
Abstract
Half-Heusler (HH) phase TmNiSb was obtained by arc-melting combined with high-pressure high-temperature sintering in conditions: p = 5.5 GPa, \(T_{HPHT}\) = 20, 250, 500, 750, and 1000 \(^{\circ }\)C. Within pressing temperatures 20–750 \(^{\circ }\)C the samples maintained HH structure, however, we observed intrinsic phase separation. The material divided into three phases: stoichiometric TmNiSb, nickel-deficient phase TmNi\(_{1-x}\)Sb, and thulium-rich phase Tm(NiSb)\(_{1-y}\). For TmNiSb sample sintered at 1000 \(^{\circ }\)C, we report structural transition to LiGaGe-type structure (P\(6_3\)mc, a = 4.367(3) Å, c = 7.138(7) Å). Interpretation of the transition is supported by X-ray powder diffraction, electron back-scattered diffraction, ab-initio calculations of Gibbs energy and phonon dispersion relations. Electrical resistivity measured for HH samples with phase separation shown non-degenerate behavior. Obtained energy gaps for HH samples were narrow (\(\le\) 260 meV), while the average hole effective masses in range 0.8–2.5\(m_e\). TmNiSb sample pressed at 750 \(^{\circ }\)C achieved the biggest power factor among the series, 13 \(\upmu\)WK\(^{-2}\)cm\(^{-1}\), which proves that the intrinsic phase separation is not detrimental for the electronic transport.
Similar content being viewed by others

Introduction
Prolific interplay of physics and chemistry inherent to the field of thermoelectrics has been gathering attention of the scientific community throughout the decades. Among many interesting groups of materials considered for this purpose, half-Heusler (HH) phases seem to be closest to real-life application, due to the fact that high energy conversion efficiency1 can be hither supplemented by relatively low price of constituent elements, good mechanical properties, as well as decent thermal stability2. Apart of thermoelectricity, HH compounds are also investigated from multiple different perspectives, e.g. magnetocaloric applications3, superconductivity coexisting with antiferromagnetism4, heavy fermion behavior5, half-metallic ferromagnetism6, and anomalous structural transitions7. Recently f-electron systems gathered special attention due to their nontrivial topological properties8.
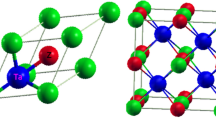
General chemical composition of HH phases is MTZ, where M denotes for early transition metal or rare-earth (RE) element, T stands for late transition metal, and Z is p-electron element. Structural prototype of HH phases is MgAgAs, space group being \(F{\bar{4}}3m\) (no. 216). The unit cell is depicted in Fig. 1a, while the atomic coordinates are gathered in Supplementary Table S1. The structure can be described as rock-salt lattice of M-Z atoms, with half of the voids in small M-Z cubes filled with T elements. The second half of 8-coordinated voids are 4d (\(\frac{3}{4}\frac{3}{4}\frac{3}{4}\)) Wyckoff position, whose in ideal structure remain unoccupied. Despite apparent simplicity of the unit cell, HH compounds are known to comprise bounty of structural peculiarities. The materials based on d-electron elements, e.g. ZrNiSn, are known for their low energy interstitial defects on nominally empty 4d site, and Zr-Sn antisites9,10. The disorder was found to be beneficial for thermoelectric energy conversion11,12. The rare-earth bearing counterparts are known to exhibit different type of dominant defects. Depending on method of synthesis they are known to comprise either vacancies on late transition metal site 4c (\(\frac{1}{4}\frac{1}{4}\frac{1}{4}\)), e.g. nickel in ScNiSb13, or split positions of 4c slot to quarterly occupied position 16e(x,x,x) with x = 0.256-0.26014,15.

Different types of crystal structures attained by equiatomic ternaries with general composition MTZ, see text for details. Figure prepared in Vesta16 software.
