esters is consistently more reactive than the quinoline ester. This follows from the fact that C-2 of the pyridine will receive a greater electron-withdrawing effect from the nitrogen atom (which will make it the most electron-deficient carbon in the ring), and this indirect electron withdrawal effect of the nitrogen atom in the 2-pyridine esters should facilitate C-O bond cleavage, while for the 8-quinoline esters, this is not the case16.
Gas phase pyrolysis has been used as a means of organic synthesis18,27,28,29,30,31,32,33. For example, flash vacuum pyrolysis of N-alkoxyphthalimides at 673–773 K and 0.02 Torr yielded the corresponding substituted aldehydes and phthalimide27. Gas-phase pyrolysis of N-(1H-benzimidazol-2-yl)-N’-arylidenehydrazines produced arylnitriles, 2-aminobenzimid- azole, 2,4,5-triphenylimidazole, 1,3-diphenyl-8H-2,3a,8-triazacyclopenta [a]indene, and 5,11-diphenyl-6H,12H-dibenzimidazo[1,2-a];1’,2’-d]pyrazine28. Also, the thermal decomposition of 1-(pyrazol-4-yl)-1H-benzotriazole derivatives gave indole and its condensed derivatives18. Furthermore, thermolysis of ethyl 2-amino-5-arylazo-6-phenylnicotinates and 5- arylazonicotinonitriles helped in synthesizing some pyridine derivatives29. El-Demerdash et al22. investigated thermal decompositions of 1-ethoxyisoquinoline (1-EisoQ), 2-ethoxyquinoline (2-EQ), and 3-ethoxyisoquinoline (3-EisoQ) to create ethylene and various tautomers in the gas phase and ethanol solution at the BMK/6–31+G(d,p) and MP2/6–311++G(2d,2p) levels of theory. The thermodynamic and kinetic stability of 2-EQ and 1-EisoQ breakdown to ethylene and keto forms is greater than that of the equivalent enols. In the gas phase with ethanol, however, the hydroxy form of 3-EisoQ is more stable than the keto tautomer22. Density functional theory (DFT) and ab initio calculations become useful techniques for obtaining information about the structure, relative stability, and other aspects of tautomers, in the sense that quantum chemical calculations can directly examine the physical properties of tautomers.
In the present work, thermochemistry and kinetics of the thermal decomposition of 1-ethoxyisoquinoline (1-EisoQ), 2-ethoxyquinoline (2-EQ), 3-ethoxyquinoline (3-EQ), 3-ethoxyisoquinoline (3-EisoQ), 4-ethoxyquinoline (4-EQ), 4-ethoxyisoquinoline (4-EisoQ), 5-ethoxyquinoline (5-EQ), 5-ethoxyisoquinoline (5-EisoQ), 8-ethoxyquinoline (8-EQ) and 8-ethoxyisoquinoline (8-EisoQ) to produce ethylene and different tautomers were studied in the gas phase at 400–1200 K and 10−6 to 10 atm using the density functional methods BMK/6–31+G(d,p), MPW1B95/6–311++G(2d,2p), M06-2X/cc-pvtz and ab initio CBS-QB3 levels.
Computational details
The density functional theory (DFT) BMK34 (Boese and Martin) method in conjunction with the 6–31+G(d,p) basis set was employed to optimize reactants, products, and transition states (all optimized structures are given in Table S1 in the supplementary file(SI)). ChemCraft software V1.835 was used to analyze vibrational frequencies that were scaled with a factor of 0.9536. Based on Hessian matrix analysis, all minima are characterized by having no imaginary frequencies while each transition state comprises only one negative eigenvalue. The located transition states were further verified through minimum energy path (MEP) analysis using intrinsic reaction coordinate (IRC) calculations at the BMK/6–31+G (d,p) level of theory in mass-weighted Cartesian coordinates37,38,39. The IRC analysis demonstrated that the transition states connect the reactants with their respective products (Table S2). In addition, to obtain more accurate results, single-point energy calculations were performed using MPW1B9540,41,42,43 and M06-2X44,45 methods with 6–311++G(2d,2p) and cc-pVTZ basis sets, respectively. MPW1B95 functional with 31% HF exchange–correlation has been reported for excellent thermo-kinetics simulations of unimolecular decomposition reactions40,41,42,43 and broad thermochemistry applications with reliable performance for hydrogen bonding and weak interaction simulations40,41,42,43. The hybrid meta-generalized gradient Minnesota functional M06-2X has a 54% HF exchange–correlation and was developed in 2006 by Zhao et al.44 to give accurate kinetic data. The hindered rotor (HR) approximation was used to treat the internal rotation of the inspected structures to obtain accurate vibrational frequencies. For locating transition states, the relaxed potential energy scans were used to obtain the lowest TSs structures achievable.
