
Abstract
Embryo development is a dynamic process and critical stages may go unnoticed with the use of traditional morphologic assessments, especially the timing of embryonic divisions and aberrant zygotic cleavage patterns. Bovine embryo development is impaired after oocyte vitrification, but little is known about the underlying morphokinetic behavior. Here, bovine zygotes from fresh (n = 708) and vitrified oocytes (n = 182) were monitored by time-lapse imaging and the timing and nature of early blastomere divisions were modeled to find associations with blastocyst development at day 8. The predictive potential of morphokinetic parameters was analyzed by logistic regression and receiver operating characteristic curve analysis to determine optimal cut-off values. Lag-phase was highly correlated with embryo development. Remarkably, 100% of zygotes that reached the blastocyst stage showed a lag-phase. Fast first cleavage increased the chance of blastocyst development to 30% with a cut-off of 32 h and 22 min. Aberrant zygotic cleavage events, including multipolar division, unequal blastomere sizes, and membrane ruffling resulted in decreased blastocyst development. Multipolar division leads to uneven blastomeres, which was associated with anuclear and multinuclear blastomeres, indicating genome segregation errors. Moreover, we described for the first time morphokinetics of embryos derived from vitrified bovine oocytes. Vitrification severely affected blastocyst development, although lower cryoprotectant concentration in equilibration solutions seems to be less detrimental for embryo yield. Impaired development was linked to slow cleavages, lower lag-phase incidence, and increased early embryonic arrest. Typically, less than 15% of the embryos produced from vitrified oocytes reached more than eight cells. Interestingly, the rate of abnormal first cleavage events was not affected by oocyte vitrification. In conclusion, time to first cleavage, the presence of a lag-phase, and the absence of aberrant zygotic cleavage were the best predictors of bovine blastocyst development for both fresh and vitrified oocytes.
Similar content being viewed by others

Introduction
In vitro embryo production (IVEP) has a major impact on improving productivity in beef and dairy cattle and has shown continuous progress and increased application over the past three decades1,2. However, there are still unresolved aspects of IVEP that limit a wider implementation of the technology1,3. First, IVEP affects implantation and pregnancy rates compared to in vivo produced embryos, because of a reduced embryo quality (reviewed by Rablagino et al.)4. Second, cryopreservation of bovine oocytes would benefit the growing in vitro embryo production programs as long-term conservation and wide-spread distribution of female genetics would become possible. However, the efficiency of IVEP following bovine oocyte cryopreservation is halved5. By consequence, the technique remains in an experimental state (reviewed by Dujíčková et al.)6.
More than one and a half million bovine embryos were produced in vitro worldwide in 20212, but 60% had to be transferred fresh due to the lack of a reliable cryopreservation method that results in favorable pregnancy rates2,7,8. Consequently, a large number of embryo recipient animals must be available, resulting in increased costs9,10. Predicting blastocyst yield and optimization of a vitrification protocol would allow to decrease the number of recipients per calf born and to optimize the logistics of the IVEP process. High quality embryos could be transferred to available recipients or vitrified and transferred later in case of a shortage of recipients10,11, embryo transfer could be delayed one day if there is a shortage of blastocysts or five days synchronization protocol could be used in case a high number of blastocysts is predicted10,12,13,14,15. Furthermore, as there is a clear correlation between embryo quality and pregnancy rate, a key challenge for cost-effective breeding is to select the best embryo to transfer following IVEP16. Traditionally, assessment of in vitro produced embryo quality has been based on the morphological evaluation of the grade and developmental stage of the embryo at the time of freezing or transfer to a recipient17. Although embryos categorized as being of excellent and good quality exhibited a higher pregnancy rate than those classified as poor16,18, embryos with high morphological scores can still contain chromosomal errors19, which are associated with reduced implantation and pregnancy rates and increased embryonic loss20,21. Therefore, in addition to morphological scoring, screening for chromosomal errors has been suggested to improve the selection of high-quality embryos19,22. Yet, genetic analyses are costly and requires careful manipulation during biopsy to preserve embryo viability23.
Morphokinetic parameters can be valuable for embryo quality evaluation but may go unnoticed when using a stereo microscope and require inspection of the embryos at specific time points24. The latter may also harm embryo development due to changes in culture conditions, such as altered pH, temperature, and humidity, resulting in additional stress25,26. Time-lapse technology enables non-invasive and continuous observation of embryo development from fertilization to transfer. This technology has been used in human studies to detect key events that predict blastocyst formation, ploidy, and embryo implantation potential and it is applied in some fertility clinics for its technical benefits and to select the best embryos for transfer (reviewed by Pennetta et al.)27,28. Morphokinetic parameters have been linked to embryo development and chromosomal or genome-wide errors29,30. For example, the pace of development of in vitro produced embryos can be linked to embryo quality, and embryos that cleave faster following fertilization are more developmentally competent than are those that cleave relatively late31,32. On the other hand, cytokinetic events diverging from the normal division of mammalian zygotes in two equally sized blastomeres have been shown to relate to embryo quality24,33,34. Multipolar division—the first division of the embryo directly into three cells29,30,33,35, ruffling membranes—motility of the oolemma before the first division36,37, the occurrence of a resting period in between the divisions or so-called lag-phase, and heterogeneous size of the blastomeres following the first division can all affect embryo development and pregnancy rates33,38. Although some time-lapse studies in cows have reported on the prevalence of abnormal cleavage patterns and on the length of cell cycles and their correlation with developmental capacity30,33,34,36,39,40,41, integration of the effect of all the morphokinetic events mentioned previously in one single study has not yet been performed. Furthermore, the ideal cut-off points for cleavage time or size differences in blastomeres remain to be determined.
Cryopreservation is nowadays a crucial tool in reproduction used for long-term storage of genetic material, and vitrification is the method of choice for cryopreservation of oocytes in several species42,43. During vitrification, cells transform into a “glassy” state by replacement of most intracellular water content with permeable cryoprotectants followed by ultra-rapid cooling44. Once vitrified specimens are warmed, cells rehydrate and cryoprotectants are removed44. Consequently, physicochemical stress can affect the oocyte spindle, cytoskeleton, mitochondria, and the zona pellucida45,46,47,48. Whether these physicochemical changes cause alterations in embryo morphokinetics is still unknown in cattle. In humans, an altered pronuclear and nucleolar activity and a delayed developmental speed were observed following vitrification49,50. However, no differences in clinical outcomes have been observed after human oocyte vitrification compared to fresh oocytes51. As it has been proven difficult to obtain transferable bovine embryos from vitrified oocytes, time-lapse imaging could help to elucidate whether vitrification causes subcellular effects that are able to alter cell division dynamics.
The implementation of morphokinetic parameters to assess embryo quality could be used in a clinical setting to improve the success rate following transfer of IVEP embryos. Moreover, knowledge on relevant morphokinetic parameters could be used to improve culture or vitrification protocols in bovine IVEP. Therefore, the aim of the study was to provide basic insight into preimplantation bovine embryo morphokinetics, using a large number of fresh oocytes and, for the first time, vitrified oocytes. The link with developmental competence provides a criterion to determine the importance of the different morphokinetic parameters. Furthermore, building on observations resulting from the first two experiments, we aimed to determine if certain morphokinetic parameters are associated with genome segregation errors using blastomere nuclear content examination following the first division as a proxy.
Results
Experiment 1
Time-lapse culture conditions support bovine embryo development but at reduced blastocyst rates
In 23 replicates including 933 zygotes (time-lapse = 351; control group = 582), cleavage rate was higher in time-lapse (90.6 ± 1.6%) than in control group culture (82.8 ± 1.7%; p = 0.001). However, culture in the Well of the Well (WOW) system, allowing time-lapse monitoring, was associated with a reduced blastocyst rate at day 8 (28.8 ± 2.4%), when compared to control group culture (40.4 ± 2.0%; p = 0.0004).
