
Abstract
Cucumber (Cucumis sativus L.), a major horticultural crop, in the family Cucurbitaceae is grown and consumed globally. Parthenocarpy is an ideal trait for many fruit and vegetables which produces seedless fruit desired by consumers. The seedlessness occurs when fruit develops without fertilization which can be either natural or induced. So far, a limited number of genes regulating parthenocarpic fruit set have been reported in several fruit or vegetable crops, most of which are involved in hormone biosynthesis or signalling. Although parthenocarpic cucumber has been widely used in commercial production for a long time; its genetic basis is not well understood. In this study, we retrieved thirty five parthenocarpy fruit-set related genes (PRGs) from bibliomic data in various plants. Thirty-five PRG homologs were identified in the cucumber genome via homology-based search. An in silico analysis was performed on phylogenetic tree, exon–intron structure, cis-regulatory elements in the promoter region, and conserved domains of their deduced proteins, which provided insights into the genetic make-up of parthenocarpy-related genes in cucumber. Simple sequence repeat (SSR) sequences were mined in these PRGs, and 31 SSR markers were designed. SSR genotyping identified three SSRs in two polymorphic genes. Quantitative real-time PCR of selected genes was conducted in five cucumber lines with varying degrees of parthenocarpic fruit set capacities, which revealed possible association of their expression with parthenocarpy. The results revealed that homologs CsWD40 and CsPIN-4 could be considered potential genes for determination of parthenocarpy as these genes showed parental polymorphism and differential gene expression in case of parthenocarpic and non-parthenocarpic parents.
Similar content being viewed by others


Introduction
Cucumber (Cucumis sativus L.) belongs to the botanical family Cucurbitaceae that also includes several other economically important crops (cucurbits) such as melon (Cucumis melo L.), watermelon (Citrullus lanatus L.), squash/pumpkin (Cucurbita spp.), bitter gourd (Momordica charantia L.) and bottle gourd (Lagenaria siceraria L.)1,2. Being a predominantly monoecious crop, a successful fruit set in cucumber depends on conditions favourable to fertilization. The yield of cucumber is reduced by either absence of pollinators or under unsuitable environmental conditions, such as diffused light, high humidity and temperature3. Breeders have considered parthenocarpy as a trait to overcome the problem of poor fruit setting caused by unfavourable pollinating conditions4.
The production of parthenocarpic fruits is an attractive technique for the development of seedless fruits independent of pollination. Seedless fruits are favoured by breeders, cultivators as well as consumers. Moreover, parthenocarpic fruits are often firmer and fleshier than their seeded counterparts5. Therefore, development of parthenocarpy or production of fruits without seeds is desirable for cucumber breeding. Parthenocarpic fruits are formed when either the ovary develops directly without fertilization or when seed abortion occurs after ovary development without producing mature seeds6. Parthenocarpy is usually driven by genetic factors; however, it can be also induced by applying different phytohormones to young inflorescences7.
Parthenocarpy is a complex trait which is controlled by various phytohormones and multiple genes regulating the synthesis, transport and signalling of those phytohormones. It can also be induced artificially via exogenous application of plant growth hormones such as auxins (2, 4-dichlorophenoxyacetic acid; naphthaleneacetic acid), cytokinins (for example, forchlorfenuron, N-(2-Chloro-4-pyridyl)-N′-phenylurea) or CCPU, and; 6-benzylaminopurine), gibberellic acids (GAs) and brassinosteroids (BRs))8. Auxin was the first phytohormone to be recognized as an inducer of parthenocarpic fruit development in citrus and strawberry4. Auxin-related parthenocarpy could be affected by genes involved in auxin biosynthesis, transport, or signalling9. GA biosynthesis and signalling play an important role in parthenocarpic fruit set10. For example, overexpression and ectopic expression of the gene for the gibberellin 20-oxidase (an enzyme involved in the synthesis of bioactive gibberellic acid) leads to the production of parthenocarpic fruit in tomato (Solanum lycopersicum) and Arabidopsis11. Cytokinins have also been reported to promote the development of parthenocarpic fruit in a variety of species including watermelon (Citrullus lanatus), pear (Pyrus pyrifolia) and kiwi (Actinidia deliciosa)12,13,14. Ethylene (ET) affects parthenocarpic fruit set by working in partnership with auxin3. Abscisic acid (ABA) may act as an antagonist of gibberellic acid or auxin to attract and maintain the sleep state of the ovaries, possibly by suppressing their transition to fruit15.
Parthenocarpic expression can also be achieved via manipulation of genes involved in hormone signalling pathways. For example, transgenic tobacco and eggplants expressing the coding region of the iaaM gene from Pseudomonas syringaepv. savastanoi, under the control of the regulatory sequences of the ovule-specific DefH9(a MADS box) gene from Antirrhinum majus, showed parthenocarpic fruit development Expression of the DefH9-iaaM chimeric transgene occurs during flower development in both tobacco and eggplant16. Similarly, Yin et al.17 demonstrated that overexpression of the DEFH9-IaaM could stimulate parthenocarpy in cucumber. Ren et al.9 reported that the overexpression of SlTIR1 resulted in parthenocarpy in tomato. Removing the function of negative regulators of auxin signalling encoded by ARF8 (AUXIN RESPONSE FACTORS) and ARF7 in Arabidopsis and tomato respectively also led to fertilization-independent fruit development18,19. Polycomb repressive complex 2 (PRC2) have been shown to contribute toward parthenocarpy10. Arabidopsis mutants defective in the PRC2-component genes have been linked to fertilization-independent seed development20. In Arabidopsis, PRC2 comprises of several genes. These genes consist of MEDEA (homolog of the Drosophila melanogaster gene Enhancer of Zeste), FIS2 (homolog of the Drosophila gene Suppressor of Zeste), FIE (homolog of Drosophila extra sex combs), and MSI1 (homolog of p55 in Drosophila)20,21,22.
Previous studies show a complex and confusing relationship between hormone responses during fruit set in cucumbers8,23,24. Recent studies on cucumber parthenocarpy have identified major loci (parth2.1, parth5.1, parth7.1, parth6.1 and parth6.2) and candidate genes (CsARF19, CsWD40, and CsEIN1)24,25,26. However, the key for assembling molecular players remains to be deciphered, and a global understanding of parthenocarpy processes is yet to be achieved. The present investigation aims to identify homologs of PRGs in cucumber with reference of PRGs which have already been reported in other crops such as Arabidopsis, tomato, fig and pear. The PRGs of various plants were retrieved from bibliomic data and used to search for PRG homologs in cucumber genome. The present investigation determination of chromosomal location, gene-structure prediction, identification of cis-regulating elements and conserved motifs, and physical and chemical analysis of the PRGs. Microsatellite markers/ simple sequence repeats (SSRs) associated with these PRGs were mined and validated in five cucumber genotypes. An expression study of the selected genes was performed through quantitative real-time PCR (qRT-PCR) in cucumber.
Results
Identification of cucumber PRGs
Based on bibliomic data, 35 PRGs were identified from various crops including tomato (Solanum lycopersicum), Arabidopsis thaliana, fig (Ficus cracia), common pear (Pyrus communis), grape (Vitis vinifera) and loquat (Eriobotrya japonica) (Table S1). The genes included SlDELLA (negative regulator of GA signalling)27, SlARFs (activation/inhibition of auxin responsive genes)18,24, SlAGAMOUS/AGL (MADS family transcription factor)28,29, SlTPL (Transducing family protein/WD40 repeat family protein)30, SlPAT (Synthesis of active gibberellic acid- natural parthenocarpy)28,31, EjYUCCA (for indole-3-pyruvate monooxygenase in auxin biosynthesis)32, FcPYR (ABA signalling pathway)8,33, FcGID1 (Gibberellin Insensitive Dwarf1—gibberellic acid receptor)34,35, , VvPISTILLATA/DEFICIENS (MADS family transcription factors- controls petal and stamen floral organ identity)10,36,37, CsLOG (Lonely Guy enzyme- conversion of nucleotide precursors into active forms)8,38, CsCKX (Cytokinin oxidase- cytokinin degradation)8,38, CsIPT (Adenylateisopentenylatetransferase-cytokinin biosynthesis)8,32, CsWD40 (WD-40 repeat family protein- cytokinin responses)24, CsCYP735A (for Cytochrome P450 monooxygenase- cytokinin biosynthesis)8,38 AtFIE (Fertilization Independent Endosperm)10,22, AtFIS (Fertilization Independent Seed)10,20, AtMEDEA (Polycomb group protein- transcriptional repression)21,39, AtMET1 (methyl transferase- methylation of symmetric CpG residues)39, and PbGA2ox (Gibberellic acid oxidase)40. The gDNA sequences of them were used as queries to identify homologous genes in the cucumber genome (9930v2.0, https://cucurbitgenomics.org/), which are listed in Table 1. Genome-wide in silico analysis revealed the 35 PRGs were distributed across all 7 cucumber chromosomes with highest number of genes on chr 3, 5, 6 (n = 7), followed by chr 4 (n = 6), chr 2 (n = 4), chr 1 (n = 3) and chr 7 (n = 1) (Fig. 1).