Energies were also refined utilizing the multistep CBS-QB346,47,48 level at the BMK geometries for accurate chemical kinetic modeling. Low-level calculations on big basis sets, mid-sized sets for second-order correlation corrections, and small basis sets for high-level correlation corrections are all part of the CBS-QB3 composite approach46,47,48. The composite CBS-QB3 method consists of five steps begins with a geometry optimization and frequency calculations at the B3LYP/6-311G(d, p) level followed by single point calculations at CCSD(T)/6–31+G (d), MP4SDQ/6–31+G (d, p), and MP2/6–311+G (2df, 2p) with CBS extrapolation. In the current study, the geometry optimization and frequency calculations steps at the B3LYP/6-311G(d, p) level were canceled and replaced by BMK/6–31+G(d,p) level geometries by using the keyword IOP(1/7 = 1,000,000) order. T1 diagnostic calculations of the CBS-QB346,47,48 approach were also employed for TSs and the generated radicals to check the existence or absence of multireference character in the estimated wavefunctions of different species.
The hindered rotor (HR) technique was utilized to tackle torsional vibration modes in 1-EisoQ, 2-EQ, 3-EQ, 3-EisoQ, 4-EQ, 4-EisoQ, 5-EQ, 5-EisoQ, 8-EQ, 8-EisoQ, and transition states. However, due to issues with the number of degrees of freedom, we encountered termination faults for several transition states. Rate constants for the successfully calculated TSs with a hampered rotor were determined and compared to those produced using a harmonic oscillator (HO). Very minimal differences were found between them, providing confidence in our conclusions derived utilizing the HO technique. All calculations were conducted with the Gaussian 16W program49.
The unimolecular thermal decomposition rate constant of 1-EisoQ, 2-EQ, 3-EQ, 3-EisoQ, 4-EQ, 4-EisoQ, 5-EQ, 5-EisoQ, 8-EQ, and 8-EisoQ were calculated employing the conventional transition state theory (TST) (Eq. 1)50,51,52,53.
For TST calculations, the dividing surface is located at s = 0.0 and the rate constant reads
where χ(T) is the tunneling correction, σ is the reaction path degeneracy, kB is the Boltzmann constant, h is the Plank’s constant, T is the temperature, QR(T) and QTS(T) are the reactant and transition state partition functions.
One-dimensional (1D) tunneling effects Eckart (Eck)54 and the fall-off regime by unimolecular Rice–Ramsperger–Kassel–Marcus (RRKM) theory at lower pressures55,56,57,58 are included in TST calculations. Both RRKM and TST theories are implemented in the Kinetic and Statistical Thermodynamical Package (KiSThelP)59.
Tunneling can play an important role in these processes because an H atom shift is involved. The tunneling correction is addressed by the Eckart tunneling adjustment in the rate equation. Furthermore, the tunneling correction χ(T) was included to account for tunneling along the reaction coordinate, which was estimated using TST and corrected using Eckart tunneling correction factors. Tunneling adjustments are used to correct TST rate coefficients for the asymmetric Eckart's 1D potential energy barrier by integrating the probability, p(E), of transmission across the associated 1D barrier at energy E and the Boltzmann distribution of energies:
where ΔHf ≠,0K represents the zero-point corrected energy barriers in the forward direction.
Using the RRKM method, the microcanonical rate coefficient k(E) is calculated for energy-dependent systems:
where G(E) is the total number of states of the transition state with energy less than or equal to E, and N(E) is the density of states of the dissociating reactant species. The thermal rate coefficient reads
where Q2 is the partition function of the active degrees of freedom of the reactant, Q‡1, and Q1 are the partition functions for adiabatic rotations of the transition state and the reactant, respectively, and E0 is the zero-point corrected threshold energy.
Moreover, the strong collision approximation was applied assuming the possible collisions deactivate with ω = βcZLJ[M] being the effective collision frequency, where βc represents the collisional efficiency, ZLJ represents the Lennard–Jones collision frequency, and [M] is the total gas concentration. A value of 0.2 was retained for βc. Using the Lennard–Jones parameter ε/kB, where ε is the energy depth of the Lennard–Jones potential and σL represents a dimensionless scale for the molecular radius, we calculated the collision frequencies (ZLJ). The Lennard–Jones potential parameters are σL = 5.476 Å and ε/kB = 367.82 K for EQ whereas σL = 3.465 Å and ε/kB = 113.5 K60 for Argon (Ar) as a diluent gas.
The accurate classical method has been used to predict the rate constants of simple bond cleavage (barrierless) reactions which have recently been used and mentioned in many advanced computational studies25,45,61,62,63,64.
Results and discussion
Figures 1 and 2 show the optimized structures of different conformers and their corresponding relative energy at different levels of theories, respectively. According to the literature14,16,65,66 and the relaxed potential scan at BMK/6–31+G(d,p) level, all inspected molecules have two different stable conformers A and B according to the rotation of ethoxy moiety group. In the case of structure A, the ethoxy group adopts trans structure, while structure B shows gauch form. Inspections of relative energies diagrams (Fig. 2) reveal that the A forms are more stable than the B forms at different levels of theories. Therefore, unless otherwise specified, the current study will be based on conformer A.