Fast first cleavage is associated with high blastocyst development
Zygotes that developed to the blastocyst stage exhibited a faster first cleavage than those which arrested (p < 0.0001). The subsequent divisions were reached at similar timepoints in both categories (Table 1).
Receiver operating characteristic analysis indicates time of first division and differences in blastomere size as predictors for embryo development
The best cut-off for timing of first cleavage to predict blastocyst development was determined by a receiver operating characteristic (ROC) curve analysis. A cut-off point of 32 h 22 min predicted day 8 blastocyst development with a sensitivity of 70%, a specificity of 64%, and an area under the curve (AUC) of 0.72 (95% confidence interval (CI): 0.65–0.78) (Fig. 1A). The second, third, and fourth cell divisions yielded a lower predictive value (Fig. 1B). Table 2 shows the ROC, AUC, cut-off (in hours) values, and the sensitivity and specificity when times to reach the first (1–2 cell), second (3–4 cell), third (5–8 cell), and fourth (9–16 cell) cell divisions were set as predictors for day 8 blastocyst development. The fifth division was not considered due to low numbers of zygotes reaching this stage.

Receiver operating characteristics (ROC) curves with blastocyst development at day 8 post insemination as the classifier (blastocyst yes vs. blastocyst no). (A) ROC curve for timing of first division. (B) Comparison of different ROC curves for each division. AUC area under the curve.
Difference in size between blastomeres following the first cleavage was also a predictor for day 8 blastocyst outcome. Using a cut-off of 23.4% difference in blastomere area at first cleavage, blastocyst development could be predicted with a sensitivity of 45%, a specificity of 76%, and an AUC = 0.63 (95% CI: 0.57–0.70).
Bovine embryos that reach the blastocyst stage display a lag-phase
The presence of a lag-phase was predictive for blastocyst development, independent from the cell division stage after which the lag-phase was detected. All of the 100 zygotes that reached the blastocyst stage exhibited a lag-phase (100%), while only 36 out of 205 arrested embryos (18.0%; p < 0.00001) showed a lag-phase. Remarkably, from the 36 arrested embryos that displayed a lag-phase, 21 reached the morula stage and 15 completed the fourth division. The rest of the embryos (n = 168) that did not exhibit a lag-phase arrested at an earlier stage (i.e., before reaching the third division) or during a stage at which the lag-phase is normally present (i.e., between third and fourth or between fourth and fifth division). For three zygotes, two that reached the blastocyst stage and one arrested, the presence or absence of a lag-phase could not be determined due to a temporary air bubble; these embryos were not included in the analysis. Blastocyst rates were similar for embryos that exhibited a lag-phase after the third (72.57 ± 4.20%) or fourth division (71.43 ± 9.90; p = 0.9; Table 3). The second division was not included in the statistical analysis due to the low number of zygotes that presented a lag-phase at this stage (n = 2). For embryos displaying a lag-phase, the average overall lag-phase length was shorter in those zygotes that developed into blastocysts (47.1 h ± 1.18) than for arrested embryos (51.7 h ± 1.93; p = 0.04), but this was not significant within one specific cell cycle (Table 3). Although a longer lag-phase was seen if it occurred after the fourth division than after the third division, this was only different for embryos developing to blastocysts (p = 0.0004) and not for arrested embryos (p = 0.5) (Table 3).
Aberrant zygotic cleavage events impair blastocyst development
In 23 replicates including 313 zygotes that cleaved, 196 (62.6%) exhibited normal cleavage (division in two cells). The most common abnormal cleavage was a direct division into three or more cells (multipolar division) (Supplementary Movie 2) for 60 zygotes (19.2%), followed by the display of a ruffling membrane (Supplementary Movie 2) in 41 zygotes (13.1%), and reverse cleavage in only 5 zygotes (1.6%) (Supplementary Movie 2). Eleven (3.5%) zygotes presented two or more of these abnormal cleavage events. All the cinematographic parameters evaluated here affected the day 8 blastocyst rate (p < 0.01) (Table 4). Reverse cleavage was not considered in the analysis due to the low number of zygotes exhibiting this phenomenon. Fast cleavers exhibited multipolar division at a similar rate than slow cleavers (21.9 vs 18.2%, respectively p = 0.32). Multipolar division tended to lead to a higher proportion of zygotes with uneven blastomeres (23.4%) than those with even blastomeres (16.5%; p = 0.05). Interestingly, fast cleavers displayed a lower percentage of ruffling membrane (4.2%) than slow cleavers (22.6%; p < 0.0001).
Experiment 2
Oocyte vitrification reduces blastocyst development
In 13 replicates, 1,776 zygotes were cultured (1,172 fresh, 604 vitrified) and cleavage and blastocyst rates were compared (Fig. 2, Supplementary Table 1).

Cleavage and day 8 blastocyst rate over zygotes starting in vitro culture after 21 h of in vitro fertilization. Different superscripts (*, **; a, b;+, ++; d, e) per group of bars represent statistical differences (p < 0.05). Results are stated as least square means ± standard errors. Control: fresh cumulus enclosed oocytes. Corona radiata (CR): fresh partially denuded oocytes enclosed by the CR only. VCR-H: CR oocytes vitrified with a protocol using high concentrations of cryoprotectants (15%) in equilibration solution. VCR-L: CR oocytes vitrified with a protocol using a low cryoprotectant (3%) concentration in equilibration solution. Numbers of zygotes in group culture (Control: n = 582, CR = 369, VCR-L = 183, VCR-H = 239) and in Well-of the-well (WOW) time-lapse (Control = 127, CR = 94, VCR-L = 91, VCR-H = 91).
Oocyte vitrification significantly affected cleavage rates. In a group culture system, only 4% of the zygotes reached the blastocyst stage when they were derived from oocytes vitrified with high (i.e., 15%) cryoprotectant concentrations in equilibration solution (VCR-H), and 7% reached the blastocyst stage when vitrified with low (i.e., 3%) cryoprotectant concentrations in equilibration solution (VCR-L). Both groups showed lower blastocyst rates than zygotes derived from fresh (i.e., non-vitrified) oocytes enclosed either by cumulus, or by corona radiata cells only (40 and 25%; p < 0.0001, respectively) (Fig. 2 and Supplementary Table 1). Partial denudation resulting in oocytes enclosed by corona radiata cells only, which is routinely performed prior to vitrification, could underly part of the impaired development, as blastocyst development was also lower in fresh corona radiata oocytes, when compared to the cumulus enclosed control group (p < 0.001) (Fig. 2 and Supplementary Table 1). Likewise, in the time-lapse system, both vitrified groups exhibited lower blastocyst rates compared with fresh cumulus enclosed oocytes (p < 0.01). However, compared with corona radiata enclosed fresh oocytes, only VCR-H showed significantly lower blastocyst development (p = 0.0022). Corona radiata fresh enclosed oocytes and VCR-L did not exhibit differences (p = 0.12) (Fig. 2 and Supplementary Table 1). In this experiment, fresh cumulus enclosed oocytes cultured in time-lapse conditions also presented a lower blastocyst rate compared to group culture (23.6 ± 3.1 vs 40.0 ± 1.6%; p = 0.0015), as observed in Experiment 1.
Morphokinetic factors associated with blastocyst development following vitrification
Morphokinetics analyses demonstrated that zygotes derived from vitrified oocytes arrested earlier than those from fresh cumulus enclosed oocytes, since only 15.6 ± 5.4% in VCR-L and 7.4 ± 3.7% in VCR-H reached the fourth division (9–16 cells), compared to 51.7 ± 4.0% in the fresh control (Table 5). Partial denudation also generated early arrest because only 32.9 ± 6.3% of the fresh corona radiata zygotes reached the fourth division stage. Compared to the fresh corona radiata, only the vitrification protocol with high concentrations of cryoprotectants resulted in lower number of zygotes reaching fourth division (Table 5).