Furthermore, selected HH compounds show phase separation (PS), i.e. their polycrystalline samples have regions strongly differing in composition, while maintaining the MgAgAs-type structure. PS was reported before for materials with general composition Ti\(_{1-x-y}\)Zr\(_x\)Hf\(_y\)MZ where M = Co, Ni, and Z = Sb, Sn17,18,19,20,21. The structural arrangement of samples was usually division into (Zr, Hf)-rich dendritic phase, and interdendritic grains enriched in titanium. The phenomenon is assumed to origin from different atomic radii of early transition metal atoms (\(r_{Ti}\) = 2.11 Å, \(r_{Zr, Hf}\) \(\approx\) 2.23 Å) and significantly distinct temperature of solidification: 1453 K for TiNiSn, 1708 K for ZrNiSn and 1760 K for HfNiSn22. In the case of d-electron HH compounds, PS was found to be beneficial for thermoelectric energy conversion due to enhanced phonon scattering with simultaneously small effect on electrical properties17,18,19,20,23. The HH samples maintain peculiar PS microstructure after extensive temperature cycling24, which is critical from perspective of future application. The phenomenon received sizable attention of theoretical part in thermoelectric community, see e.g. Refs.25,26,27. PS was found to be important also for other groups of TE materials: GeTe28,29,30, PbTe31,32, AgPb\(_m\)SbTe\(_{m+2}\)33,34 and SnSe35.
At elevated temperatures and/or high pressures HH compounds undergo interesting phase transitions. TiPtGe was reported to transform at 885 \(^{\circ }\)C from cubic MgAgAs-type structure to orthorhombic TiNiSi-type cell with large volume contraction of 10%, despite increase in temperature7. Similar type of phase transition was observed for YbPdSb36. In the TiNiSi-type structure, Ni and Si form 6-atoms puckered rings, while Ti forming zig-zag chains along [100] direction, filling the Ni-Si tunnels. The reader is referred to Fig. 1b, and Fig. 1f for the TiNiSi-type unit cell in top and side view, respectively. The related atomic positions are gathered in Supplementary Table S1. On the path from MgAgAs-type to TiNiSi-type transformation, the authors of Ref.7 suggested presence of metastable ZrBeSi-type structure, space group P63/mc. Experimentally, ZrBeSi-like structure was stabilized in HH phases VCoSb and VFeSb, crystallizing in MgAgAs-type structure at atmospheric pressure, while hexagonal unit cell was observed after synthesis in extreme conditions: 5.0 GPa and 900 \(^{\circ }\)C37,38. Similarly to TiNiSi-type structure, ZrBeSi-type cell also hosts form 6-atoms rings, here made from Be and Si atoms. The reader is referred to Fig. 1c for top view of the structure and Fig. 1g for the side view. Interestingly, the so-formed Be-Si planes are here spatially separated from Zr layers, so the crystal structure might be considered as quasi 2-dimensional, c.f. Fig. 1g. ZrBeSi-type structure is sometimes also referred as ordered version of Ni\(_2\)In-type cell, or AlB\(_2\)-type. The latter is in principle binary subcell of ZrBeSi. See Ref.39 for exhaustive description of these crystal structures. In the series RENiSb, materials with light rare-earth elements RE = La, Ce, Pr, Nd, Sm crystallize with hexagonal ZrBeSi-type structure, while heavier counterparts with RE = Tb, Dy, Ho, Er, Tm, Lu, attain HH structure40. GdNiSb being in the middle between those two groups can be obtained in both polymorphs40. Last, but not the least CaAuBi41,42 and YbAuBi43 crystallize in MgAgAs-type and LiGaGe-type (sg. P63mc), see Fig. 1d, h. The latter prototype can be viewed as distorted version of ZrBeSi-type cell. It attains majority of features characteristic for ZrBeSi-type structure, but all atomic positions have relaxed z coordinates, which leads to puckering of Ga-Ge rings; see Fig. 1h for the side view.
Recently, thermoelectric figure of merit ZT = 1.5 (\(ZT=\frac{S^2}{\rho \kappa }T\), where S denotes thermopower, \(\rho\) stands for electrical resistivity, while \(\kappa\) is thermal conductivity) at elevated temperatures was reached for several HH compositions: TaFeSb-44, NbFeSb-45, and ZrCoBi-based materials46. Good thermoelectric performance of HH compounds results from values of the power factor (PF = \(S^{2}/\rho\)) as high as 50 \(\upmu\)WK\(^{-2}\)cm\(^{-1}\). Relatively large thermal conductivity (\(\kappa \ge\) 3 WK\(^{-1}\)m\(^{-1}\)), however, is still an obstacle to overcome. Preliminary research shown that f-electron HH phases can exhibit lower \(\kappa\) than the d-electron counterparts, where pristine compounds are compared: thermal conductivities near room temperature for exemplary rare-earth (RE) based HH phases were e.g. 5 WK\(^{-1}\)m\(^{-1}\) for HoPdSb47 and in 3-4 WK\(^{-1}\)m\(^{-1}\) range for TmNiSb48,49, while for d-electron compounds \(\kappa\) at 300 K was 9 WK\(^{-1}\)m\(^{-1}\) for ZrCoBi46, 13 WK\(^{-1}\)m\(^{-1}\) for FeVSb47, 6 WK\(^{-1}\)m\(^{-1}\) for HfNiSn, and 8 WK\(^{-1}\)m\(^{-1}\) for ZrNiSn50. Suppressed phonon transport in RE-containing materials results from their lower Debye temperatures, high density and sizable intrinsic atomic disorder51.