Optimized structures of different conformers (A, B) at the BMK/6–31+G(d,p) level of theory level.

Relative stabilities of different conformers (kcal mol−1, energies calculated relative to conformer A) at BMK, MPW1B95, M06-2X, and CBS-QB3 levels.
The unimolecular decomposition reaction of ethoxyquinolines and ethoxyisoquinolines takes place via the breaking of the bond connecting the oxygen atom with the ethyl group along with H atom transfer from the CH3 group (1,5- or 1,3-H atom shift) to either nitrogen or oxygen atoms giving rise to keto or enol tautomers with ethylene elimination. The optimized structures of different ethoxyisoquinolines, ethoxyquinolines (1-EisoQ, 2-EQ, 3-EQ, 3-EisoQ, 4-EQ, 4-EisoQ, 5-EQ, 5-EisoQ, 8-EQ, and 8-EisoQ) and their decomposition products are shown in Fig. 3, while Fig. 4. shows their encounter transition states. The investigated complex bond fission reactions (reactions with barriers, R1-R13) and simple bond fission reactions (barrierless reactions, R14-R28) decomposition reactions are summarized as follows:
-
(a)
Complex bond fission reactions (reactions with barriers)


Optimized structures of ethoxyisoquinolines, ethoxyquinolines, and their corresponding decomposition products calculated at BMK/6–31+G(d,p) level. Bond lengths and angles are given in Ångström and degree, respectively.

Optimized transition state structures for the thermal decomposition reactions of ethoxyisoquinolines, ethoxyquinolines calculated at BMK/6–31+G(d,p) level. Bond lengths and angles are given in Ångström and degree, respectively.
1-EisoQ → 1-isoQenol + C2H4 | (R1) |
1-EisoQ → 1-isoQketo + C2H4 | (R2) |
2-EQ → 2-Qenol + C2H4 | (R3) |
2-EQ → 2-Qketo + C2H4 | (R4) |
3-EisoQ → 3-isoQenol + C2H4 | (R5) |
3-EisoQ → 3-isoQketo + C2H4 | (R6) |
3-EQ → 3-Qenol + C2H4 | (R7) |
4-EisoQ → 4-isoQenol + C2H4 | (R8) |
4-EQ → 4-Qenol + C2H4 | (R9) |
5-EisoQ → 5-isoQenol + C2H4 | (R10) |
5-EQ → 5-Qenol + C2H4 | (R11) |
8-EisoQ → 8-isoQenol + C2H4 | (R12) |
8-EQ → 8-Qenol + C2H4 | (R13) |
-
(b)
Simple bond fission reactions (barrierless reactions)
1-EisoQ → 1-iso•OQ + •C2H5 | (R14) |
1-EisoQ → 1-•CH2OisoQ + •CH3 | (R15) |
1-EisoQ → 1-CH3•CHOisoQ + •H | (R16) |
1-EisoQ → 1-•CH2CH2OisoQ + •H | (R17) |
1-EisoQ → 1-isoQ• + •OC2H5 | (R18) |
2-EQ → 2-•OQ + •C2H5 | (R19) |
2-EQ → 2-•CH2OQ + •CH3 | (R20) |
2-EQ → 2-CH3•CHOQ + •H | (R21) |
2-EQ → 2-•CH2CH2OQ + •H | (R22) |
2-EQ → 2-Q• + •OC2H5 | (R23) |
3-EisoQ → 3-•OisoQ + •C2H5 | (R24) |
3-EisoQ → 3-•CH2OisoQ + •CH3 | (R25) |
3-EisoQ → 3-CH3•CHOisoQ + •H | (R26) |
3-EisoQ → 3-•CH2CH2OisoQ + •H | (R27) |
3-EisoQ → 3-isoQ• + •OC2H5 | (R28) |
The reactions R1–R13 pass through transition states symbolized as TS1enol, TS2keto, TS3enol, TS4keto, TS5enol, TS6keto, TS7enol, TS8enol, TS9enol, TS10enol, TS11enol, TS12enol, and TS13enol, respectively.
Enthalpies of formation
Thermochemical data are critical for comprehending and estimating the stability, reaction pathways, and kinetics of a specific chemical system59,67,68. The computed structures, moments of inertia, vibration frequencies, symmetry, electron degeneracy, and known mass of each molecule are used to calculate entropy and heat capacity contributions as a function of temperature59,67,68. The computational results of fundamental quantities including entropies (S°298), enthalpies of formation (ΔHf°298), and heat capacities (Cp298) are presented in Table 1 for the major species which are depicted in Fig. S1 (in the SI file) during combustion processes. The enthalpies of formation for some selected oxygenates (1-butanol, ethyl propanoate, methyl formate, formic, acetic, and propanoic acids) have been calculated by El-Nahas et al.43. El-Nahas et al.43 found that BMK, MPW195 functionals, and CBS-QB3 have root mean square errors (RMSE) in enthalpies of formation no larger than 4 kcal/mol when compared to the experiment. At 298 K and 1 atm, the S°298 and Cp298 values for each molecule were computed. Even though Cp values are independent of the ab initio procedure, they are generated from the statistical mechanics treatment59,67,68. Our results demonstrated excellent agreement with earlier experimental14,64 and computational66 results, giving confidence in the unknown thermochemical characteristics of the radicals generated during 1-EisoQ, 2-EQ, 3-EQ, 3-EisoQ, 4-EQ, 4-EisoQ, 5-EQ, 5-EisoQ, 8-EQ, and 8-EisoQ decomposition.