The two main parameters associated with blastocyst development, namely speed of first cleavage and percentage of embryos that exhibited lag-phase were affected by vitrification and partial denudation (Table 6). Speed to reach first cleavage was similarly affected in vitrified and fresh corona radiata oocytes compared with cumulus enclosed control oocytes (p < 0.002) (Table 6). Average time to complete the first three divisions was also affected in fresh corona radiata and vitrified oocytes. Time to complete all divisions in the different groups is displayed in Supplementary Table 2. A lower percentage of zygotes derived from fresh partiality denuded oocytes and both groups of vitrified oocytes exhibited a lag-phase, compared with zygotes from cumulus enclosed oocytes (p < 0.04). However, for this parameter, oocyte cryopreservation with high amounts of cryoprotectants resulted also in a significantly lower number of zygotes that exhibited lag-phase compared with fresh corona radiata oocytes (p = 0.01) (Table 6). Vitrification did not affect the occurrence of abnormal events around first cleavage, nor the blastomere size (Table 6).
Experiment 3
Multipolar zygotic division is associated with nuclear abnormalities and unequal blastomere sizes
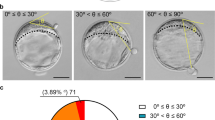
Following analysis of 43 blastomeres from 22 bipolar zygotes, and 65 blastomeres from 20 multipolar zygotes collected immediately after the zygotic division and analyzed for their nuclear content and size, embryos presenting a bipolar cleavage were found to have a greater number of blastomeres with a normal nuclear content (89.7 ± 5.82%) than embryos presenting a multipolar division (70.3 ± 9.31%; p = 0.03) (Fig. 3). Notably, no anuclear blastomeres were detected following bipolar zygotic division. In addition, zygotes presenting a bipolar division did not present differently sized mononuclear (5962 ± 306 µm2) or multinuclear (6535 ± 707 µm2) blastomeres (p = 0.39). In contrast, zygotes presenting multipolar division, contained smaller sized anuclear blastomeres (3022 ± 357 µm2) compared to mononuclear (4049 ± 208 µm2; p = 0.019) and multinuclear blastomeres (4831 ± 356 µm2; p = 0.0006), respectively. Also, the blastomere size of multinuclear blastomeres tended to be larger compared to mononuclear blastomeres (p = 0.10) in zygotes presenting a multipolar division, as shown in Supplementary Figure 1.

Number and percentages of blastomeres found to have a normal, mononuclear and aberrant (anuclear or multinuclear) content following bipolar (B) or multipolar zygotic divisions (A).
Discussion
Morphokinetics of bovine embryos can be used to predict blastocyst development in a non-invasive manner. In this study, we used ROC curves to determine reliable cut-offs for known predictive parameters, including timing of the first cleavage and size equality of the resulting blastomeres. The effect of abnormal events during zygotic cleavage, was further explored and a link was found between multipolar division, blastomere size, and nuclear content. Finally, for the first time we evaluated morphokinetics of zygotes derived from vitrified mature bovine oocytes using two vitrification protocols. Fast first cleavage, the presence of a lag-phase, and the absence of abnormal zygotic cleavage were elementary for blastocyst development in for both vitrified and fresh bovine oocytes.
Both timing and nature of the first embryonic division are linked to subsequent embryo development34,52,53. Our results confirm previous studies demonstrating that zygotes which cleave faster present higher developmental competence38,54,55. However, in the present study, we used the gold standard method to discriminate between these two categories (ROC curve). As such, we determined the ideal cut-off point in time (32 h and 22 min) to differentiate between fast and slow cleaving embryos, as a predictor for blastocyst development. This time-point differs from the traditional way to separate fast and slow, which is making use of the average time of first division for zygotes that become a blastocyst33,36,41. By using ROC analysis, we determined the optimal threshold with the highest summation of sensitivity and specificity to predict blastocyst development32,56. These results may be applicable for commercial purposes in which fast and slow cleavers need to be differentiated. Some studies have suggested to avoid selecting fast cleaving embryos, since they may be prone to loss of genome imprinting57,58. We did not evaluate genomic imprinting here and a similar percentage of multipolar division was seen, but we showed that faster zygotes exhibit higher blastocyst rates. In our hands, the average time to reach the first division for fresh cumulus enclosed oocytes in serum free culture was slightly higher (33 h) than in other studies (~ 25 h)33,36,39,41, which may be related to the use of fetal calf serum in those studies, as this has been demonstrated to accelerate embryo development59,60. On the other hand, our results also matched with studies in bovine34,41 and humans61 which showed that differences in blastomere size after zygotic division decrease subsequent developmental potential. Our ROC curve results revealed the ideal cut-point for differentiating between even and uneven blastomeres (< 24% difference in area). Additionally, we showed that the presence of smaller or larger blastomeres in cleaved embryos (uneven cleavage) occurs more frequently following multipolar division, and it tends to be associated with anuclear or multinuclear content following multipolar or bipolar division, respectively. These results coincide with human results, in which a significantly higher degree of aneuploidy (29.4 vs 8.5%) and multinuclear rate (21.1 vs 2.1%) was found in blastomeres from uneven embryos61. Hence, there seems to be an association between blastomere size, nuclear content, and multipolar division.
With regard to abnormal cleavage events, we found lower developmental capacity in zygotes with multipolar cleavage or ruffling membrane, which is in line with human studies29,37 and recent publications in human and bovine zygotes that showed how multipolar division leads to lower embryo development compared to normal cleavers30,34. As supported by the higher number of nuclear abnormalities following multipolar zygotic division, multipolar zygotic division seems to be associated with genome segregation errors in embryos leading to a higher frequency of mixoploidy, and chromosomal abnormalities in the resultant embryos33,39,41 and embryonic arrest30.31,36,38. Surprisingly, Somfai et al.41 did not find differences in developmental rates between zygotes with multipolar and bipolar division, although a numerical difference was present in the developmental rate to the expanded blastocyst stage between normal cleavers and multipolar cleavers (51 vs 35% respectively). Ruffling membrane and reverse cleavage are abnormal events described in human embryos known to affect their developmental capacity27,37. Both events have been described in cattle recently34,36. Although our findings confirm that a ruffling membrane decreases the developmental capacity of such zygotes, the incidence we found for both events differs from the study of Magata et al.36 who reported a lower number of zygotes with ruffling membrane (6 vs 13%) and the contrary for reverse cleavage (17 vs 1.6%). Yaacobi-Artzi et al.34 reported an intermediated proportion of zygotes with reverse cleavage (11%) and none of those zygotes reached the blastocyst stage. Due to the low number of zygotes presenting reverse cleavage in the present study, we did not perform statistical analysis with these data. We hypothesized that any discrepancies might be due to a sperm driven factor62 or to differences in medium composition63.
Our findings confirm a solid correlation between the presence of a lag-phase and blastocyst development64, because almost all embryos that reached the blastocyst stage presented a lag-phase. Most of the zygotes displayed this lag-phase after the third division, which is also agreement with previous studies33,64. The lag-phase was first described by Grisart et al.64 in a small number of embryos and the association with high developmental potential was confirmed later by Sugimura et al.33 and by the present study. As this lag-phase occurs around the embryonic genome activation, it has been suggested that these events are linked64,65. Embryonic genome activation is a critical step in blastocyst development since embryos must overcome a state of transcription repression to initiate the transcriptional activation of the genome after maternal clearance of mRNA and proteins necessary for oocyte maturation65,66,67. The cell cycle length is related to their transcriptomic activity68,69, and the extended duration of the lag-phase could be caused by a prolongation of the G2 phase, when chromatin remodeling occurs for the zygotic genome activation70,71,72.