Graphical (scaled) representation of physical locations for parthenocarpy genes on cucumber chromosomes (numbered 1–7). Different colours of circles represent different genes.
Intron–exon structure of PRGs
Intron–exon structure of each PRG was predicted via the GSDS2.0 tool. The intron–exon organisation of each cucumber PRG and corresponding reference gene used as query is depicted in Fig. 2. In cucumber, the shortest genes included CsMADS and CsAGL (< 1 kb) while the longest one was CsARF8 (> 22 kb). CsTPL had the most exons (22) while three genes (CsDELLA, CsPYR1 and CsMADS) were intron-free with a single exon. The gene CYP78A6 from Arabidopsis (AtCYP78A6) exhibited similar intron–exon organization as that of CsCYP78A6. The rest of the genes from Arabidopsis (AtARF8, AtFIE, AtFIS2 and AtMEDEA) showed great variation as compared with their cucumber homologs. The fig genes (FcGA20OX2 and FcGID1) had the same number of exons as CsGA20OX2 and CsGID1 despite having dissimilar lengths. All the genes from tomato (except SlDELLA) showed significant variation in intron–exon structures as compared to cucumber homologs. The PISTILLATA gene had 6 intron in cucumber (CsPISTILLATA) while 7 introns in grape (VvPISTILLATA).

Intron–exon structures of cucumber PRGs and their reference genes from different crops. Exons and introns are shown by blue rectangles and thick black curved lines, respectively. Lengths of exons are fit to scale (At: Arabidopsis thaliana; Fc: Ficus cracia; Sl: Solanum lycopersicum; Vv: Vitiv vinifera).
Cis-regulatory element (CRE) analysis and identification of conserved motifs in PRGs
The CREs responsible for parthenocarpy was examined in previous studies41,42. The promoter regions (> 300 bp) of the PRGs were analysed for CREs. Eight motifs were identified including the CAAT box, CArG box, G-box, Box-4, GARE box, ABRE box, Box-II and IBOX. The abundance and distribution of CREs in promoters of the PRGs are shown in Fig. 3. The CAAT box which is considered a core promoter element43 was the most abundant CRE present in all genes (63%). It helps in influencing the frequency of transcription initiation44. GARE and ABRE are gibberellin and ABA response elements respectively which had also been identified as TALE (three amino acid loop extension) gene members in pomegranate. G-box helps in regulating transcription of multiple genes45.

Distribution of various CREs on each gene (Insert: Abundance of each CRE).
A motif sequence is a set of conserved amino acid residues which play an important role in protein functioning and are located within a certain distance from each other. These motifs help in elucidating the functions of uncharacterised proteins46. The conserved motifs were analysed via the MEME suite. In total, 8 motifs were predicted, as represented in Fig. 4 by solid blocks. The sequences of the motifs are given in Table 2. The most frequently occurring motif was motif 1 (CYYTCTYTTHTTTTYYTTTYTTYTYTT) and the least occurring motif was motif 5 (BCTSCRGCTCCWKCTGMTGC). The genes CsPIN-4, CsSEP1, CsARF8 and CsIAA9 had all eight motifs and the gene CsYUCCA had only one motif (motif 2). The gene CsARF7 and CsCKX2 had motifs only on the positive sense strand while the genes CsYUCCA, CsFIS2, CsIPT and CsAGL6 had motifs only on the negative sense strand. The functional analysis of the motifs identified was performed using GoMo (Table 2). The GO (Gene ontology) terms were assigned to the motifs with high specificity (> 80%) except motifs 6 and 8. The motifs were categorized under molecular function and biological process under different GO terms. Based on motif annotation, motif 1 was annotated to be involved in polarity specification of adaxial/abaxial axis (GO:0,009,944), primary shoot apical meristem (GO:0,010,072) and ATP binding (GO:0,005,524). As parthenocarpy is closely regulated by plant hormones, the motifs were assigned GO terms in relation to general plant metabolism and phytohormone regulating pathways. The results indicated that these motifs may play roles in biological processes and metabolic functions such as ATP binding, transcriptional activity and cytokinin mediated signalling pathway (Table 2).

Conserved motifs in nucleotide sequences of PRGs in cucumber predicted using MEME suite. Different motifs are shown in different colours.
Phylogenetic analysis
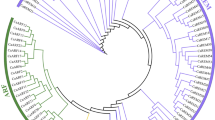
A phylogenetic tree was constructed using CDS sequences of 35 PRGs each from Arabidopsis, melon, cucumber, tomato and citrus (total 175 PRGs). The phylogenetic tree was grouped in five homology groups on the basis of maximum likelihood in different species. In general, most of the cucumber genes were clustered with melon homologs and least related to tomato homologs. Based on the phylogeny, the genes were divided into five groups (I–V) with 47, 41, 20, 41 and 26 in each group respectively (Fig. 5a).

(a) Phylogenetic trees (homology groups I–V) showing relationship among 175 PRGs from Arabidopsis (At, blue), melon (Cm, black), citrus (Cr, green), cucumber (Cs, pink), and tomato (Sl, red) (b) Distribution of cucumber PRGs among three categories (cellular component, molecular functions and biological processes) via gene ontology analysis.
Gene ontology
The protein sequences of the 35 PRGs were functionally annotated. The annotation was categorized into three categories based on three aspects: biological process, molecular function, and cellular component (Fig. 5b). The majority of the proteins belonged to the category biological process (GO:0,008,150) (Table S2). The proteins were involved in functions of metabolic process (GO:0,008,152) (n = 30), cellular process (GO:0,009,987) (n = 30), followed by cellular metabolic process (GO:0,044,237) (n = 29) and organic substance metabolic process (GO:0,071,704) (n = 26). The GO terms associated with molecular function (GO:0,003,674) predicted several categories including binding (GO:0,005,488) (n = 28) followed by catalytic activity (GO:0,003,824) (n = 21), organic cyclic compound binding (GO:0,097,159) (n = 22) and heterocyclic compound binding (GO:1,901,363) (n = 22). The cellular component category (GO:0,005,575) exhibited occurrence of proteins in various sub-cellular locations such as cellular anatomical entity (GO:0,110,165) (n = 25) followed by intracellular anatomical structure (GO:0,005,622) (n = 22). The functional enrichment of the genes was performed using Bonferroni method with threshold value of 0.05. The analysis categorized the genes into 4 categories as molecular function (n = 9), biological process (n = 40), cellular component (n = 1) and KEGG (Kyoto encyclopedia of genes and genomes; n = 2) (Fig S1). The detailed results of functional enrichment analysis have been provided in Table S3. Forty GO IDs related to biological process, nine GO IDs related to molecular function and one GO ID related to cellular component were identified. The highly enriched GO ID under biological process was biological regulation (GO:0,065,007) (n = 20) followed by regulation of macromolecule metabolic process (GO:0,060,255) and regulation of metabolic process (GO:0,019,222) with n = 14 each. In metabolic process, the maximum number of genes were assigned to double-stranded DNA binding (GO:0,003,690) with n = 7. Fourteen genes were placed under single GO ID of nucleus (GO:0,005,634) under cellular component. The KEGG pathway included plant hormone signal transduction and diterpenoid biosynthesis pathways.
KEGG pathway
The KEGG pathway analysis revealed that the most genes were involved in plant hormone signal transduction and biosynthesis of secondary metabolites (Table 3). The genes encoding proteins cytokinin oxidase (CKX) and gibberellin oxidase (GAox) were involved in metabolic pathways and gene encoding indole acetic acid (IAA) and auxin response factors (ARF) were involved in plant hormone signal transduction. The gene CsPAT had a role in biosynthesis and metabolism of amino acids (tyrosine, phenylalanine) and alkaloid biosynthesis. The diterpenoid biosynthesis pathway involved the gibberellin oxidase genes and zeatin biosynthesis pathway involved the cytokinin oxidase and cytochrome-P450 monoxygenase genes. The detailed metabolic pathways are shown in Supplementary Fig. S2 (a-d). Among them, those involved in GA biosynthesis/signalling pathways included DELLA, GA20OX, GA2OX, PAT and GID. The metabolism of cytokinin is regulated by genes IPT, CYP735A, LOG, RR and CKX. The auxin pathways included genes such as YUCCA and ARF. The detailed functions of these genes were elucidated via functional enrichment and homology modelling.
Physical and chemical properties and homology modelling of PRG proteins
The physical and chemical properties of proteins encoded by the 35 PRGs were analysed including chromosomal location, length, PI (isoelectric point), molecular weight, instability, instability index, aliphatic index and GRAVY (Grand Average of Hydropathicity) index (Table 1). The length of proteins encoded by gene CsMET1 was the highest while that of CsGAST1 was the shortest. The proteins had an average PI value of 6.847 (ranged from 4.78 to 9.24). All the proteins had molecular weight higher than 2 kDa. The average molecular weight was 82,429.405 Da. The percent composition of essential amino acids in the proteins is given in Table S6. Some of the proteins appeared unstable in nature based on instability index of ProtParamExpasy > 40 except those encoded by CsYUCCA, CsPIN-4, CsGA20OX, CsCKX1, CsCKX2, CsGA20OX2, CsGA2OX1, CsGA2OX2, CsIAA, CsAGL6, CsTPL and CsPAT. The average aliphatic and GRAVY index were observed to be between 82.45 and − 0.3131, respectively.