Energetics
Potential energies profiles for the unimolecular decomposition of 1-EisoQ, 2-EQ, 3-EQ, 3-EisoQ, 4-EQ, 4-EisoQ, 5-EQ, 5-EisoQ, 8-EQ, and 8-EisoQ at CBS-QB3//BMK/6–31+G(d,p) are illustrated in Figs. 5 and 6. Gibbs free energies for the investigated reactions calculated at the same level of theory are collected in Table 2. Keto formation passes through six-membered ring transition states while producing the corresponding enol tautomer is accomplished via a four-membered ring transition state. Therefore, the former reaction requires less energy than the latter as a result of the stability of the six-membered ring relative to the four-membered transition state. For example, using CBS-QB3 energies, the production of keto and enol tautomers from 1-EisoQ needs free energy barriers of 47.2 and 65.5 kcal/mol, respectively with reaction enthalpy change of 11.5 and 15.9 kcal/mol. Therefore, the decomposition of 1-EisoQ to yield the keto form is thermodynamically and kinetically more preferable than the formation of the enol tautomer. Similarly, the formation of 2-quinolone (2-Qketo) is kinetically and thermodynamically more favorable compared to its enol (2-Qenol) by 17.7 for barrier energy and reaction energy difference of 3.5 kcal/mol. From Table 2, the decomposition of 4-EQ to enol form and ethylene is the least endothermic channel and most thermodynamically favored reaction with a higher degree of spontaneity ΔG = − 28.3 kcal/mol, where the hydrogen bond between the H atom of the hydroxyl group with the nitrogen atom plays a significant role. This is clear when we compare the endothermicity and spontaneity of forming 8-hydroyisoquinoline with that of 8-hydroxyquinoline.

Potential energy profile for the dominated complex fission reactions (R1–R13) during the unimolecular decomposition of 1-EisoQ, 2-EQ, 3-EisoQ, (∆E0, ∆E0‡, kcal/mol) at CBS-QB3//BMK/6–31+G(d,p) level.

Potential energy profile for the dominated barrierless reactions (R14–R28) during the unimolecular decomposition of 1-EisoQ, 2-EQ, and 3-EisoQ (∆E0, ∆E0‡, kcal/mol) at CBS-QB3//BMK/6–31+G(d,p) level.
When enols are formed from ethoxyquinoline or ethoxyisoquinoline where the ethoxy group is not adjacent to the nitrogen atom (3-EQ, 4-EQ, 4-EisoQ, 5-EQ, 5-EisoQ, 8-EQ, and 8-EisoQ), the energy barrier for 1,3-H atom shift is lowered by 3–5 kcal/mol. The same finding has been noticed in the H-atom shift in a series of some six-membered carbo- and heterocyclic compounds69.
-
(a)
Complex bond fission reactions
-
(b)
Simple bond fission reactions
Rate constants
Rate constants calculations (k1–13) for all complex fission reactions were calculated using the conventional transition state theory (TST) combined with Eckart tunneling (TST/Eck) as well as the statistical Rice–Ramsperger–Kassel–Marcus (RRKM) theories over a temperature range of 400–1200 K at pressure 1 atm at CBS-QB3//BMK/6–31+G(d,p) level. The employed temperature range was selected for two reasons: (i) it covers the experimental kinetic measurements; (ii) the rates at this temperature range allow comparisons among the present systems and other related compounds.
Detailed rate constants calculated from TST and RRKM theories along with tunneling corrections (Eck) for the H-atom transfer reactions (R1–R13) at CBS-QB3//BMK/6–31+G(d,p) level in the temperature range 400–1200 K and 1 atm are listed in Tables S3–S15, while the calculated rate coefficients for simple bond fission reactions (R14–R18) were collected in SI file. At a low temperature of 400 K, Eck tunneling gives higher contributions of 5.13, 5.92, 5.64, 7.56, 8.11, 6.27, 9.50, 9.57, 8.40 for R1, R3, R5, R7, R8, R9, R10, R11, and R12, respectively, than other reactions. Variation of Eckart tunneling correction against temperature change 400–1200 K was drawn in Fig. 7.

Variation of Eckart tunneling factor against temperature.
In general, the contribution of tunneling is marginal for all reactions at temperatures ≥ 1000 K. The tunneling contribution decreases with the rising of temperature until reaching 1.2 at T > 1000 K. From Tables S16 and S17, Investigations of different simple bonds fissions reactions (R14–R28) show that only R15, R20, and R25 reactions that encounter CH3 removal can be considered important above 1100 K, while the rest of reactions can be ignored.