For the first time, morphokinetic analysis of the embryos generated from bovine vitrified oocytes was performed. These analyses demonstrate that a lower proportion of zygotes derived from vitrified oocytes reach the third (5–8 cells) and fourth (9–16 cells) division, which occur around the time of embryonic genome activation73. Likewise, we showed that oocytes cryopreserved with low or high cryoprotectant concentrations had a tendency to cleave slower or even exhibited a significantly slower cleavage, respectively. In humans, where vitrification of in vivo matured oocytes is a standard clinical procedure, the time of the first zygotic division, and the time from 2 cells to blastulation was delayed for around 1 h49,74, and the overall blastocyst development was delayed50. It remains unclear whether these alterations have consequences for the developmental potential of human zygotes, because no impact on implantation and clinical outcome was observed49,50. In the present study, vitrification of in vitro matured bovine oocytes was performed. It has been demonstrated that in vivo matured oocytes have a higher developmental potential than those vitrified at the immature stage or after in vitro maturation, which is in conformity with the reduced developmental competence observed here75,76,77. Interestingly, in our study, a similar incidence of abnormal cleavage was observed in zygotes from fresh and vitrified oocytes. Yet, more embryos need to be analyzed to confirm this finding.
Partial denudation of oocytes has been proposed for oocyte vitrification, since full cumulus enclosed oocytes exhibit lower maturation after vitrification, probably because of reduced permeability to cryoprotectants78,79,80 and higher volumes of vitrification solution surrounding large cumulus oocyte complexes (COCs)76. By leaving only corona radiata cells, some layers of cumulus cells are maintained, which are necessary during maturation and fertilization, while the vitrification procedure is facilitated and the cooling rate increased due to the consistent, small sizes of the COCs78,81,82,83,84. However, in the present study, partial removal of cumulus cells resulted in slower cleavage and impaired developmental potential of the fresh oocytes too, which is in accordance with previous findings78,79,80. During fertilization, cumulus cells attract, trap, select spermatozoa, induce changes in sperm physiology, and prevent premature hardening of the zona pellucida, allowing fertilization and increasing fertility85,86,87,88. The reduced number of cumulus cell layers in partially denuded oocytes might be the reason for the lower fertilization rates and consequently lower blastocyst yield. Vitrification itself had an additional negative effect on cleavage and blastocyst rates, especially when high concentrations of cryoprotectants (15%) were used in the equilibration solution. Less zygotes reached the fourth division and showed a lag-phase, which may be related to changes in the oocyte’s cytoskeleton, hardening of the zona pellucida, or mitochondrial and spindle damage48,89,90,91.
In conclusion, after comparing several predictors of blastocyst development, we found that the speed of the first division is the best predictor of blastocyst development in early embryo development followed by the absence of abnormal cleavage. Presence of lag-phase in later cleavage stages is crucial to reach the blastocyst stage, and both speed of first cleavage and lag-phase are affected after oocyte vitrification, reducing the developmental capacity of bovine oocytes. Moreover, aberrant genome segregation, as indicated by multinuclear and anuclear blastomeres in the resultant embryos, seems to be correlated with multipolar division. In future experiments, this can be further linked to embryo quality in terms of pregnancy and calving rates.
Materials and methods
No prior ethical review was required for this study since ovaria from cows were gathered in a commercial slaughterhouse for human consumption.
Experimental design
An overview of the experimental design is depicted in Fig. 4 and detailed explanation is provided in supplementary data as annex 1.

Experimental set-up. Experiment 1: After in vitro maturation (IVM) and in vitro fertilization (IVF), presumed zygotes derived from fresh cumulus enclosed oocytes were divided for in vitro culture (IVC) between traditional group culture (n = 582) and Well of the Well (WOW) time-lapse system (n = 351) to evaluate developmental potential and morphokinetics during all culture (8 days). Experiment 2: After IVM, oocytes were divided in 4 groups, Control: fresh cumulus enclosed oocytes (n = 709); Corona radiata (CR): fresh oocytes partially denuded to leave only the CR (463). VCR-H: CR oocytes vitrified with a protocol using high concentrations of cryoprotectants (15%) in equilibration solution (n = 274); VCR-L: CR oocytes vitrified with a protocol using a low cryoprotectants concentration (3%) in equilibration solution (n = 330). After IVF they were split in Group culture and WOW time-lapse for developmental and morphokinetics analyses. Experiment 3: Presumptive zygotes were cultured in WOW time-lapse systems, and collected immediately after the first cleavage, they were classified as bipolar (cleaved in two cells; n = 22) or multipolar (cleaved directly in three or more; n = 20) division, zona pellucida was removed and individual blastomeres were measured, fixed, and stained with Hoechst 33342 for nuclear evaluation.
Media and reagents
Earle’s and Hank’s tissue culture medium (TCM)-199-medium, gentamicin, and kanamycin were purchased from Life Technologies Europe (Ghent, Belgium) and all other reagents were obtained from Sigma (Schnelldorf, Germany), unless otherwise mentioned. All media were filter-sterilized before use with a 0.22 μm filter (Pall Corporation, Ann Arbor, MI, USA).
Collection and in vitro maturation of oocytes
Oocyte collection and in vitro maturation was performed as previously described by Sidi et al.92. Briefly, bovine ovaries from the slaughterhouse were rinsed in physiological saline supplemented with kanamycin (25 mg/mL). Cumulus-oocyte complexes were obtained from 2 to 8 mm follicles with an 18-gauge needle attached to a 10-mL syringe, and the follicular fluid was collected in 2.5 mL of Hepes-albumin-pyruvate-lactate Tyrode's solution (HEPES—TALP). Those COCs with uniformly granulated cytoplasm and surrounded by at least three compact layers of cumulus cells were grouped per 60 and matured in 500 μL of maturation medium (TCM-199 Earle’s salts, supplemented with 50-µg/mL gentamicin and 20-ng/mL epidermal growth factor) at 38.5 °C in 5% CO2 in air for 20–22 h, depending on the experimental group.
Vitrification and warming
After 20 h of IVM, COCs randomly assigned to vitrification were partially denuded by gentle pipetting in HEPES-TALP medium leaving only the CR, and subsequently vitrified by two methods78,83. The composition of the vitrification and warming solutions is summarized in Table 7. A custom adapted cryodevice as described previously was utilized to vitrify and store the oocytes in liquid nitrogen (LN2)81,93.
Protocol with high cryoprotectant concentration in equilibration solution (VCR-H)
Oocytes with only CR were vitrified as described by Ortiz-Escribano et al.78. Briefly, oocytes were partially denudated and kept in 4 mL of base solution (BS) (Hepes-TCM-199 Hank’s salts supplemented with 20% fetal bovine serum (BS20) (Greiner Bio-One)) covered with paraffin oil. All handlings were performed on a thermal plate at 39 °C. Oocytes were equilibrated in 500 μL of equilibration solution (ES) for 10–15 min. After oocytes recovered their original volume, they were transferred into three consecutive 100 μL droplets of vitrification solution (VS). The time between placement of the oocytes in the VS and immerging of the cryodevice in liquid nitrogen was 30–45 s. Four to five oocytes were loaded onto the cryodevice. Oocytes were warmed in a 35 mm × 10 mm petri dish with 4 mL of warming solution, followed by a three-step wash-out in 500 μL solutions of the hyperosmolar sucrose reducing from 1 to 0.5 M, then 0.25 M, and 0 M in BS20, covered by oil. Oocytes were washed in BS20 twice and kept in BS20 until all oocytes for one replicate were warmed (~ 30 min). After that, they were placed in maturation medium for 1–3 h to recover and complete 22 h of maturation.