The structure of PRG proteins was predicted via homology modelling in Phyre2, which uses the alignment of Hidden Markov Models via HMM-HMM search to significantly improve the accuracy of alignment47. The template proteins used for modelling along with percentange of confidence for homology and conformational states are given in Table S4 and S5. The essential amino acid composition of the proteins has been provided in Table S6. Of the total proteins, structures of 27 proteins exhibited 100% confidence (Fig. 6). The prediction of the secondary structure of PRG by the protein homology revealed that the structures of the proteins predominantly comprised of α-helices (8.38–69.23%), extended strands (2.27–23.78%), β-turns (0–9.19%) and random coils (15.53–66.02%) (Table S5). The only protein without any β-turn was encoded by gene CsSEP1. The proteins encoded by genes CsGA2ox1 and CsGA20OX2; and CsARF7 and CsARF8 shared similar structure (composition of α and β structures) but were different in their essential amino acid composition (Table S6). The functional role of the proteins was also determined during the homology modelling. The proteins were identified under several PDB header and PDB molecules (Table S4). Most of the proteins showed the functions in components of either plant development and signalling pathways such as sepallata, or auxin response factors or as constituents of enzymes involved in metabolism of plant hormones such as gibberellins, cytokinins and indole-acetic acid. The overall secondary structures of PGR proteins gave insights into the different domains such as catalytic domain, binding domain, N-terminal and C-terminal along with the presence of α and β structures. The homology modelling might help in the future to develop point mutation, and identifying master regulator for regulation. These PRG proteins could help to achieve specific targets by their use in genetic engineering tools such as CRISPR and RNAi (RNA interference) studies. Hence, all the predicted protein structures could be considered highly reliable offering a preliminary basis for understanding the molecular function of parthenocarpy-related proteins along with regulation by other factors.

Predicted structures of proteins with 100% confidence level for the genes (a) CsYUCCA (b) CsDELLA (c) CsFIS2 (d) CsLOG (e) CsFIE (f) CsGA20OX (g) CsARF8 (h) CsPYR1 (i) CsGA2ox1 (j) CsGA20OX2 (k) CsCKX2 (l) CsARF7 (m) CsCYP735A1 (n) CsEIN1 (o) CsIAA (p) CsPIN-4 (q) CsIPT (r) CsIAA (s) CsCKX1 (t) CsWD40 (u) CsMET1 (v) CsGH3 (w) CsCYP78A6 (x) CsGAST1 (y) CsGID1 (z) CsPAT (aa) CsTPL.
Protein–protein interaction (PPI) network of PRG homologs
The PPI network for the PRGs was retrieved through STRING and clustered via k-means clustering. Three interconnected networks were identified (Fig. 7). The network constituted 35 nodes and 22 edges with average node degree of 1.26 and interaction score > 0.4. Most of the interactions generated were based on either text mining or experimentally derived depicted by green and pink lines respectively. The other node colours represented various interactions which included teal (from curated databases), blue (gene co-occurrence), dark green (co-expression) and lilac (protein homology). The clustering divided the proteins into five clusters with average local clustering coefficient of 0.41. The proteins in the same cluster shared similar biological function, such as red (response to gibberellin), yellow (WD repeat-containing domain superfamily), green (cytokinin metabolism), cyan (phosphoproteins) and blue (proteins without any significant clustering co-efficient). The results indicated that the proteins encoded by genes CsDEFICIENS and CsDELLA occupied the central positions in two different clusters. The CsDEFICIENS interacted with CsPISTILLATA, CsSEP1 and CsFIE. Both PISTILLATA and SEP1 (SEPALLATA) are MADS box transcription factor proteins48,49. The protein encoded by CsDELLA was related to proteins having functions as gibberellin oxidase (encoded by CsGA20OX, CsGA2OX, CsGA20OX2), cytokinin oxidase (encoded by CsCKX2, CsCYP735A2), Gibberellin Insensitive Dwarf receptor (encoded by CsGID) and lonel guy enzyme (encoded by CsLOG). The CsAux/IAA and CsARF8 were connected with CsIAA9 and CsGH3 respectively.

Protein–protein interactions (PPI) of parthenocarpy related genes constructed using Cytoscape. Dotted lines represent edges between clusters.
Evaluation of SSR markers
Simple sequence repeats (SSRs) were mined in the genomic, coding and cDNA sequences of PRGs. One hundred and four SSRs were identified, which are presented in Table S7. The distribution of the 104 SSRs across genomic, cDNA and coding sequences are shown in Fig. 8a. Most SSRs are harboured in genomic sequences which included mono-, di-, tri-, tetra- and penta-nucleotide repeats and in compound formation (Fig. 8b). The most abundant form of SSRs was as mononucleotide repeats followed by dinucleotide repeats and compound SSRs. The distribution of SSRs across individual PRG in case of genomic, cDNA and coding sequences is shown in Fig. 8c. The maximum number of SSRs were detected in PISTILLATA followed by WD40 and ARF8. The SSR markers were validated via PCR amplification and product separation via PAGE (Polyacrylamide gel electrophoresis). A total of 31 pairs of primers were designed based on these markers (Table S8). Out of the total, three primers for the genes CsPIN-4 and CsWD40 showed polymorphism within the parental genotypes i.e. Punjab Kheera-1, Gy-14, PBRK5, Punjab Naveen and AVCU1303 (Fig. 9).

(a) Distribution of SSR markers among genomic, cDNA and coding sequences of PRGs (b) Abundance of mono-, di-, tri-, tetra-, penta-nucleotide repeats and compound SSRs in genomic sequences of PRGs. (c) Number of SSRs discovered from genomic, cDNA and coding sequences of various PRGs.

PAGE image showing parental polymorphism (L: 100 bp ladder; 1: Punjab Naveen; 2: Gy-14; 3: AVCU1303; 4: Punjab Kheera-1; and 5: PBRK5). Polymorphic markers are shown in red box.
Expression analysis of PRGs in five cucumber lines
We examined the expression of nine PRGs (CsPIN-4, CsIAA, CsMEDEA, CsDEFICIENS, CsCKX2, CsWD40, CsDELLA, CsPISTILLATA, and CsCYP78A6) in five genotypes of cucumber including Gy-14 (gynoecious and non-parthenocarpic), AVCU1303 (sub-gynoecious and non-parthenocarpic), Punjab Naveen (monoecious and non-parthenocarpic), PBRK5 (monoecious and weak parthenocarpic) and Punjab Kheera-1 (gynoecious and parthenocarpic) using qRT-PCR (Fig. 10). Leaf samples were collected from each variety at 7, 14 and 21 days after flowering (DF). The nine PRGs chosen for validation were selected based on their function. All the genes selected were components of different pathways (phytohormone metabolism and signalling, reproductive development and regulation of biological processes) and had fallen under different GO terms (Table S2). The leaf samples were chosen for the study as a study conducted by Wang et al.40 revealed that the tissue specific expression of GA20ox2 was the maximum in leaf sample of pear. CsPIN-4 and CsDEFECIENS were down-regulated in parthenocarpic cucumber ‘Punjab Kheera-1’ with high fold changes (~ 2) (Fig. 10a and d). The genes CsIAA, CsCKX2, CsWD40, CsDELLA, CsPISTILLATA and CsCYP78A6 were down-regulated in both parthenocarpic and non-parthenocarpic genotypes; however, the decrease in expression level was less in parthenocarpic as compared to non-parthenocarpic ones. The gene CsMEDEA was positively regulated in all genotypes except Gy14 (non-parthenocarpic) and Punjab Kheera-1(parthenocarpic) with slightly negative gene expression.

Fold changes in gene expression level of different parthenocarpy related genes at different days after flowering in cucumber (a) CsPIN-4 (b) CsIAA (c) CsMEDEA (d) CsDEFICIENS (e) CsCKX2 (f) CsWD40 (g) CsDELLA (h) CsPISTILLATA (i) CsCYP78A6.
The fold change was the highest at 21 DF for all the genes except CsPISTILLATA in case of non-parthenocarpic genotype. The expression level was high at 7 DF, which decreased at 14 DF and again increased up to 21 DF. The inconsistency in the patterns followed by the expression level of the PRGs was consistent with the results of Li et al.23, Su et al.24 and Wu et al.8. In case of CsPISTILLATA gene, the expression level increased gradually from 7 to 21 DF. The genotype PBRK5 (parthenocarpic and monoecious) had all the genes positively expressed except CsCKX2 with slight negative fold change (− 0.12) at 21 DF. Thus, the genes CsPIN-4, CsWD-40 and CsPISTILLATA showed greater degree of fold change i.e. enhanced expression in parthenocarpic genotypes as compared to non-parthenocarpic genotypes. The intron–exon organization of the three genes showed high degree of variation in their genetic structure (Fig. 2). The genes CsPIN-4, CsWD-40 and CsPISTILLATA were 2 kbp, 16 kbp and 3 kbp in length with 10, 18 and 6 exons respectively (Fig. 2). Moreover, the genes CsPIN-4 and CsPISTILLATA were located under the homology group I and the gene CsWD-40 was located in homology group IV tree (Fig. 5a). The genes CsWD40 and CsPIN-4 also showed parental polymorphism (Fig. 9) making them putative candidate genes for parthencarpy in cucumber.