Plots of TST/Eck and RRKM rate constants (k1–k13) against temperature as depicted in Figs. 8 and 9 illustrate an Arrhenius behavior. Inspection of these Figures reveals positive temperature dependence with rate coefficients (frequency factor (A) and activation energy (Ea)) fit with two-parameter equations (k = A exp(−Ea/RT)).

TST/Eck rate constants for unimolecular decomposition reactions of the investigated ethoxyquinolines and ethoxyisoquinolines (R1–R13) at (T = 400–1200 K, p = 1 atm) calculated at CBS-QB3//BMK/6–31+G(d,p) level.

RRKM rate constants for unimolecular decomposition reactions of the investigated ethoxyquinolines and ethoxyisoquinolines (R1–R13) at (T = 400–1200 K, p = 1 atm) calculated at CBS-QB3//BMK/6–31+G(d,p) level.
The results obtained using MPW1B95 and M06-2X levels were compared against the experiment and the accurate cost-effective CBS-QB3 ab initio multilevel computational method48,49,50. Figures 10, 11 and Table S18 show a comparison of Arrhenius plots for the main dominant pathways of reaction decomposition (R2, R4, R6) at 650 K and 1 atm and from Al-Awadi et al.14,16 The results indicate comparable results between rate constants using CBS-QB3 energies and the experimental.


The calculated A and Ea values as derived from the displayed Arrhenius plots are collected in Table 3 and reasonably agree with the reported experimental data of Al-Awadi et al.14,15,16,17,18,19,20 for quinolines, isoquinolines, and other aromatic systems bearing ethoxy group.
The pressure dependence of the studied unimolecular H-atom transfer thermal decomposition reactions is calculated employing RRKM theory at a low-pressure range of 10−6 to 10 atm at 800 K and is sketched in Fig. 12 and summarized in Table S19.

Rate constants k1−13 (s−1) for unimolecular decomposition reactions of the investigated ethoxyquinolines and ethoxyisoquinolines (R1–R13) obtained from pressure-dependent RRKM theory based on CBS-QB3//BMK/6–31+G(d,p) energies, (T = 800 K, P = 10−6–10 atm).
As can be seen in Fig. 12, the rate coefficients for reactions (R1–R13) over the applied range of pressure (10−6–10 atm), are pressure dependent at the applied temperature.
Conclusion
This paper describes the thermochemistry and kinetics of the unimolecular gas phase thermal decomposition reactions of ten ethoxy- and ethoxyisoquinolines (1-EisoQ, 2-EQ, 3-EQ, 3-EisoQ, 4-EQ, 4-EisoQ, 5-EQ, 5-EisoQ, 8-EQ, and 8-EisoQ) at MPW1B95, M06-2X, and CBS-QB3 methods. The rate constants for these reactions were calculated using conventional transition state theory combined with Eckart tunneling (TST/Eck) and the statistical Rice–Ramsperger–Kassel–Marcus (RRKM) theories. The obtained results can be summarized as follows:
-
1.
Formation of ethylene and keto form is preferred kinetically and thermodynamically.
-
2.
Quinolones and isoquinolones are lower in energy than the corresponding enols except in the case of 3-isoquinolone where no aromatic ring is present.
-
3.
Removal of ⋅the CH3 radical is the only important simple fission reaction and became significant at higher temperatures (T ≥ 1100 K).
-
4.
The investigated decomposition reactions show clear and significant temperature- and pressure-dependent rate constants in the considered ranges.
Data availability
All data generated through this study are included in this manuscript and the Supporting Information file.
References
Chen, M., Chen, H., Ma, J., Liu, X. & Zhang, S. Synthesis and anticancer activity of novel quinoline–docetaxel analogues. Bioorg. Med. Chem. Lett. 24, 2867–2870 (2014).
Nayak, N., Ramprasad, J. & Dalimba, U. Synthesis and antitubercular and antibacterial activity of some active fluorine containing quinoline–pyrazole hybrid derivatives. J. Fluor. Chem. 183, 59–68 (2016).
Guo, L.-J., Wei, C.-X., Jia, J.-H., Zhao, L.-M. & Quan, Z.-S. Design and synthesis of 5-alkoxy-[1, 2, 4] triazolo [4, 3-a] quinoline derivatives with anticonvulsant activity. Eur. J. Med. Chem. 44, 954–958 (2009).
Pinz, M. et al. 4-Phenylselenyl-7-chloroquinoline, a new quinoline derivative containing selenium, has potential antinociceptive and anti-inflammatory actions. Eur. J. Pharmacol. 780, 122–128 (2016).
Vandekerckhove, S. & D’hooghe, M. Quinoline-based antimalarial hybrid compounds. Bioorg. Med. Chem. 23, 5098–5119 (2015).