Protocol with low concentrations of cryoprotectants in equilibration solution (VCR-L)
Oocytes with only CR were vitrified as described by Ishii et al.83 with minor modifications. Briefly, oocytes were partially denudated were kept in 4 mL of base solution BS (Hepes-TCM-199 Hank’s salts supplemented with 10% fetal bovine serum (BS10) (Greiner Bio-One)) covered with paraffin oil. All handlings were performed on a thermal plate at 25 °C, except the thawing step which was performed at 37˚C. Oocytes were equilibrated in 500 μL of ES for 10–15 min. After oocytes recovered their original volume, they were transferred into three consecutive 100 μL droplets of VS. The time between placement of the oocytes in the VS and immerging of the cryodevice in liquid nitrogen was 30–45 s. Four to five oocytes were loaded onto the cryodevice. Oocytes were warmed in a 35 mm × 10 mm petri dish with 4 mL of warming solution, followed by a three-step wash-out in 500 μL solutions of the hyperosmolar sucrose reducing from 0.5 to 0.25 M, then 0.125 M, then 0.0625 and finally 0 M in BS10, covered by oil. Oocytes were washed in BS10 twice and kept in BS20 until all oocytes for one replicate were warmed (~ 30 min). After that, they were placed in maturation medium for 1–3 h to recover and complete 22 h of maturation.
In vitro fertilization and culture
In vitro fertilization and culture were carried out as previously described by Wydooghe et al.94 and Sidi et al.92. In brief, after IVM of fresh oocytes or post warming IVM recovery culture for vitrified/warmed oocytes, IVF was performed in the same manner. Frozen-thawed sperm of a previously tested fertile bull was used. Spermatozoa were separated using a discontinuous Percoll gradient (45 and 90%; GE Healthcare Biosciences, Uppsala, Sweden), and the semen concentration was adjusted to 1 × 106 spermatozoa/mL. Matured oocytes were incubated with spermatozoa in 500 μL in IVF-TALP for 21 h at 38.5 °C in 5% CO2 in humidified air. Consequently, presumptive zygotes were vortexed in 2.5 mL of TALP for three min to remove cumulus cells, and intact zygotes (intact plasma membrane) were transferred to traditional group culture or WOW time-lapse. Group culture was performed in all treatments, with 15–25 zygotes in a 50 μL droplet of synthetic oviduct fluid (SOF) medium enriched with non-essential and essential amino acids (SOFaa) and ITS (5 μg/mL insulin; 5 μg/mL transferrin; 5 ng/mL selenium) under paraffin oil. For time-lapse, 9 or 16 presumed zygotes were placed in individual wells and placed under single droplet of 30 μL covered by 2.5 mL of paraffin oil. The dishes were placed under a time-lapse imaging system and remained in the same incubator than group culture.
Time-lapse imaging system
A compact, digital inverted microscope (Primo VisionTM; Vitrolife, Göteborg, Sweden) was placed into a culture incubator (5% CO2, 5% O2 and 90% N2). The WOW dishes were placed in the microscope's sample holder. All embryos were placed in the field of view and the focus was manually set. Analysis was performed during all the length of the embryo culture (8 days = 192 h) and pictures were taken every 5 min without any disturbance of the embryos. For the time-lapse analyses, the first, second, third, fourth and fifth divisions were defined by reaching the 2 cell-, 3–4 cells-, 5–8 cell-, 9–16 cell- or more than 16 cell-stage, respectively were detected. Blastocyst-stage was defined by the start of blastocoel formation. After the fifth division, the embryos keep dividing, but the exact timing of the subsequent divisions and the number of cells could not be identified unequivocally due to the superposition of cells and the dark nature of the bovine cytoplasm. For those zygotes that experienced multipolar division, the time of first division was considered as a missing value. For all embryos, in the case it was not possible to properly evaluate specific cleavage events, the data points were treated as missing data. In some cases, following the second division, the individual blastomeres could not be count clearly. Therefore, the time of the following divisions was defined when the visible blastomeres cleaved or when the embryonic mass moved due to the cleavage of non-visible blastomeres, rather than counting the exact number of blastomeres. After the first cleavage, the longest and shortest diameter of individual blastomeres were measured to estimate the area of the blastomere using the formula described in the statistical analyses section. The lag-phase was defined as the time in between subsequent divisions, when it exceeded at least twice the duration of the preceding cell cycle64, due to a temporary developmental arrest of the embryo characterized by a period with no evidence of any blastomere division, and barely any movement of the cells33 (Supplementary Movie 1). The blastocyst stage was marked by the first appearance of the blastocoel. Ruffing membrane was defined as a strong motility of the oolemma before the first division (Supplementary Movie 2). Reverse cleavage was defined as the zygote dividing completely into two or more blastomeres followed by refusion of those blastomeres into one cell (Supplementary Movie 2). Figure 5 summarizes the parameters evaluated in the WOW time-lapse system.

Schematic representation of embryo evaluation and parameters analyzed during time-lapse assessment.
Multipolar and chromosomal segregation
Zygotes produced from oocytes collected from five different cows were monitored by time-lapse imaging in three replicates. Single blastomeres were dissociated immediately upon the normal, bipolar zygotic division (in two blastomeres) and the multipolar zygotic division (more than two blastomeres). To this end, pronase (0.1% protease from S. griseus in TCM-199 hank’s salt) was used to dissolve the zona pellucida in the embryos, and subsequently, embryos were washed in TCM-199 with 10% FBS and Ca+2/Mg+2-free PBS with 0.05% BSA to promote blastomere separation. Next, embryos were pipetted in Ca+2/Mg+2-free PBS with 0.1% polyvinylpyrrolidone (PVP) using a STRIPPER pipet holder and a 135-μm capillary (Origio, Cooper Surgical, CT, US) for blastomere dissociation followed by three straight washings steps in same medium. Cells without a distinct cell membrane, having an irregular form, and having a tiny diameter were classified as a fragment. Blastomeres and fragments were fixed overnight in 4% paraformaldehyde and DNA was stained with Hoechst 33,342 (1:1000 dilution in Ca + 2/Mg + 2-free PBS with 1% PVP at room temperature × 10 min). The blastomeres and fragments were examined the next day using bright field and fluorescent microscopy on a Leica DM 5500 B microscope with an excitation filter of BP 450/90 nM. The longest and shortest diameter of individual blastomeres were measured to estimate the area of the blastomere using the formula described in the statistical analyses section. The nuclear content of individual blastomeres was evaluated as follows: blastomere nuclear content was considered normal when one nucleus or metaphase plate (mononuclear) was observed and abnormal when no nuclear content (anuclear) or more than one nucleus and/or metaphase plate (multinuclear) were detected.
Statistical analyses
General
Collection of data was performed in Microsoft Excel (Microsoft Corp., Redmond, WA) work file. Data organization was prepared using PivotTables function (Microsoft Excel) and the statistical analyses were completed in R version 4.0.5 (R Core Team, Vienna, Austria). The replicate was set as random for all the logistic and linear regression models, except for experiments on the association of morphokinetic parameters with nuclear abnormalities, where the cow used within the replicate and the embryo of origin were set as random. In all cases, model residuals were tested by Shapiro–Wilk’s test and if there was no normal distribution (p < 0.05), the outcome variable was transformed by log10. The residuals were normally distributed for all transformed variables (Shapiro–Wilk’s p > 0.05), and assumptions for homoscedasticity and linearity were accomplished in all cases. Results are stated as least squares means and standard errors. Significance was set at p < 0.05 and tendency levels were set at p < 0.1.
Group control versus time-lapse development
Logistic regression models were built to evaluate the effect of presumptive zygotes conventionally cultured in groups (n = 25) or zygotes cultured in 9- or 16-microwells (time-lapse) on embryo development (cleavage and day 8 blastocyst rates).
Morphokinectic parameters
Logistic regression models were built to compare the prevalence of morphokinetic events between groups. The prevalence of morphokinetics events in zygotes derived from fresh oocytes were expressed as a proportion (event/total zygotes).