TmNiSb is representative of rare-earth bearing HH compounds13,14,15,49 crystallizing with lattice parameter a = 6.241 Å, as reported by Dwight et al.52 and 6.225 Å by Pecharski and coworkers53. Sportouch et al. investigated its thermoelectric properties below room temperature, which resulted in the lowest thermal conductivity at 300 K among other RENiSb (3 WK\(^{-1}\)m\(^{-1}\)), and PF = 1 \(\upmu\)WK\(^{-2}\)cm\(^{-1}\)48. Our reports on thermoelectric properties of RENiSb compounds in wider temperature range confirmed low thermal conductivity of TmNiSb49,51 and shown that at around 700 K the compound can attain high power factor, depending on method of synthesis: 11 \(\upmu\)WK\(^{-2}\)cm\(^{-1}\) for arc-melting sample49, and 17 \(\upmu\)WK\(^{-2}\)cm\(^{-1}\) for specimen synthesized by combination of arc-melting and spark plasma sintering49.
Other interesting method for preparation of thermoelectric materials is high-pressure high-temperature sintering (HPHT), which employs rapid compression of elemental constituent in pressures of several GPa and elevated temperatures54. Up to date the technique has been used for improving thermoelectric performance of PbTe54, Bi\(_2\)Te\(_3\)55, skutterudites56, and SnSe57. Main influence of HPHT process on thermoelectric transport is enhanced phonon scattering due to lattice defects introduced on different size scales58. In our previous work, we studied thermoelectric properties of Sc\(_{1-x}\)Tm\(_x\)NiSb half-Heusler phases. Formation of solid solution lead to decrease of thermal conductivity in the alloy space with respect to parent compounds and the best performance was achieved for Sc\(_{0.75}\)Tm\(_{0.25}\)NiSb (PF = 12 \(\upmu\)WK\(^{-2}\)cm\(^{-1}\) at 630 K)59. This study, however, was not focused on optimization of synthesis conditions; the samples were pressed uniformly in 5.5 GPa and 750 \(^{\circ }\)C.

(a) XRD patterns of TmNiSb specimen prepared at 1000 \(^{\circ }\)C with Rietveld fit using ZrBeSi-type and MgAgAs-type phases. Panel (b) present Rietveld fit for the same sample with LiGaGe-type and MgAgAs-type phases. Black ovals mark Bragg maxima for which the most significant differences were noticed between the two refinements, while triangles denote Bragg peaks of impurity phase. Miller indices are given for the most pronounced peaks of ZrBeSi-type and LiGaGe-type phases.
Inspired by the previous reports on HPHT synthesis and curious about behavior of RE HH compounds under pressure, we explore influence of different HPHT synthesis conditions on TmNiSb. Extraordinary structural features were noticed depending on temperature of sintering. We observed division of the HH compound through the intrinsic phase separation (PS) into TmNiSb, TmNi\(_{1-x}\)Sn, and Tm(NiSb)\(_{1-y}\) phases. Furthermore, sample prepared at 1000 \(^{\circ }\)C shown structural phase transition to LiGaGe-type structure. The study is complemented by thermoelectric characterization, which shows that pressure-driven phase separation might be useful for optimization of the performance. This work is part of larger project aimed at investigation of structural, thermoelectric and magnetic properties of rare-earth bearing HH compounds60,61,62,63,64,65,66,67.
Results and discussion
Powder X-ray diffraction
Powder X-ray diffraction patterns of samples prepared at temperatures 20–750 \(^{\circ }\)C are displayed in Supplementary Figure S1a. All significant Bragg maxima can be indexed to MgAgAs-type structure. Lattice parameters obtained from Rietveld refinement are: 6.237(1) Å, 6.223(2) Å, 6.244(1) Å, and 6.242(1) Å, for samples prepared at 20, 250, 500, and 750 \(^{\circ }\)C, respectively. Value of the lattice parameter oscillate closely around those from the previous literature52. Bragg peaks in our XRD patterns are rather wide and with slightly irregular profile, see Supplementary Figure S1b for exemplary profile of (422) maximum. This feature might be explained e.g. by large strain and some percentage of nonstoichiometric HH grains with slightly differing lattice parameters. Densities measured by Archimedes method were in 8.5–9.3 g/cm\(^3\) range (89–97 % of theoretical single phase density), with the lowest for sample pressed at room temperature and the highest for specimen prepared at 750 \(^{\circ }\)C. Minuscule impurity peaks marked by asterisks on Supplementary Figure S1a. stem mostly from presence of Ni\(_{1-x}\)Sb\(_x\) solid solutions and small amount of Tm\(_2\)O\(_3\).