Discussion
Parthenocarpy comprises an important horticultural trait in many commercially grown fruit and vegetable crops. Due to its complex mechanism regulated by various genetic and environmental factors, the process of parthenocarpy is not completely understood in cucumber. Recent studies pertaining to parthenocarpy in cucumber included reports of major QTLs linked to parthenocarpy24,25,26 and transcriptome analysis of phytohormone biosynthesis and signal transduction genes8. The current study focused on the identification of genes regulating parthenocarpy via various pathways previously identified in other crop plants. A total of 35 PRG homologs were identified in cucumber which were distributed along all the seven chromosomes. Of all the genes, CsPAT and CsMET1 were mapped in the regions of two cucumber parthenocarpy QTL, parth4.1 and parth5.1, respectively26. This point lays down the foundation that there are many other genes which might plays significant role in regulation of parthenocarpy.
The comprehensive phylogenetic analysis was performed using Mega X software to understand the evolutionary significance of PRGs which clustered the genes into five homology groups on the basis of sequence similarity (Fig. 5a). The phylogenetic analysis showed that genes DEFICIENS, PISTILLATA, SEP1, AGL6, MADS, AGAMOUS, FIE, GH3, PIN4, and MET1 were clustered in homology group I except AtPISTILLATA, AtAGL6 and SlAGL6. The AtPISTILLATA was present in homology group IV along with IAA and CYP78A6 genes and AtAGL6 and SlAGL6 were clustered in homology group V. The genes belonging to cucumber present in group I showed homology with the same genes of other plants, except CsAGL6 which showed homology with MADS of melon, citrus and tomato. In previous reports, the MADS showed putative function in relation to flowering41. The MADS genes are considered homeotic genes and primarily their function was involved in determination of identification of flower concentric whorls50. Similarly CsMEDEA closely shared common clade with CsLOG gene whose function had been identified in expression of cytokinin biosynthesis genes related to anthesis51. It could be concluded that CsAGL6 might have direct role in fertilization that related with seed development. The phylogenetic analysis showed that homology groups I and V were mainly related to fertilization independent seed formation and embryogenesis (Fig. 5a). However groups II and IV were related with genes activated with some other modification such as methytransferases and group III genes were related with auxin regulation. Previously, Wang et al.34 performed the phylogenetic analysis of GA20ox2 gene in pear with that of Arabidopsis, apple, tomato, citrus, rice and grape depicting the GA20ox2 to be closely linked with fertilisation. Similarly in current study revealed that GA20ox2 gene in cucumber was closely linked to melon placed under group V (CsFIS2, CsGID1, CsGA20OX, CsGA2ox1) which implies this group might directly play role in fertilization and help to achieve the parthenocarpy in cucumber.
Intron–exon structure helps in identifying evolutionary changes. The exon–intron pattern of the DNA sequence was explored and plotted with the phylogenetic tree to provide some insight into the evolutionary gene structure. Further to understand the genic level structure, the genetic organization of the candidate group (homology group I and IV) was determined by analysis of intron and exon structure. The current study indicated that CsSEP1 gene in cucumber (group I) comprised of seven exons (Fig. 2, and 5a). Yu et al.52 analysed the gene structure by studying the exon and intron pattern of SEP1 and SEP3 genes for agronomical traits, inferring that except for SEP1/2-like genes in Brassicaceae, all the genes had eight exons. AGL6 subfamily (group I; Fig. 5a) consisted of two exons in cucumber (Fig. 2). The number of introns in coding sequences of ARF genes of Vitis vinifera and other family members range from one to three. In cucumber, CsARF7 and CsARF19 genes were predicted to have 12 introns. The result also revealed some variation in exons and introns number and length in different branches representing PRGs of homology groups I to V. In the homology group I, the number of exons and introns of cucumber PRGs ranged from 0 to 13 and 1 to 12 respectively (Fig. 2). They were also connected with the same common direct ancestor from melon and Arabidopsis (model plant) (Fig. 5a). The number of exons and introns gradually increased with increase in nodes and in homology group I, CsFIE had 13 exons and 12 introns. The overall analysis of phylogenetic tree and intron exon pattern revealed that group IV and V are highly impactful for the further study. The intron–exon structure pattern showed modification in the genes during evolution and the shuffling of intron in genes supported the neo-functionalization of genes across various taxa52.
The further evolutionary pattern and footprints were also checked by identifying conserved motifs. The conserved motifs were identified in cucumber PRGs (Fig. 4) which represented conserved sequences of amino acids across different genes whose function was assigned via GOMO analysis (Table 2). The result showed that the cucumber PRGs containing all the conserved protein motifs (motif 1–8) were present in homology groups I and IV (Table 2; Fig. 5a). According to phylogenetic analysis the groups I and IV, the genes CsSEP1, CsPIN-4, CsIAA9 and CsARF8 contained all eight types of motif pattern. However, the group III consisted of a few motifs with CsYUCCA having a single motif showing the loss of the protein motifs during evolution. The functional analysis of the motifs assigned their role in general plant metabolism except motif 4 (HARRAAAARAAAAGAAAAGRAAARRARA) which was involved in cytokinin mediated signalling pathway (GO:0,009,736). The motif was discovered in all genes except CsIAA, CsAGL6, CsCYP78A6, CsYUCCA, CsMEDEA, CsDEFICIENS, CsIAA, CsARF7 and CsLOG (Fig. 4). Thus, most of the genes were involved in phytohormone signalling pathways. The previous studies by Li et al.53, Fu et al.54, Su et al.8 and Sun et al.55 identified various conserved motifs in genes controlling parthenocarpy in plants such as Arabidopsis, grapevine and cucumber.
The transcriptional regulation can be better understood by the Cis-regulatory elements which are essential transcriptional regulatory units present in the promoter region of the sequence (Fig. 3). In the present study, 8 CREs were identified including CAAT box, G-box, BOX-4, GARE box, ABRE box, CArG box, IBOX and BOX-II. The CAAT box was the most abundant sequence that was present in all genes with the basic function of CAAT box being in endosperm or seed development56. The presence of CAAT box in all the homologs confirms that the motifs are conserved in plants. The G-box is one of the best characterized CREs in plants57,58,59. It plays an important role in fruit specific expression and has been identified in diverse set of unrelated genes, such as those regulated by visible and ultraviolet light60, ABA61, methyl-jasmonate and anaerobiosis and has a role in ethylene induction as well as in seed-specific expression. It is also known as ABRE (abscisic acid -responsive element)62. Studies have indicated that the G-box elements cannot act alone and require additional CREs for their function63,64. This statement supports the fact that the promoter region of cucumber PRGs contains a number of CREs required for the high and specific expression of the gene in fruit tissues65. Our study found that the G-box was located in CsDELLA, CsFIE, CsMET1, CsARF19, CsPYR1, CsCKX2, CsAGL6, CsTPL and CsGAST1. Their presence indicates the environmental factors such as light may play a role during parthenocarpic fruit formation. ABRE (CCACGTGG) motifs had been reported to be involved in abscisic acid regulation and are regulated by calcium66,67. ABRE related elements had also been detected in A. thaliana pathogenesis related sequences68. GARE motifs are gibberellin-responsive elements present68. The presence of ABRE and GARE motifs in the PRGs indicated that role of plant hormone signals’ crosstalk in the regulation of parthenocarpy. CArG constituted potential MADS domain protein binding sites regulating gynoecium development69. In vitro and in vivo assays had shown that MADS proteins bind as dimers to CArG boxes, with the consensus sequence CCA[A/T]6GG (SRF-type) or C[A/T]8G (MEF2-type)70. Certain MADS proteins such as AGAMOUS-LIKE-15 (AGL15) preferred longer MEF2- type binding site71. Besides CAAT box, the presence of such CREs which are regulated by light and hormonal interactions indicated that plant hormones and environmental factors interact with each other during fruit ripening process72. The findings suggest a complex network of regulation of parthenocarpy in cucumber.