The best cut-off to predict blastocyst development
Multiple ROC and AUC were constructed to find the optimal cut-off point value with the greatest summation of sensitivity and specificity to predict blastocyst development based on zygote morphokinetic data acquired via time-lapse cinematography. For all the ROC curves, the blastocyst development at day 8 post insemination was set as the classifier (blastocyst yes vs. blastocyst no). A first ROC curve was fitted using the cleavage time (hours) as the predictor. To do so, cleavage was referred to as the zygotic division, irrespectively on the number of resultant blastomeres. Furthermore, multiple ROC curves were fitted by including predictors such as time (hours) to reach the first (1–2 cell), second (3–4 cell), third (5–8 cell), and fourth (9–16 cell) cell division, and the difference in blastomere area at cleavage. In case of multipolar division, for the difference in blastomere area at cleavage, the difference in area between the biggest and the second biggest blastomere area was set up as the predictor.
For determining the area, measurement of the longest and shortest length of the blastomere was made at the time of the first cleavage and the following formula was applied:
In which L: measure of longest side of the blastomere in μm. S: measure of shortest side of the blastomere in μm.
Consequently, the delta between the biggest and second biggest blastomere was calculates as follow:
Morphokinetic associated with blastocyst development
Logistic regression models were used to fit the effects of morphokinetic parameters on blastocyst development. The responsive variable for blastocyst development was the day 8 blastocyst outcome (blastocyst yes vs. blastocyst no). The fixed effects tested were, speed of the blastomere division (fast- vs. slow-dividing embryos; cut-off based on the results of the ROC curve as described earlier), type of cleavage (bipolar vs. multipolar), ruffling membrane (yes vs. no), lag-phase (yes vs. no), and the difference in the blastomere area at cleavage (even vs. uneven; cut-off based on the results of the ROC curve as described earlier).
Effect of oocyte vitrification on morphokinetic parameters
Logistic regression models were used to evaluate the effect of different protocols of oocyte vitrification on the morphokinetic and morpho-metric dynamics, and developmental parameters of cleaved zygotes. First, we tested the effects of oocyte vitrification protocols on the percentage of zygotes that reached first (1–2 cell), second (3–4 cell), third (5‐8 cell), and fourth (9‐16 cell) cell division. Moreover, we assessed the time (in hours) when cell divisions occurred for each vitrification protocol. Lastly, we evaluated the effects of vitrification on speed of cleavage (cut-off based on the results of the ROC curve as described earlier), type of cleavage, ruffling membrane, lag-phase, difference in the blastomere area at cleavage (cut-off based on the results of the ROC curve as described earlier), and day 8 blastocyst rate.
The association of multipolar zygotic division and blastomere size with nuclear abnormalities
To determine the impact of the type of cleavage (bipolar vs. multipolar) on the number of nuclei (mononuclear vs. anuclear or multinuclear) in the resulting blastomeres, logistic regression models were constructed. To ascertain the impact of the nuclear content (mononuclear vs. anuclear vs. multinuclear) on the area of the resulting blastomeres, linear mixed models were used. Due to a fixed effect resulting from the type of division on the size of the blastomere (p < 0.001), the linear model was built separately for blastomeres resulting from bipolar zygotic division and blastomeres resulting from multipolar zygotic divisions.
Data availability
All data generated or analyzed during this study were included in the manuscript and its supplementary information files. Raw data are available from the corresponding author on reasonable request.
References
Ferré, L. B. et al. Review: Recent advances in bovine in vitro embryo production: Reproductive biotechnology history and methods. Animal 14, 991–1004 (2020).
IETS Data Retrieval Committee. 2020 Statistics of embryo production and transfer in domestic farm animals World embryo industry grows despite the Pandemic. Embryo Technol. Newsl. 39, 17 (2021).
Ealy, A. D., Wooldridge, L. K. & McCoski, S. R. BOARD INVITED REVIEW: Post-transfer consequences of in vitro-produced embryos in cattle. J. Anim. Sci. 97, 2555 (2019).
Rabaglino, M. B. et al. Application of multi-omics data integration and machine learning approaches to identify epigenetic and transcriptomic differences between in vitro and in vivo produced bovine embryos. PLoS ONE 16, e0252096 (2021).
Mogas, T. Update on the vitrification of bovine oocytes and invitro-produced embryos. Reprod. Fertil. Dev. 31, 105 (2019).
Dujíčková, L., Makarevich, A. V., Olexiková, L., Kubovičová, E. & Strejček, F. Methodological approaches for vitrification of bovine oocytes. Zygote https://doi.org/10.1017/S0967199420000465 (2020).
Do, V. H. et al. Vitrification of in vitro-derived bovine embryos: targeting enhancement of quality by refining technology and standardising procedures. Reprod. Fertil. Dev. 31, 837–846 (2019).
Valente, R. S., Marsico, T. V. & Sudano, M. J. Basic and applied features in the cryopreservation progress of bovine embryos. Anim. Reprod. Sci. 239, 106970 (2022).
Contreras, D. A., Galina, C. S. & Chenoweth, P. Prospects for increasing the utilization of cattle embryo transfer by small-scale milk and meat producers in tropical regions. Reprod. Domest. Anim. 56, 1479–1485 (2021).
Beltrame, R. T., Barioni, L. G., Maestri, B. D. & Quirino, C. R. Economic optimization of the number of recipients in bovine embryo transfer programs. Sci. Agric. 64, 221–226 (2007).
Lundin, K. & Park, H. Time-lapse technology for embryo culture and selection. Ups. J. Med. Sci. 125, 77 (2020).
Ferré, L. B., Kjelland, M. E., Taiyeb, A. M., Campos-Chillon, F. & Ross, P. J. Recent progress in bovine in vitro-derived embryo cryotolerance: Impact of in vitro culture systems, advances in cryopreservation and future considerations. Reprod. Domest. Anim. 55, 659–676 (2020).
Gomez, E. et al. Metabolites secreted by bovine embryos in vitro predict pregnancies that the recipient plasma metabolome cannot, and vice versa. Metabolites 11, 162 (2021).
Sala, R. V. et al. Optimization of a 5-day fixed-time embryo transfer (FTET) protocol in heifers I. Manipulation of circulating progesterone through reutilization of intravaginal progesterone devices during FTET. Theriogenology 156, 171–180 (2020).
Hansen, P. J. The incompletely fulfilled promise of embryo transfer in cattle—why aren’t pregnancy rates greater and what can we do about it?. J. Anim. Sci. 98, 1–20 (2020).
Bó, G. A. & Mapletoft, R. J. Evaluation and classification of bovine embryos. Anim. Reprod. 10, 344–348 (2013).
Perkel, K. J., Tscherner, A., Merrill, C., Lamarre, J. & Madan, P. The ART of selecting the best embryo: A review of early embryonic mortality and bovine embryo viability assessment methods. Mol. Reprod. Dev. 82, 822–838 (2015).
Hasler, J. F., McCauley, A. D., Lathrop, W. F. & Foote, R. H. Effect of donor-embryo-recipient interactions on pregnancy rate in a large-scale bovine embryo transfer program. Theriogenology 27, 139–168 (1987).
Silvestri, G. et al. Preimplantation genetic testing for aneuploidy improves live birth rates with in vitro produced bovine embryos: A blind retrospective study. Cells 10, 2284 (2021).
Werner, M. D. et al. Clinically recognizable error rate after the transfer of comprehensive chromosomal screened euploid embryos is low. Fertil. Steril. 102, 1613–1618 (2014).
Blue, N. R., Page, J. M. & Silver, R. M. Genetic abnormalities and pregnancy loss. Semin. Perinatol. 43, 66 (2019).
Tutt, D. A. R. et al. Analysis of bovine blastocysts indicates ovarian stimulation does not induce chromosome errors, nor discordance between inner-cell mass and trophectoderm lineages. Theriogenology 161, 108–119 (2021).
de Sousa, R. V. et al. Biopsy of bovine embryos produced in vivo and in vitro does not affect pregnancy rates. Theriogenology 90, 25–31 (2017).