Fiorito, J. et al. Synthesis of quinoline derivatives: Discovery of a potent and selective phosphodiesterase 5 inhibitor for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 60, 285–294 (2013).
Hu, Y. et al. Inhibition of Tpl2 kinase and TNFα production with quinoline-3-carbonitriles for the treatment of rheumatoid arthritis. Bioorg. Med. Chem. Lett. 16, 6067–6072 (2006).
Arjunan, V., Mohan, S., Balamourougane, P. & Ravindran, P. Quantum chemical and spectroscopic investigations of 5-aminoquinoline. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 74, 1215–1223 (2009).
Ibrahim, S., Makhlouf, M. T., Abdel-Hafez, A. & Moharram, A. Some transition metal chelates of 8-hydroxyquinoline-5-sulfonamides as possible drugs. J. Inorg. Biochem. 28, 57–65 (1986).
Taylor, P. J., van der Zwan, G. & Antonov, L. Tautomerism: Introduction, history, and recent developments in experimental and theoretical methods. In Tautomerism: Methods and Theories (ed. Antonov, L.) 1–24 (Wiley-VCH: Weinheim, Germany, 2013).
Senthilkumar, K. & Kolandaivel, P. Quantum chemical studies on tautomerism of barbituric acid in gas phase and in solution. J. Comput. Aided Mol. Des. 16, 263–272 (2002).
Jinshan, L., Heming, X. & Haishan, D. DFT and ab initio studies on stability and isomerization of benzofuroxan. Chin. J. Chem. Phys. 13, 55–60 (2000).
Jingchang, Z., Sen, Z., Zuoyin, Y. & Weiliang, C. A theoretical study on the reaction mechanism of epoxidation reaction of the [4-(bromomethyl) bicyclo [4.4. 1] undeca-1, 3, 5, 7, 9-pentaen-3-yl] methanol. Chin. J. Chem. Phys. 18, 167–173 (2005).
Al-Awadi, N. & Taylor, R. The mechanism of thermal eliminations. Part 21. Rate data for pyrolysis of 2-ethoxyquinoline, 1-and 3-ethoxyisoquinoline, and 1-ethoxythiazole: Correlation of reactivities with π-bond order of the C [double bond, length as m-dash] N bond. J. Chem. Soc. Perkin Trans. 2, 1589–1592 (1986).
Al-Awadi, N. & Taylor, R. The mechanism of thermal eliminations. Part 18. Relative rates of pyrolysis of 2-ethoxypyrazine, 3-ethoxypyridazine, 2-and 4-ethoxypyrimidine, 3-chloro-6-ethoxypyridazine, and 2-chloro-4-ethoxypyrimidine: The effect of the aza ‘substituent’ and π-bond order on the elimination rate. J. Chem. Soc. Perkin Trans. 2, 1255–1258 (1986).
Al-Awadi, N. A., Kaul, K. & Dib, H. H. Gas-phase pyrolytic reactions of esters of 2-pyridine and 8-quinoline sulfonic acid. Int. J. Chem. Kinet. 32, 771–775 (2000).
Dib, H., Ibrahim, M., Al-Awadi, N., Ibrahim, Y. & Al-Awadi, S. Gas-phase pyrolytic reaction of 3-phenoxy and 3-phenylsulfanyl-1-propanol derivatives: Kinetic and mechanistic study. Int. J. Chem. Kinet. 40, 51–58 (2008).
Dib, H. H., John, E., El-Dusouqui, O. M., Ibrahim, Y. A. & Al-Awadi, N. A. Gas-phase pyrolysis of 1-(pyrazol-4-yl)-1H-benzotriazoles. Pyrolytic approach to indole and condensed indole derivatives of potential synthetic and biological applications. J. Anal. Appl. Pyrolysis 124, 403–408 (2017).
Al-Azemi, T. F., Dib, H. H., Al-Awadi, N. A. & El-Dusouqui, O. M. Selective pyrolysis of bifunctional compounds: Gas-phase elimination of carbonate–ester functionalities. Tetrahedron 64, 4126–4134 (2008).
Ahmad, Z., Al-Awadi, N. & Al-Sagheer, F. Thermal degradation studies in poly (vinyl chloride)/poly (methyl methacrylate) blends. Polym. Degrad. Stab. 93, 456–465 (2008).
Ibrahim, Y. A., Al-Awadi, N. A. & Kual, K. Flash vacuum pyrolysis of azo and nitrosophenols: New routes towards hydroxyarylnitrenes and their reactions. Tetrahedron 59, 5425–5430 (2003).
El-Demerdash, S. H., El-Gogary, T. M. & El-Nahas, A. M. A thermochemistry and kinetic study on the thermal decomposition of ethoxyquinoline and ethoxyisoquinoline. Int. J. Chem. Kinet. 50, 604–611 (2018).
El-Nahas, A. M. et al. Enthalpies of formation, bond dissociation energies and reaction paths for the decomposition of model biofuels: ethyl propanoate and methyl butanoate. J. Phys. Chem. A 111, 3727–3739 (2007).