Furthermore, the GO analysis was performed to categorize genes according to their origin/function. Biological process defines a gene based on its biological objective to which the gene or its product contributes; molecular function is defined as the biochemical activity of a gene product and cellular component refers to the cellular location where a gene product is active73. The genes CsGA20OX and CsGA20OX2 were involved in gibberellin-20-oxidase activity (GO:0,045,544) (Table S3). Besides these two genes, CsGA2ox1 was involved in gibberellin metabolic process (GO:0,009,685). The genes CsCKX1 and CsCKX2 were involved in cytokinin dehydrogenase activity (GO:0,019,139). The gibberellic acid (GA) synthesis or signaling genes have important roles in development of parthenocarpic fruit. In Arabidopsis, the overexpression of GA2ox induces seed abortion74. The gene is known to encode an enzyme which inactivates GA75. It has been previously reported that a deficiency of GAs leads to reduced seed growth due to poor utilization of assimilates76. The CKX genes involved in degradation of cytokinin were expressed less in highly parthenocarpic cucumber as compared to weak parthenocarpic cucumber which indicated that downregulation of CKX induced parthenocarpy8. Backiyarani et al.77 carried the GO analysis in Musa for parthenocarpy related genes. They showed that the majority of the genes were involved in regulation of cellular macromolecule biosynthesis process and transcriptional regulatory activity. In case of PRGs in zucchini (Cucurbita pepo L.), metabolic process and cellular component were the most represented groups78. Chen et al.79 performed GO analysis of the differentially expressed genes involved in parthenocarpy in case of eggplant. The majority of the genes belonged to membrane-bound organelle, DNA integration, RNA-directed DNA polymerase activity, nucleic and metabolic process, plasma membrane, and nucleic acid binding categories.
The expression profiles of various genes were studied. The genes CsPIN-4, CsDEFICIENS and CsWD-40 were negatively expressed in Punjab Kheera-1 (Fig. 10a, d, f). Similar results were reported by Ong-Abdullah et al.80 who showed that loss-of-function mutation in DEFICIENS gene in Elais guineensis resulted in parthenocarpy. DEFICIENS had similar function to PISTILLATA; it is a B class MADS-box gene regulating petal/stamen identity in snapdragon81. Similarly, the loss of function of tomato DEFICIENS resulted in parthenocarpy, together with abnormal stamen differentiation82. The gene PI (PISTILLATA) is associated with parthenocarpic fruit development in apple (Malus domestica) but not in Arabidopsis83. The gene CsDELLA was negatively expressed at later stages in parthenocarpic genotypes (Punjab Kheera-1 and PBRK-5) (Fig. 10g). In tomato, the loss-of-function of DELLA gene (procera (pro) mutation) corresponding to a single non-synonymous substitution in the GRAS domain of the SlDELLA displayed enhanced gibberellic acid phenotypes including parthenocarpy84. The PIN-FORMED (PIN) protein family is responsible for auxin efflux transport and the PIN genes are involved in various developmental processes including embryogenesis, shoot and root morphogenesis, gravitropism, and phototropism85. In Arabidopsis stem cells, PIN regulates the expression of the WUSCHEL transcription factor, which indicates the importance of critical auxin gradient/transport to control vital root and shoot stem cell regulators86. Silencing of SlPIN4 had been reported to cause precocious ovary development resulting in parthenocarpic fruit in tomato87. The gene CsMEDEA was positively regulated in all non-parthenocarpic genotypes and negatively regulated at intermediate stage in parthenocarpic genotypes. The gene encodes a polycomb group protein which is directly associated with promoter region of PHE1 which is a MADS-box gene21. The MEDEA mutants had shown suppression in seed abortion indicating the expression of MEDEA as an essential regulator in seed development21. The WD40 repeat proteins play multiple roles in cellular processes, including cell cycle regulation, cell apoptosis, autophagy, gene transcription, signal transduction, histone modification, DNA damage repair, RNA modification, cytoskeletal assembly, and chromatin assembly88. The gene CsWD40 has been described as an ortholog of WD40 in Arabidopsis which plays important role in cytokinin response and has been described as a promising candidate gene related to parthenocarpy89,90. The reports were consistent with our study. The genes CsWD-40 and CsPIN-4 showing expression level also showed parental polymorphism using SSR markers. Thus, the genes CsPIN-4 and CsWD-40 could be considered as potential candidate genes to determine parthenocarpy.
Conclusions
Although parthenocarpy is an important agronomic trait and has been used in production for a long time, the mechanisms of parthenocarpic fruit set seem complex. Though regulated by phytohormones, the mechanism is difficult to understand and the phenotypic demarcation is difficult as environmental factors play an important role in regulating fruit set. Thus, the study was carried out to identify the genes involved in parthenocarpy in cucumber. A total of 35 genes were identified via homology based approach in cucumber. Majority of the genes were involved in phytohormone synthesis, regulation and signalling. Phylogenetic analysis grouped the parthenocarpy related genes in different genera into five major homology groups clustering genes based on their functioning and phylogeny. The genes CsDEFICIENS, CsPISTILLATA, CsWD40 and CsPIN-4 were negatively expressed with high fold changes (~ 2) in parthenocarpic genotypes. Moreover, the genes CsWD-40 and CsPIN-4 also exhibited parental polymorphism. Thus these two genes could be used as candidate genes for determining parthenocarpy in cucumber.
Materials and methods
Identification and sequence retrieval of parthenocarpy related genes in cucumber
We reviewed the literature and identified PRGs from various fruit or vegetable crops that are either directly or indirectly involved in regulation of parthenocarpy. The crop plants included Cucumis sativus L. (cucumber)8,24,26, Solanum lycopersicum L. (tomato)30,74,81,82,83,84, Pyrus communis L. (pear)44,85,86, Ficus carica L. (fig)33,87; and across various other taxa10,21,32,39,47,88. Genomic DNA sequences of those PRGs in the cucumber genome were obtained through BLASTn in several databases Ensembl Plants, Cucurbits Genome Database and NCBI , which were further cross-verified by using BLASTp with default settings (expected threshold 0.05) and percentage identity more than 80% and e-value less than zero on query sequences using Ensembl Plant database (https://plants.ensembl.org/Multi/Tools/Blast). Only top hits were selected. The genes were plotted onto the seven chromosomes of cucumber in an orderly manner from the short-arm to the long-arm telomere using Phenogram Plot (http://visualization.ritchielab.org/phenograms/plot).
Intron–exon gene structure of PRGs
Positions of exons and introns of these cucumber PRGs were determined based on genomic information. Full length genomic (gDNA) and coding sequences (CDS) of cucumber PRGs retrieved from EnsemblPlant (https://plants.ensembl.org/Multi/Tools/Blast) were further utilized for the determination of exon–intron organizations of these genes using Gene-Structure Display Server GSDS2.0 (https://gsds.cbi.pku.edu.cn)91.
Cis-regulatory element analysis and identification of conserved motifs
Cis-regulating elements (CREs) of PRGs were analysed to explore the DNA binding domains in the promoter region. The genomic sequence of each gene (> 300 bp) upstream of the transcription start site was retrieved from NCBI database (https://www.ncbi.nlm.nih.gov/). The analysis of both sense and anti-sense strands of promoter sequences was carried out using Plant CARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)92 and PLACE (https://www.dna.affrc.go.jp/PLACE/?action=newplace)93. The conserved motifs were discovered using the MEME suite (https://meme-suite.org/meme/tools/meme)94 with parameters motif width ranging from 6 to 50 and number of sites in sequences for each motif ranging from 2 to 200. The maximum number of motifs to be found was set at 8. The function of each motif was further elucidated by submitting the motif sequence to GoMo (Gene ontology for motifs) version 5.5.0 (https://meme-suite.org/meme/tools/gomo)95 and significant threshold and number of scores shuffling rounds were set at 0.05 and 1000 respectively for annotation of the motifs.
Phylogenetic analysis
The PRGs for phylogenetic analysis were considered from 5 different plants (Arabidopsis, melon, cucumber, tomato and citrus) each having 35 PRGs (total 175 PRGs). The nucleotide sequences of PRGs from Arabidopsis, melon, cucumber, tomato and citrus were aligned with gap opening and gap extension penalties of 10 and 0.1, respectively, using ClustalW. A Maximum-Likelihood method was used to develop a cladogram of all the sequences. The associated taxa clustered together in the bootstrap test of 1000 replicates. The phylogenetic tree was constructed using MEGA X software96 and visualized through iTOL Interactive Tree of Life (https://itol.embl.de/).
Gene Ontology (GO) analysis and KEGG pathway annotation
The functional prediction of PRGs and the analysis of annotation data were done using BLAST2GO tool (https://www.blast2go/com/)97. The amino acid sequences of parthenocarpic genes were imported into BLAST2GO program to follow these three steps i.e., (i) BLASTP against protein database of NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (ii) mapping and retrieval of gene ontology terms associated with BLAST search (iii) annotation of GO terms associated with each query to relate the sequences to known protein function. The Gene Annotation (GO) was categorized into three classes: cellular components, biological processes and molecular functions. The functional enrichment of the genes was performed using GProfiler (https://biit.cs.ut.ee/gprofiler/gost) using Bonferroni correction method with user threshold of 0.05 and numeric IDs treated as ENTREZGENE. Additionally, the KEGG mapping (https://www.kegg.jp/kegg/mapper/) was done to display enzymatic functions in the context of the metabolic pathways in which they participate98.