Mandawala, A. A., Harvey, S. C., Roy, T. K. & Fowler, K. E. Time-lapse embryo imaging and morphokinetic profiling: Towards a general characterisation of embryogenesis. Anim. Reprod. Sci. 174, 2–10 (2016).
Campagna, C., Sirard, M. A., Ayotte, P. & Bailey, J. L. Impaired maturation, fertilization, and embryonic development of porcine oocytes following exposure to an environmentally relevant organochlorine mixture. Biol. Reprod. 65, 554–560 (2001).
Urrego, R., Rodriguez-Osorio, N. & Niemann, H. Epigenetic disorders and altered gene expression after use of Assisted Reproductive Technologies in domestic cattle. Epigenetics 9, 803–815 (2014).
Pennetta, F., Lagalla, C. & Borini, A. Embryo morphokinetic characteristics and euploidy. Curr. Opin. Obstet. Gynecol. 30, 185–196 (2018).
McQueen, D. B. et al. Can embryo morphokinetic parameters predict euploid pregnancy loss?. Fertil. Steril. 115, 382–388 (2021).
Zhan, Q., Ye, Z., Clarke, R., Rosenwaks, Z. & Zaninovic, N. Direct unequal cleavages: Embryo developmental competence, genetic constitution and clinical outcome. PLoS ONE 11, e0166398 (2016).
de Coster, T. et al. Parental genomes segregate into distinct blastomeres during multipolar zygotic divisions leading to mixoploid and chimeric blastocysts. Genome Biol. 23, 1–29 (2022).
van Soom, A., van Vlaenderen, I., Mahmoudzadeh, A. R., Deluyker, H. & de Kruif, A. Compaction rate of in vitro fertilized bovine embryos related to the interval from insemination to first cleavage. Theriogenology 38, 905–919 (1992).
Bartolacci, A. et al. Early embryo morphokinetics is a better predictor of post-ICSI live birth than embryo morphology: Speed is more important than beauty at the cleavage stage. Zygote https://doi.org/10.1017/S0967199421000253 (2021).
Sugimura, S. et al. Promising system for selecting healthy in vitro-fertilized embryos in cattle. PLoS ONE 7, e36627 (2012).
Yaacobi-Artzi, S., Kalo, D. & Roth, Z. Association between the morphokinetics of in-vitro-derived bovine embryos and the transcriptomic profile of the derived blastocysts. PLoS ONE 17, e0276642 (2022).
Rubio, I. et al. Limited implantation success of direct-cleaved human zygotes: A time-lapse study. Fertil. Steril. 98, 1458–1463 (2012).
Magata, F. et al. Growth potential of bovine embryos presenting abnormal cleavage observed through time lapse cinematography. Theriogenology https://doi.org/10.1016/J.THERIOGENOLOGY.2019.04.031 (2019).
Barrie, A. et al. Preliminary investigation of the prevalence and implantation potential of abnormal embryonic phenotypes assessed using time-lapse imaging. Reprod. Biomed. Online 34, 455–462 (2017).
Lundin, K., Bergh, C. & Hardarson, T. Early embryo cleavage is a strong indicator of embryo quality in human IVF. Hum. Reprod. 16, 2652–2657 (2001).
Sugimura, S. et al. Time-lapse cinematography-compatible polystyrene-based microwell culture system: A novel tool for tracking the development of individual bovine embryos. Biol. Reprod. 83, 970–978 (2010).
Suzuki, R., Okada, M., Nagai, H., Kobayashi, J. & Sugimura, S. Morphokinetic analysis of pronuclei using time-lapse cinematography in bovine zygotes. Theriogenology 166, 55–63 (2021).
Somfai, T. et al. Relationship between the length of cell cycles, cleavage pattern and developmental competence in bovine embryos generated by in vitro fertilization or parthenogenesis. J. Reprod. Dev. 56, 200–207 (2010).
Vajta, G. Vitrification of the oocytes and embryos of domestic animals. Anim. Reprod. Sci. 60–61, 357–364 (2000).
Tharasanit, T. & Thuwanut, P. Oocyte cryopreservation in domestic animals and humans: principles, techniques and updated outcomes. Animals (Basel) 11, 10 (2021).
Arav, A. Cryopreservation of oocytes and embryos. Theriogenology 81, 96–102 (2014).
Morató, R., Mogas, T. & Maddox-Hyttel, P. Ultrastructure of bovine oocytes exposed to taxol prior to OPS vitrification. Mol. Reprod. Dev. 75, 1318–1326 (2008).
Gutnisky, C. et al. Morphological, biochemical and functional studies to evaluate bovine oocyte vitrification. Theriogenology 143, 18–26 (2020).
Marques, C. C. et al. Bovine oocyte membrane permeability and cryosurvival: Effects of different cryoprotectants and calcium in the vitrification media. Cryobiology 81, 4–11 (2018).
Yan, C.-L. et al. Mitochondrial behaviors in the vitrified mouse oocyte and its parthenogenetic embryo: effect of Taxol pretreatment and relationship to competence. Fertil. Steril. 93, 959–966 (2010).
Cobo, A. et al. Effect of oocyte vitrification on embryo quality: time-lapse analysis and morphokinetic evaluation. Fertil. Steril. 108, 491-497.e3 (2017).
de Gheselle, S., de Sutter, P. & Tilleman, K. In-vitro development of embryos derived from vitrified-warmed oocytes is delayed compared with embryos derived from fresh oocytes: A time-lapse sibling oocyte study. Reprod. Biomed. Online 40, 82–90 (2020).
Rienzi, L. et al. Oocyte, embryo and blastocyst cryopreservation in ART: systematic review and meta-analysis comparing slow-freezing versus vitrification to produce evidence for the development of global guidance. Hum. Reprod. Update 23, 139–155 (2017).
Sugimura, S., Akai, T. & Imai, K. Selection of viable in vitro-fertilized bovine embryos using time-lapse monitoring in microwell culture dishes. J. Reprod. Dev. 63, 353–357 (2017).
Fenwick, J., Platteau, P., Murdoch, A. P. & Herbert, M. Time from insemination to first cleavage predicts developmental competence of human preimplantation embryos in vitro. Hum. Reprod. 17, 407–412 (2002).
Isom, S. C., Li, R. F., Whitworth, K. M. & Prather, R. S. Timing of first embryonic cleavage is a positive indicator of the in vitro developmental potential of porcine embryos derived from in vitro fertilization, somatic cell nuclear transfer and parthenogenesis. Mol. Reprod. Dev. 79, 197–207 (2012).
Lonergan, P. et al. Effect of time interval from insemination to first cleavage on the developmental characteristics, sex ratio and pregnancy rate after transfer of bovine embryos. J. Reprod. Fertil. 117, 159–167 (1999).
Nahm, F. S. Receiver operating characteristic curve: overview and practical use for clinicians. Korean J. Anesthesiol. 75, 25 (2022).
Velker, B. A. M., Denomme, M. M. & Mann, M. R. W. Loss of genomic imprinting in mouse embryos with fast rates of preimplantation development in culture. Biol. Reprod. 86, 143–144 (2012).
Gutierrez-Adan, A., White, C. R., van Soom, A. & Mann, M. R. W. Why we should not select the faster embryo: Lessons from mice and cattle. Reprod. Fertil. Dev. 27, 765–775 (2015).
van Langendonckt, A. et al. Effects of supplementation with fetal calf serum on development of bovine embryos in synthetic oviduct fluid medium. J. Reprod. Fertil. 109, 87–93 (1997).
Rizos, D. et al. Bovine embryo culture in the presence or absence of serum: implications for blastocyst development, cryotolerance, and messenger RNA expression. Biol. Reprod. 68, 236–243 (2003).
Hardarson, T., Hanson, C., Sjögren, A. & Lundin, K. Human embryos with unevenly sized blastomeres have lower pregnancy and implantation rates: indications for aneuploidy and multinucleation. Hum. Reprod. 16, 313–318 (2001).