El-Nahas, A. M., Heikal, L. A., Mangood, A. H. & El-Shereefy, E.-S.E. Structures and energetics of unimolecular thermal degradation of isopropyl butanoate as a model biofuel: Density functional theory and ab initio studies. J. Phys. Chem. A 114, 7996–8002 (2010).
Shiroudi, A. et al. A computational study on the kinetics of pyrolysis of isopropyl propionate as a biodiesel model: DFT and ab initio investigation. Fuel 281, 118798 (2020).
El-Gogary, T. M., Heikal, L. A., Abdel-Rahman, M. A. & El-Nahas, A. M. First-principle kinetic studies of unimolecular pyrolysis of isopropyl esters as biodiesel surrogates. Theor. Chem. Acc. 140, 1–15 (2021).
Al-Etaibi, A. M., Al-Awadi, N. A., Ibrahim, M. R. & Ibrahim, Y. A. Gas-phase pyrolysis of N-alkoxyphthalimides to functionally substituted aldehydes: Kinetic and mechanistic study. ARKIVOC 10, 149–162 (2010).
Ibrahim, M. R., Al-Azemi, T. F., Al-Etabi, A., El-Dusouqui, O. M. & Al-Awadi, N. A. Gas-phase pyrolysis of benzimidazole derivatives: Novel route to condensed heterocycles. Tetrahedron 66, 4243–4250 (2010).
Al-Awadi, N. A., Ibrahim, Y. A., Elnagdi, M., Adam, A. Y. & John, E. Gas-phase pyrolysis of arylazonicotinates and nicotinonitriles: Routes towards new aminopyridine and pyrido [3, 4-c] cinnoline derivatives. J. Anal. Appl. Pyrol. 124, 602–609 (2017).
Al-Qallaf, M. A., Dib, H. H., Al-Awadi, N. A. & El-Dusouqui, O. M. Arylidenepyridylhydrazines: Synthesis, and kinetics and mechanism of their gas-phase pyrolysis. J. Anal. Appl. Pyrol. 124, 446–453 (2017).
Ibrahim, Y. A., Al-Awadi, N. A. & Ibrahim, M. R. Gas-phase thermolysis of thieno [3, 2-e][1, 2, 4] triazines. Interesting routes towards heterocyclic ring systems. Tetrahedron 60, 9121–9130 (2004).
Al-Etaibi, A., Makhseed, S., Al-Awadi, N. A. & Ibrahim, Y. A. A novel simple pyrolytic approach towards anhydronucleosides. Tetrahedron Lett. 46, 31–35 (2005).
Dib, H. H., Al-Awadi, N. A., Ibrahim, Y. A. & El-Dusouqui, O. M. Gas-phase thermolysis of benzotriazole derivatives. Part 2: Synthesis of benzimidazo [1, 2-b] cinnolines, a novel heterocyclic ring system, by pyrolysis of benzotriazole derivatives. Kinetic and mechanistic study. Tetrahedron 59, 9455–9464 (2003).
Boese, A. D. & Martin, J. M. Development of density functionals for thermochemical kinetics. J. Chem. Phys. 121, 3405–3416 (2004).
Chemcraft v. 1.8 (http://www.Chemcraftprog.com).
Merrick, J. P., Moran, D. & Radom, L. An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A 111, 11683–11700 (2007).
Gonzalez, C. & Schlegel, H. B. An improved algorithm for reaction path following. J. Chem. Phys. 90, 2154–2161 (1989).
Gonzalez, C. & Schlegel, H. B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 94, 5523–5527 (1990).
Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 14, 363–368 (1981).
Mahmoud, M. A. et al. Structures, energetics, and kinetics of H-atom abstraction from methyl propionate by molecular oxygen: Ab initio and DFT investigations. Comput. Theor. Chem. 1196, 113119 (2021).
Zhao, Y. & Truhlar, D. G. Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: The MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J. Phys. Chem. A 108, 6908–6918 (2004).
Zhao, Y. & Truhlar, D. G. Design of density functionals that are broadly accurate for thermochemistry, thermochemical kinetics, and nonbonded interactions. J. Phys. Chem. A 109, 5656–5667 (2005).
El-Nahas, A. M. et al. Assessment of hybrid, meta-hybrid-GGA, and long-range corrected density functionals for the estimation of enthalpies of formation, barrier heights, and ionisation potentials of selected C1–C5 oxygenates. Mol. Phys. 113, 1630–1635 (2015).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Al-Otaibi, J. S. et al. Thermo-kinetic theoretical studies on pyrolysis of dimethoxymethane fuel additive. Fuel 290, 119970 (2021).
Montgomery, J. A. Jr., Frisch, M. J., Ochterski, J. W. & Petersson, G. A. A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J. Chem. Phys. 110, 2822–2827 (1999).
Montgomery, J. A. Jr., Frisch, M. J., Ochterski, J. W. & Petersson, G. A. A complete basis set model chemistry. VII. Use of the minimum population localization method. J. Chem. Phys. 112, 6532–6542 (2000).