Physical and chemical properties, homology modelling and protein–protein interaction network of PRG proteins
The physical and chemical properties of the proteins involved in parthenocarpy were examined using ProtParamExPasy server (https://web.expasy.org/protparam/)99. The properties included length, molecular weight, instability index, PI value, aliphatic index and Grand Average of Hydropathicity index (GRAVY). The sub-cellular location of the proteins was determined through ProtComp version 9.0 server (http://www.softberry.com/) and the Pfam domains were predicted via Pfam 35.0(http://pfam.xfam.org/) based on profile Hidden Markov Models100. The amino acid sequences of all the proteins were fed in Phyre2 (Protein Homology/analogY Recognition Engine; http://www.sbg.bio.ic.ac.uk/phyre2) for predicting the protein structure by homology modelling under ‘expert’ mode using HH-search alignment algorithm101. The search was performed in normal mode of Phyre2. The protein structure of all the proteins modelled at > 90% confidence. The conformational states of the proteins were predicted using SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html) with output width 70, similarity threshold 8 and window width 17. The amino acid sequences were submitted to STRING v11.5 (https://string-db.org/), a pre-computed database for the exploration of protein–protein interaction (PPI) using STRING network type with medium confidence (0.400) and 5% false discover rate stringency. The PPI network was retrieved using k-means clustering with the maximum number of clusters set to five.
SSR mining and evaluation
The SSRs were mined using MISA web tool (https://webblast.ipk-gatersleben.de/misa/)102. The coding sequences in FASTA format were uploaded in the MISA-web tool. A specific project name was specified and SSR search parameters were set as present by default. The file output parameter was generated as Misa. The primers for the SSRs were designed using PolyMorphPredict web-tool (http://webtom.cabgrid.res.in/polypred/)103. The list of the primers is provided in supplementary Table S1. The primes were procured from Integrated DNA Technologies Inc., USA. The amplification was performed using profile: initial denaturation at 95 °C for 5 min followed by 35 cycles of denaturation at 95 °C for 40 s, annealing for 40 s, extension at 72 °C for 40 s and final extention at 72 °C for 7 min and hold at 4 °C.
qRT-PCR analysis of cucumber PRGs
Five cucumber lines with varying degrees of parthenocarpic fruit-set capacities were used to evaluate the association of the expression of PRGs. These genotypes included Gy-14 (gynoecious and non-parthenocarpic), AVCU1303 (sub-gynoecious and non-parthenocarpic), Punjab Naveen (monoecious and non-parthenocarpic), PBRK5 (monoecious and weak parthenocarpic) and Punjab Kheera-1 (gynoecious and parthenocarpic). The plants were grown under poly-house conditions (average temperature 30–35 °C). Three biological replicates for each genotype were taken for RNA isolation. The relative expression of selected PRGs was examined using quantitative real-time PCR (qRT-PCR). The leaf samples from test materials were collected at different time intervals: before flowering (control) and 7, 14 and 21 DF (Days after flowering). Total RNA was extracted using the Trizol™ reagent method and stored at − 80 °C. The cDNA was synthesized using the Thermo Scientific First Strand cDNA Synthesis Kit following manufacturer’s protocol. The quality and integrity of the total RNA and cDNA was checked via agarose gel electrophoresis and spectroscopic method using NanoDrop 2000D (NanoDrop Technologies, Wilmington, DE, USA).
The primers used for qRT-PCR were designed using Primer3 tool (https://bioinfo.ut.ee/primer3/) and validated for hairpin formation via OligoCalC (http://biotools.nubic.northwestern.edu/OligoCalc.html). Information of all primers used in this study is provided in supplemental Table S9. The cucumber 18S rRNA gene (GenBank ID: X51542.1), was used as an internal control104. qRT-PCR was performed with the KAPA SYBR FAST qPCR Master Mix kit (Kapa Biosystems). The relative gene expression was calculated using the 2−ΔΔCT method following Livak and Schmittgen105. For each sample, there were three biological and three technical replicates.
Data availability
The datasets generated and/or analysed during the current study have been provided as either in the text or supplemental materials. The genomic DNA, cDNA sequences or deduced protein sequences are publicly available in the cucurbit genomics (https://www.cucurbitgenomics.org/) website.
References
Renner, S. S. & Schaefer, H. Phylogeny and evolution of the cucurbitaceae. In Genetics and Genomics of Cucurbitaceae (eds Grumet, R. et al.) 13–23 (Springer International Publishing, 2017). https://doi.org/10.1007/7397_2016_14.
Chomicki, G., Schaefer, H. & Renner, S. S. Origin and domestication of Cucurbitaceae crops: insights from pylogenies, genomics and archaeology. New Phytol. 226, 1240–1255. https://doi.org/10.1111/nph.16015 (2020).
Martinez, C., Manzano, S. & Megías, Z. Involvement of ethylene biosynthesis and signalling in fruit set and early fruit development in zucchini squash (Cucurbita pepo L.). BMC Plant Biol. 13(1), 139. https://doi.org/10.1186/1471-2229-13-139 (2013).
Gustafson, F. G. Auxin distribution in fruits and its significance in fruit development. Am. J. Bot. 26, 189–194 (1939).
Fabrice, R. B., Michel, D. & Patrick, G. Less is better: new approaches for seedless fruit production. Biotopics 18, 233–242 (2000).
Picarella, M. E. & Mazzucato, A. The occurance of seedlessness in higher plants; insights on roles and mechanisms of parthenocarpy. Front. Plant Sci. https://doi.org/10.3389/fpls.2018.01997 (2019).
Schwabe, W. W. & Mills, J. J. (1981) Hormones and parthenocarpic fruit set: a literature survey. Hort. Abstracts 51, 661–698 (1981).
Su, L. et al. Cytokinin and auxin modulate cucumber parthenocarpy fruit development. Sci. Hort. 282, 110026. https://doi.org/10.1016/j.scienta.2021.110026 (2021).
Ren, Z. et al. The auxin receptor homologue in Solanum lycopersicum stimulates tomato fruit set and leaf morphogenesis. J. Exp. Bot. 62, 2815–2826 (2011).
Joldersma, D. & Liu, Z. The making of virgin fruit: the molecular and genetic basis of parthenocarpy. J. Exp. Bot. 69(5), 955–962 (2018).
García-Hurtado, N. et al. The characterization of transgenic tomato overexpressing gibberellin 20-oxidase reveals induction of parthenocarpic fruit growth, higher yield, and alteration of the gibberellin biosynthetic pathway. J. Exp. Bot. 63, 5803–5813 (2012).
Hayata, Y. & Niimi, Y. Synthetic cytokinin-1-(2=chloro=4=pyridyl)-3-phenylurea (CPPU)-promotes fruit set and induces parthenocarpy in watermelon. J. Am. Hort. Soc. 120, 997–1000 (1995).
Lewis, D. H., Burge, G. K., Hopping, M. E. & Jameson, P. E. Cytokinins and fruit development in the kiwifruit (Actinidia deliciosa). II. Effects of reduced pollination and CPPU application. Physiol. Plant. 98, 187–95 (1996).
Kadota, M. & Niimi, Y. Effects of cytokinin types and their concentrations on shoot proliferation and hyperhydricity in in vitro pear cultivar shoots. Plant Cell Tissue Organ Culture 72, 261–265 (2003).
Pascual, L. et al. Transcriptomic analysis of tomato carpel development reveals alterations in ethylene and gibberellin synthesis during pat3/pat4 parthenocarpic fruit set. BMC Plant Biol. 9(1), 67. https://doi.org/10.1186/1471-2229-9-67 (2009).
Rotino, G. L., Perri, E., Zottini, M., Sommer, H. & Spena, A. Genetic engineering of parthenocarpic plants. Nat. Biotechnol. 15, 1398–1401 (1997).
Yin, Z. et al. The DefH9-iaaM-containing construct efficiently induces parthenocarpy in cucumber. Cell. Mol. Biol. Lett. 11, 279–290 (2006).
Goetz, M. et al. Expression of aberrant forms of AUXIN RESPONSE FACTOR8 stimulates parthenocarpy in Arabidopsis and tomato. Plant Physiol. 145(2), 351–366. https://doi.org/10.1104/pp.107.104174 (2007).
de Jong, M., Wolters-Arts, M., Feron, R., Mariani, C. & Vriezen, W. H. The Solanum lycopersicum auxin response factor 7 (SlARF7) regulates auxin signaling during tomato fruit set and development. Plant J. 57, 160–170 (2009).
Chaudhury, A. M. et al. Fertilization-independent seed development in Arabidopsis thaliana. Proc. Nat. Acad. Sci. 94, 4223–4228 (1997).
Kohler, C. et al. The Polycomb-group protein MEDEA regulates seed development by controlling expression of the MADS-box gene PHERES1. Genes Dev. 17, 1540–1553 (2003).
Ohad, N. et al. A mutation that allows endosperm development without fertilization. Proc. Nat. Acad. Sci. 93, 5319–5324 (1996).
Li, J. et al. Proteomic insights into fruit set of cucumber (Cucumis sativus L.) suggest the cues of hormone-independent parthenocarpy. BMC Genomics 18, 896 (2017).