Nikolova, S. et al. Impact of sperm characteristics on time-lapse embryo morphokinetic parameters and clinical outcome of conventional in vitro fertilization. Andrology 8, 1107–1116 (2020).
Burruel, V., Klooster, K., Barker, C. M., Pera, R. R. & Meyers, S. Abnormal early cleavage events predict early embryo demise: sperm oxidative stress and early abnormal cleavage. Sci. Rep. 4, 1–10 (2014).
Grisart, B., Massip, A. & Dessy, F. Cinematographic analysis of bovine embryo development in serum-free oviduct-conditioned medium. Reproduction 101, 257–264 (1994).
Paulson, E. E., Fishman, E. L., Schultz, R. M. & Ross, P. J. Embryonic microRNAs are essential for bovine preimplantation embryo development. Proc. Natl. Acad. Sci. USA 119, e221942119 (2022).
Meirelles, F. V. et al. Genome activation and developmental block in bovine embryos. Anim. Reprod. Sci. 82–83, 13–20 (2004).
Lee, M. T., Bonneau, A. R. & Giraldez, A. J. Zygotic genome activation during the maternal-to-zygotic transition. Annu. Rev. Cell Dev. Biol. 30, 581 (2014).
Barnes, F. L. & First, N. L. Embryonic transcription in in vitro cultured bovine embryos. Mol. Reprod. Dev. 29, 117–123 (1991).
Frei, R. E., Schultz, G. A. & Church, R. B. Qualitative and quantitative changes in protein synthesis occur at the 8–16-cell stage of embryogenesis in the cow. Reproduction 86, 637–641 (1989).
de Sousa, P. A., Watson, A. J., Schultz, G. A. & Bilodeau-Goeseels, S. Oogenetic and zygotic gene expression directing early bovine embryogenesis: A review. Gamete Res. https://doi.org/10.1002/(SICI)1098-2795(199809)51:1 (1998).
Thompson, E. M., Legouy, E., Christians, E. & Renard, J. P. Progressive maturation of chromatin structure regulates HSP70.1 gene expression in the preimplantation mouse embryo. Development 121, 3425–3437 (1995).
Barnes, F. L. & Eyestone, W. H. Early cleavage and the maternal zygotic transition in bovine embryos. Theriogenology 33, 141–152 (1990).
Graf, A. et al. Genome activation in bovine embryos: review of the literature and new insights from RNA sequencing experiments. Anim. Reprod. Sci. 149, 46–58 (2014).
Cobo, A. & Diaz, C. Clinical application of oocyte vitrification: A systematic review and meta-analysis of randomized controlled trials. Fertil. Steril. 96, 277–285 (2011).
Brambillasca, F. et al. The current challenges to efficient immature oocyte cryopreservation. J. Assist. Reprod. Genet. 30, 1531–1539 (2013).
Cao, Y.-X. & Chian, R.-C. Fertility Preservation with Immature and in Vitro Matured Oocytes. Semin. Reprod. Med. 27, 456–464 (2009).
Chian, R. C. & Cao, Y. X. In vitro maturation of immature human oocytes for clinical application. Methods Mol. Biol. 1154, 271–288 (2014).
Ortiz-Escribano, N. et al. Role of cumulus cells during vitrification and fertilization of mature bovine oocytes: Effects on survival, fertilization, and blastocyst development. Theriogenology 86, 635–641 (2016).
Papis, K., Shimizu, M., Saha, S., Izaike, Y. & Modliński, J. A. Effects of vitrification of partially denuded bovine immature oocytes. Anim. Sci. Pap. Rep. 31, 14 (2013).
Ševelová, H. & Lopatářová, M. Closed system for bovine oocyte vitrification. Acta Vet. Brno 81, 201–206 (2012).
Ortiz-Escribano, N. et al. An improved vitrification protocol for equine immature oocytes, resulting in a first live foal. Equine Vet. J. 50, 391–397 (2018).
Fujihira, T., Nagai, H. & Fukui, Y. Relationship between equilibration times and the presence of cumulus cells, and effect of taxol treatment for vitrification of in vitro matured porcine oocytes. Cryobiology 51, 339–343 (2005).
Ishii, T., Tomita, K., Sakakibara, H. & Ohkura, S. Embryogenesis of vitrified mature bovine oocytes is improved in the presence of multi-layered cumulus cells. J. Reprod. Dev. 64, 95–99 (2018).
dos Santos-Neto, P. C. et al. Cumulus cells during in vitro fertilization and oocyte vitrification in sheep: Remove, maintain or add?. Cryobiology 92, 161–167 (2020).
van Soom, A., Tanghe, S., de Pauw, I., Maes, D. & de Kruif, A. Function of the cumulus oophorus before and during mammalian fertilization. Reprod. Domest. Anim. 37, 144–151 (2002).
Kidder, G. M. & Mhawi, A. A. Gap junctions and ovarian folliculogenesis. Reproduction 123, 613–620 (2002).
Eisenbach, M. Sperm chemotaxis. Rev. Reprod. 4, 56–66 (1999).
Cox, J. F., Hormazábal, J. & Santa María, A. Effect of the cumulus on in vitro fertilization of bovine matured oocytes. Theriogenology 40, 1259–1267 (1993).
Rusciano, G. et al. Raman-microscopy investigation of vitrification-induced structural damages in mature bovine oocytes. PLoS ONE 12, e0177677 (2017).
Morató, R., Izquierdo, D., Paramio, M. T. & Mogas, T. Embryo development and structural analysis of in vitro matured bovine oocytes vitrified in flexipet denuding pipettes. Theriogenology 70, 1536–1543 (2008).
Tsutsumi, R. & Huang, T. Effects of vitrification on mitochondrial distribution and spindle configuration during in vitro maturation of human germinal vesicle-stage oocytes. Fertil. Steril. 98, S90 (2012).
Sidi, S. et al. Lycopene supplementation to serum-free maturation medium improves in vitro bovine embryo development and quality and modulates embryonic transcriptomic profile. Antioxidants 11, 344 (2022).
Angel-Velez, D. et al. New alternative mixtures of cryoprotectants for equine immature oocyte vitrification. Animals 11, 3077 (2021).
Wydooghe, E. et al. Replacing serum in culture medium with albumin and insulin, transferrin and selenium is the key to successful bovine embryo development in individual culture. Reprod. Fertil. Dev. 26, 717 (2014).
Acknowledgements
The authors thank Petra Van Damme for her technical assistance. This project was funded by Bijzonder Onderzoeksfonds GOA (Geconcerteerde onderzoeksacties) 2018000504 (GOA030-18 BOF) and by by European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement number 860960. D. A.-V. was supported by funding COLFUTURO and the Ministerio de Ciencia Tecnología Innovación of Colombia. T. D.C., K.C.P. and O.B.P. are supported by the Research Foundation Flanders (FWO) (Grant Numbers 1139820N, 1228821N and 12Y5220N).
Author information
Authors and Affiliations
Contributions
Conceptualization: D.A.-V., T. D.C., O.B.P. and K.S. Data curation and formal analysis: D.A.-V., T.D.C., and O.B.P. Funding Acquisition: A.V.S. and K.S. Investigation and Methodology: D.A.-V., T.D.C., N.A.-D., A.F.-M., C.B., K.C.P. Visualization: D.A.-V., T.D.C., and A.F.-M. Writing original draft: D.A.-V., T.D.C., A.F.-M., O.B.P. and K.S. Reviewing the final manuscript: All authors read the final draft and agreed to its submission for publication. A.F.-M. designing and drew Figs. 2, 4 and 5.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary Video 1.
Supplementary Video 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Angel-Velez, D., De Coster, T., Azari-Dolatabad, N. et al. Embryo morphokinetics derived from fresh and vitrified bovine oocytes predict blastocyst development and nuclear abnormalities. Sci Rep 13, 4765 (2023). https://doi.org/10.1038/s41598-023-31268-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-31268-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