Casasnovas, R. et al. Simplification of the CBS-QB3 method for predicting gas-phase deprotonation free energies. Int. J. Quantum Chem. 110, 323–330 (2010).
Frisch, M. et al. Gaussian 16, Revision A. 03, Gaussian. Inc., Wallingford CT 3 (2016).
Pelzer, H. & Wigner, E. The speed constansts of the exchange reactions. Z. Phys. Chem. B 15, 445–552 (1932).
Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 3, 107–115 (1935).
Evans, M. G. & Polanyi, M. Some applications of the transition state method to the calculation of reaction velocities, especially in solution. Trans. Faraday Soc. 31, 875–894 (1935).
Glasstone, S., Laidler, K. J. & Eyring, H. The Theory of Rate Processes; The Kinetics of Chemical Reactions, Viscosity, Diffusion and Electrochemical Phenomena (McGraw-Hill Book Company, 1941).
Eckart, C. The penetration of a potential barrier by electrons. Phys. Rev. 35, 1303 (1930).
Marcus, R. A. & Rice, O. The kinetics of the recombination of methyl radicals and iodine atoms. J. Phys. Chem. 55, 894–908 (1951).
Rice, O. K. & Ramsperger, H. C. Theories of unimolecular gas reactions at low pressures. J. Am. Chem. Soc. 49, 1617–1629 (1927).
Kassel, L. S. Studies in homogeneous gas reactions. I. J. Phys. Chem. 32, 225–242 (2002).
Marcus, R. A. Unimolecular dissociations and free radical recombination reactions. J. Chem. Phys. 20, 359–364 (1952).
Canneaux, S., Bohr, F. & Henon, E. KiSThelP: A program to predict thermodynamic properties and rate constants from quantum chemistry results. J. Comput. Chem. 35, 82–93 (2014).
Mourits, F. M. & Rummens, F. H. A critical evaluation of Lennard-Jones and Stockmayer potential parameters and of some correlation methods. Can. J. Chem. 55, 3007–3020 (1977).
El-Nahas, A. M., Mangood, A. H., Takeuchi, H. & Taketsugu, T. Thermal decomposition of 2-butanol as a potential nonfossil fuel: A computational study. J. Phys. Chem. A 115, 2837–2846 (2011).
Abdel-Rahman, M. A., Al-Hashimi, N., Shibl, M. F., Yoshizawa, K. & El-Nahas, A. M. Thermochemistry and kinetics of the thermal degradation of 2-methoxyethanol as possible biofuel additives. Sci. Rep. 9, 4535 (2019).
Abdel-Rahman, M. A. et al. A W1 computational study on the kinetics of initial pyrolysis of a biodiesel model: Methyl propanoate. New J. Chem. 45, 19531–19541 (2021).
Abdel-Rahman, M. A., Shiroudi, A., Kaya, S. & El-Nahas, A. M. Theoretical investigations on the unimolecular decomposition mechanisms of isopropyl acetate. J. Mol. Struct. 1262, 133006 (2022).
da Silva, M. A. R., Matos, M. A. R. & Monte, M. J. S. Enthalpies of combustion, vapour pressures, and enthalpies of sublimation of 2-hydroxyquinoline, 4-methyl-2-hydroxyquinoline, 4-hydroxyquinoline, and 2-methyl-4-hydroxyquinoline. J. Chem. Thermodyn. 22, 609–616 (1990).
Meshhal, M. M., El-Demerdash, S. H. & El-Nahas, A. M. A thermochemical computational study on hydroxyquinolines and their azulene analogues. J. Mol. Struct. 1183, 70–77 (2019).
Mahmoud, M. A., El-Demerdash, S. H., Mangood, A. H. & El-Nahas, A. M. Modeling of thermochemistry of thermal decomposition of methyl propionate and ethyl acetate biodiesel. Int. J. Adv. Sci. Tech. Res. 2, 190–202 (2017).
Moc, J. & Simmie, J. M. Hydrogen abstraction from n-butanol by the hydroxyl radical: High level ab initio study of the relative significance of various abstraction channels and the role of weakly bound intermediates. J. Phys. Chem. A 114, 5558–5564 (2010).
Alabugin, I. V., Manoharan, M., Breiner, B. & Lewis, F. D. Control of kinetics and thermodynamics of [1, 5]-shifts by aromaticity: A view through the prism of Marcus theory. J. Am. Chem. Soc. 125, 9329–9342 (2003).
Acknowledgements
This paper is dedicated to the soul of the late Prof. Dr. Ahmed El-Nahas, without whom this project would never have been possible.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
M.A.A.-R., M.F.S., and M.A.M.M contributed equally to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdel-Rahman, M.A., Shibl, M.F. & Mahmoud, M.A.M. Pyrolytic elimination of ethylene from ethoxyquinolines and ethoxyisoquinolines: a computational study. Sci Rep 13, 6248 (2023). https://doi.org/10.1038/s41598-023-33272-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-33272-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