Wu, Z. et al. Identification of a stable major-effect QTL (Parth 2.1) controlling parthenocarpy in cucumber and associated candidate gene analysis via whole genomere-sequencing. BMC Plant Biol. 16, 182 (2016).
Gou, C. X. et al. Evaluation and genetic analysis of parthenocarpic germplasms in cucumber. Genes 13, 225. https://doi.org/10.3390/genes1302022 (2022).
Lietzow, C. D., Zhu, H. Y., Pandey, S., Havey, M. J. & Weng, Y. QTL mapping of parthenocarpic fruit set in North American processing cucumber. Theor. App. Genet. 129, 2387–2401 (2016).
Shinozaki, Y. et al. Identification and functional study of a mild allele of SlDELLA gene conferring the potential for improved yield in tomato. Sci. Rep. 8, 12043. https://doi.org/10.1038/s41598-018-30502-w (2018).
Takisawa, R. et al. The parthenocarpic gene Pat-k is generated by a natural mutation of SIAGL6 affecting fruit development in tomato (Solanum lycopersicum L.). BMC Plant Biol. 18, 72 (2018).
Klap, C. et al. Tomato facultative parthenocarpy results from SlAGAMOUS-LIKE 6 loss of function. Plant Biotechnol. J. 15, 634–647 (2017).
He, M. et al. SITPL1 silencing induces facultative parthenocarpy in tomato. Front. Plant Sci. 12, 672232. https://doi.org/10.3389/fpls.2021.672232 (2021).
Fos, M., Nuez, F. & García-Martínez, J. L. The gene pat-2, which induces natural parthenocarpy, alters the gibberellin content in unpollinated tomato ovaries. Plant Physiol. 122(2), 471–480. https://doi.org/10.1104/pp.122.2.471 (2000).
Mesejo, C., Reig, C., Martínez-Fuentes, A. & Agustí, M. Parthenocarpic fruit production in loquat (Eriobotrya japonica Lindl.) by using gibberellic acid. Sci. Hortic. 126, 37–41 (2010).
Chai, L., Chai, P., Chen, S., Flaishman, M. A. & Ma, H. Transcriptome analysis unravels spatiotemporal modulation of phytohormone-pathway expression underlying gibberellin-induced parthenocarpic fruit set in San Pedro-type fig (Ficus carica L.). BMC Plant Biol. 18, 100. https://doi.org/10.1186/s12870-018-1318-1 (2018).
Sun, T. P. et al. Molecular mechanism of gibberellin signalling in plants. Annu. Rev. Plant Biol. 55, 197–223 (2004).
Chai, P. et al. Cytokinin induced parthenocarpy of San Pedro type fig (Ficus carica L.) main crop: explained by phytohormone assay and transcriptomic network comparison. Plant Mol. Biol. 99, 329–346 (2019).
Fernandez, L., Chaib, J., Martinez-Zapater, J. M., Thomas, M. R. & Torregrosa, L. Mis-expression of a PISTILLATA-like MADS box gene prevents fruit development in grapevine. Plant J. 73, 918–928 (2013).
Zhang, H. et al. Downstream of GA4, PbCYP78A6 participates in regulating cell cycle-related genes and parthenogenesis in pear (Pyrus bretshneideri Retd.). BMC Plant Biol. 21, 292 (2021).
Sharif, R. et al. Hormonal interactions underlying parthenocarpic fruit formation in horticultural crops. Hort. Res. https://doi.org/10.1093/hr/uhab024 (2022).
Schmidt, A. et al. The polycomb group protein MEDEA and the DNA methyltransferase MET1 interact to repress autonomous endosperm development in Arabidopsis. Plant J. 73, 776–787 (2013).
Wang, H. et al. PbGA20ox2 regulates fruit set and induces parthenocarpy by enhancing GA4 content. Front. Plant Sci. 11, 113. https://doi.org/10.3389/fpls.2020.00113 (2020).
Molesini, B., Dusi, V., Pennisi, F. & Pandolfini, F. How hormones and MADS-box transcription factors are involved in controlling fruit set and parthenocarpy in tomato. Genes 11, 1441 (2020).
Sharma, N., Russell, S. D., Bhalla, P. L. & Singh, M. B. Putative cis-regulatory elements in genes highly expressed in rice sperm cells. BMC Res. Notes 4, 319. https://doi.org/10.1186/1756-0500-4-319 (2011).
Lee-Huang, S. et al. The human erythropoietin-encoding gene contains a CAAT box, TATA boxes and other transcriptional regulatory elements in its 5’ flanking region. Gene 128, 227–236. https://doi.org/10.1016/0378-1119(93)90567-M (1993).
Kusnetsov, V., Landsberger, M., Meurer, J. & Oelmüller, R. The assembly of the CAAT-box binding complex at a photosynthesis gene promoter is regulated by light, cytokinin, and the stage of the plastids. J. Biol. Chem. 274, 36009–36014. https://doi.org/10.1074/jbc.274.50.36009 (1999).
Menkens, A. E., Schindler, U. & Cashmore, A. R. The G-box: a ubiquitous regulatory DNA element in plants bound by the GBF family of bZIP proteins. Trends Biochem. Sci. 20, 506–510. https://doi.org/10.1016/S0968-0004(00)89118-5 (1995).
Galperin, M. Y. & Frishman, D. Towards automated prediction of protein function from microbial genomic sequences. In Methods in Microbiology (eds Craig, A. G. & Hoheisel, J. D.) 245–263 (Academic Press, 1999).
Kelley, L. A. & Sternberg, M. J. E. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 (2009).
Seymour, G. B. et al. A SEPALLATA gene is involved in the development and ripening of strawberry (Fragaria×ananassa Duch.) fruit, a non-climacteric tissue. J. Exp. Bot. 62, 1179–88. https://doi.org/10.1093/jxb/erq360 (2011).
Liu, L. et al. Histological, hormonal and transcriptomic reveal the changes upon gibberellin-induced parthenocarpy in pear fruit. Hort. Res. 5, 1 (2018).
Coen, E. S. & Meyerowitz, E. M. The war of the whorls: genetic interactions controlling flower development. Nature 353, 31–37 (1991).
Matsuo, S., Kikuchi, K., Fukuda, M., Honda, I. & Imanishi, S. Roles and regulation of cytokinins in tomato fruit development. J. Exp. Bot. 63(15), 5569–5579 (2012).
Yu, X. et al. Prevalent exon-intron structural changes in the APETALLA1/FRUITFULL, SEPALLATA, AGAMOUS-LIKE6, and FLOWERING LOCUS C MADS-box gene subfamilies provide new insights into their evolution. Front. Plant Sci. https://doi.org/10.3389/fpls.2016.00598 (2016).
Li, Y. et al. Identification and expression analysis of miR160 and their target genes in cucumber. Biochem. Genet. 60, 127–152 (2022).
Fu, F. Q. et al. A role of brassinosteroids in early fruit development in cucumber. J. Exp. Bot. 59(90), 2299–2308 (2008).
Sun, H. et al. Comprehensive analysis of cucumber gibberellin oxidase family genes and functional characterization of CsGA20ox1 in root development in Arabidopsis. Int. J. Mol. Sci. 19(10), 3135. https://doi.org/10.3390/ijms19103135 (2018).
Mejia, N. et al. Molecular, genetic and transcriptional evidence for a role of VvAGL11 in stenospermocarpic seedlessness in grapevine. BMC Plant Biol. 11, 5 (2011).
Kumar, P. et al. Pivotal role of bZIPs in amylose biosynthesis by genome survey and transcriptome analysis in wheat (Triticum aestivum L.) mutants. Sci. Rep. 8, 17240. https://doi.org/10.1038/s41598-018-35366-8 (2018).
Kumar, P. et al. Genome-wide identification and expression profiling of basic leucine zipper transcription factors following abiotic stresses in potato (Solanum tuberosum L.). PLoS One 16(3), e0247864. https://doi.org/10.1371/journal.pone.0247864 (2021).
Kumar, P. et al. Understanding the regulatory relationship of abscisic acid and bZIP transcription factors towards amylose biosynthesis in wheat. Mol. Biol. Rep. 48(3), 2473–2483. https://doi.org/10.1007/s11033-021-06282-4 (2021).
Hartmann, U., Sagasser, M., Mehrtens, F., Stracke, R. & Weisshaar, B. Differential combinatorial interactions of cis-acting elements recognized by R2R3-MYB, BZIP, and BHLH factors control light-responsive and tissue-specific activation of phenylpropanoid biosynthesis genes. Plant Mol. Biol. 57, 155–171 (2005).
Maniatis, T., Goodbourn, S. & Fischer, J. A. Regulation of inducible and tissue-specific gene expression. Science 236, 1237–1245 (1987).
Siberil, Y., Doireau, P. & Gantet, P. Plant bZIP G-box binding factors modular structure and activation mechanisms. Eur. J. Biochem. 268(22), 5655–5666 (2001).
Krieger, E. K., Allen, E., Gilbertson, L. A. & Roberts, J. K. The Flavr Savr tomato, an early example of RNAi technology. Hort. Sci. 43, 962–964 (2008).
Martineau, B. First Fruit: The Creation of the Flavr Savr Tomato and the Birth of Biotech Foods (McGraw Hill companies, 2001).
Unni, S. C., Vivek, P. J., Maju, T. T., Varghese, R. T. & Soniya, E. V. Molecular cloning and characterization of fruit specific promoter from Cucumis sativus L.. Am. J. Mol. Biol. 2, 132–139 (2012).
Pla, M. et al. The cis-regulatory element CCACGTGG is involved in ABA and water-stress responses of the maize gene rab28. Plant Mol. Biol. 21, 259–266 (1993).
Whalley, H. J. et al. Transcriptomic analysis reveals calcium regulation of specific promoter motifs in Arabidopsis. Plant Cell 23, 4079–4095 (2013).
Kaur, A., Pati, P. K., Pati, A. M. & Nagpal, A. K. In-silico analysis of cis-acting regulatory elements of pathogenesis-related proteins of Arabidopsis thaliana and Oryza sativa. PLoS One 12(9), e0184523. https://doi.org/10.1371/journal.pone.0184523 (2017).
Sehra, B. & Franks, R. G. Redundant CArG Box Cis-motif activity mediates SHATTERPROOF2 transcriptional regulation during Arabidopsis thaliana gynoecium development. Front. Plant Sci. 8, 1712. https://doi.org/10.3389/fpls.2017.01712 (2017).
Shore, P. & Sharrocks, A. D. The MADS-box family of transcription factors. Eur. J. Biochem. 229, 1–13. https://doi.org/10.1111/j.1432-1033.1995.tb20430.x (1995).
Tang, W. & Perry, S. E. Binding site selection for the plant MADS domain protein AGL15: an in vitro and in vivo study. J. Biol. Chem. 278, 28154–28159 (2003).
Dutt, M., Dhekney, S., Soriano, L., Kandel, R. & Grosser, J. W. Temporal and spatial control of gene expression in horticultural crops. Hortic. Res. 1, 14047. https://doi.org/10.1038/hortres.2014.47 (2014).
Ashburner, M. et al. Gene ontolgy: tool for the unification of biology. Gene Ontol. Consortium. Nat. Genet. 25(1), 25–29. https://doi.org/10.1038/75556 (2000).
Singh, D. P. et al. Overexpression of gibberellin inactivation gene alters seed development, KNOX gene expression, and plant development in Arabidopsis. Physiol. Plant. 138(1), 74–90 (2010).
Thomas, S. G., Phillips, A. L. & Hedden, P. Molecular cloning and functional expression of gibberellin 2-oxidases, multifunctional enzymes involved in gibberellin deactivation. Proc. Nat. Acad. Sci. 96, 4698–4703 (1999).
Swain, S. M., Reid, J. B. & Kamiya, Y. Gibberellins are required for embryo and seed development in pea. Plant J. 12, 1329–1338 (1997).
Backiyarani, S., Sasikala, R., Sharmiladevi, S. & Uma, S. Decoding the molecular mechanism of parthenocarpy in Musa spp. through protein-protein interaction network. Sci. Rep. 11, 14592 (2021).
Pomares-Viciana, T. et al. First RNA-seq approach to study fruit set and parthenocarpy in zuchhini (Cucurbita pepo L.). BMC Plant Biol. 19, 61 (2019).
Chen, X. et al. Comparative transcriptome analysis provides insights into molecular mechanisms for parthenocarpic fruit development in eggplant (Solanum melongena L.). PLoS One 12(6), e0179491. https://doi.org/10.1371/journal.pone.0179491 (2017).
Ong-Abdullah, M., Ordway, J. M. & Jiang, N. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nat. 525, 533–537. https://doi.org/10.1038/nature15365 (2015).
Sommer, H. et al. Deficiens, a homeotic gene involved in the control of flower morphogenesis in Antirrhinum majus: the protein shows homology to transcription factors. EMBO J. 9, 605–613 (1990).
Ampomah-Dwamena, C., Morris, B. A., Sutherland, P., Veit, B. & Yao, J. L. Down regulation of TM29, a tomato SEPALLATA homolog, causes parthenocarpic fruit development and floral reversion. Plant Physiol. 130, 605–617 (2002).
Yao, J. L., Dong, Y. H. & Morris, B. Parthenocarpic apple fruit production conferred by transposon insertion mutations in a MADS-box transcription factor. Proc. Natl. Acad. Sci. USA 98, 1306–1311 (2001).
Bassel, G. W., Mullen, R. T. & Bewley, J. D. Procera is a putative DELLA mutant in tomato (Solanum lycopersicum): effects on the seed and vegetative plant. J. Exp. Bot. 59, 585–593 (2008).
Paponov, I. A., Teale, W. D., Trebar, M., Blilou, I. & Palme, K. The PIN auxin efflux facilitators: evolutionary and functional perspectives. Trends Plant Sci. 10, 170–177 (2005).
Blilou, I. et al. The PIN auxin efflux facilitator network controls growth and patterning in Arabidopsis roots. Nature 433, 39–44 (2005).
Mounet, F. et al. Down-regulation of a single auxin efflux transport protein in tomato induces precocious fruit development. J. Exp. Bot. 63(13), 4901–4917. https://doi.org/10.1093/jxb/ers167 (2012).
Liu, Z. et al. The WD40 gene family in potato (Solanum Tuberosum L.): genome-wide analysis and identification of anthocyanin and drought-related WD40s. Agronomy 10(3), 401. https://doi.org/10.3390/agronomy10030401 (2020).
Kiba, T. et al. Combinatorial microarray analysis revealing arabidopsis genes implicated in cytokinin responses through the His→Aspphosphorelay circuitry. Plant Cell Physiol. 46, 339–355. https://doi.org/10.1093/pcp/pci033 (2005).
Ding, J. et al. Cytokinin-induced parthenocarpic fruit development in tomato is partly dependent on enhanced gibberellin and auxin biosynthesis. PLoS One 8(7), e70080 (2013).
Hu, B. et al. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31(8), 1296–1297 (2015).
Lescot, M. et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30(1), 325–327 (2002).
Higo, K., Ugawa, Y., Iwamoto, M. & Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 27(1), 297–300 (1999).
Bailey, T. L., Johnson, J., Grant, C. E. & Noble, W. S. The MEME Suite. Nucleic Acids Res. 43(1), 39–49 (2015).
Buske, F. A., Boden, M., Bauer, D. C. & Bailey, T. L. Assigning roles to DNA regulatory motifs using comparative genomics. Bioinformatics 26(7), 860–866 (2010).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Kanehisa, M. & Sato, Y. KEGG mapper for inferring cellular functions from protein sequences. Protein Sci. 29, 28–35 (2020).
Gasteiger, E. et al. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook (ed. Walker, J. M.) (Springer Protocols Handbooks Humana Press, 2005).
Mistry, J. et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 49(D1), D412–D419 (2021).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. https://doi.org/10.1038/nprot.2015.053 (2015).
Beier, S., Theil, T., Munch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33(16), 2583–2585 (2017).
Das, R. et al. PolyMorphPredict: a universal web-tool for rapid polymorphic microsatellite marker discovery from whole genome and transcriptome data. Front. Plant Sci. https://doi.org/10.3389/fpls.2018.01966 (2019).
Baloglu, M. C., Eldem, V., Hajyzadeh, M. & Unver, T. Genome-wide analysis of the bZIP transcription factors in cucumber. PLoS One 9(4), e96014. https://doi.org/10.1371/journal.pone.0096014 (2014).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 25, 402–408 (2001).
Serrani, J. C., Rivero, O. R., Fos, M. & Martinez, J. L. G. Auxin-induced fruit-set in tomato is mediated in part by gibberellins. Plant J. 56(6), 922–934 (2008).
Acknowledgements
The authors are highly thankful to the Department of Science and Technology for providing funds under the Science and Engineering Research Board, SERB-POWER Grant (SPG/2021/000999).
Author information
Authors and Affiliations
Contributions
H.K. performed bioinformatics study and wet lab experiments using qRT-PCR and SSR markers, P.M. designed the in silico and wet-lab experiments and wrote the manuscript, P.K. performed bioinformatics study, R.K.D. provided with plant material required for the study, P.C. and Y.W. reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaur, H., Manchanda, P., Kumar, P. et al. Genome-wide identification and characterization of parthenocarpic fruit set-related gene homologs in cucumber (Cucumis sativus L.). Sci Rep 13, 2403 (2023). https://doi.org/10.1038/s41598-023-29660-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-29660-3
This article is cited by
-
Heterosis and combining ability for yield and quality traits in monoecious, gynoecious and parthenocarpic parental lines of cucumber (Cucumis sativus L.) under tropical condition
Euphytica (2024)
-
In-Silico Identification, Characterization and Expression Analysis of Genes Involved in Resistant Starch Biosynthesis in Potato (Solanum tuberosum L.) Varieties
Molecular Biotechnology (2024)
-
Utilizing novel parthenocarpic gynoecious cucumber (Cucumis sativus L.) inbreds for exploiting their heterotic potential under poly-house conditions
Euphytica (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