For sample synthesized at 1000 \(^{\circ }\)C diffraction pattern changed significantly. Fig. 2a shows our preliminary fit with two phases: MgAgAs-type and ZrBeSi-type structures, where the latter was assumed by analogy to closely related isostructural analogue GdNiSb40. The obtained lattice parameter for HH phase is a = 6.22(2) Å, while for ZrBeSi-type a = 4.367(3) Å, c = 7.139(7) Å. The volume fraction is 5.43(4) % for HH and 94.6(9)% for the hexagonal phase, with global fit quality parameter GOF = 3.16. All the atomic coordinates are fixed due to symmetry requirements in these phases; see Supplementary Table S1. Due to visible discrepancies in several maxima intensity (see black ovals in Fig. 2a) we attempted another Rietveld fit with LiGaGe-type phase instead of the ZrBeSi-type. The results are shown in Fig. 2b. Fit quality was visibly enhances, GOF decreased to 1.96. Obtained lattice parameters are virtually equal to those resultant from previous refinement: 6.22(2) Å for HH phase and a = 4.366(3) Å, c = 7.136(7) Å for LiGaGe-type compound. The refined atomic coordinates allowing puckering of NiSb sheets (see Fig. 1h) are: \(z_{Tm}\) = 0.516(2), \(z_{Ni}\) = 0.680(1), \(z_{Sb}\) = 0.285(1). According to expectations, volume fraction of two respective phases is rather similar to the previous fit: 93.9(7) % for LiGaGe-type phase and 6.1(2) % for HH phase. Experimental density of TmNiSb specimen is 9.76 gm/cm\(^3\), i.e. higher than HH samples synthesized at temperatures 20–750 \(^{\circ }\)C.
We suggest, that maxima marked by triangles in 2\(\Theta\) = 32–38 \(^{\circ }\) range might correspond to the orthorhombic modification of TmNiSb compound in TiNiSi-type structure. This polymorphic modification was observed previously for other HH compounds7,36. However its small relative volume in the currently studied samples combined with low symmetry made the precise structure identification impossible. In future we plan to proceed with differential scanning calorimetry and in situ synchrotron diffraction experiment aimed on resolving the plausible second phase transition in TmNiSb.

SEM images of TmNiSb samples sintered at temperatures: (a) 20 \(^{\circ }\)C, (b) 250 \(^{\circ }\)C, (c) 500 \(^{\circ }\)C, (d) 750 \(^{\circ }\)C, (e) 1000 \(^{\circ }\)C.

Chemical maps of TmNiSb specimens prepared at temperatures 20–1000 \(^{\circ }\)C. For results of quantitative analysis see Table 1. Enumeration of point is according to X.Y system, where X denotes captured phase (1—TmNiSb, 2—TmNi\(_{1-x}\)Sb, 3—Tm(NiSb)\(_{1-y}\), 4—NiSb, 5—Tm, 6—Ni rich precipiTtation), and Y is the ordinal number of particular point.
Scanning electron microscopy, energy dispersive X-ray spectroscopy
SEM images of sample microstructure after sintering under different preforming temperatures are presented in Fig. 3. In order to enhance visibility of morphological features, we tilted the image plate by 70 \(^{\circ }\) with respected to the detector. As apparent from the figure, preparation of samples at low temperatures (20 \(^{\circ }\)C, 250 \(^{\circ }\)C) results in porous structure. After increasing temperature to 500-1000 \(^{\circ }\)C, the specimens appear to be more dense, almost indistinguishable from perspective of SEM imaging. Results of energy dispersive X-ray spectroscopy (EDS) analysis are displayed in Fig. 4 and quantified in Table 1. The stoichiometric half-Heusler phase TmNiSb is marked as 1.X on maps, where X is the ordinal number of a point in which EDS analysis was performed. As a result of phase separation there are visible also: non-stoichiometric HH phase depleted in nickel TmNi\(_{1-x}\)Sb (2.X) and HH phase enriched in thulium Tm(NiSb)\(_{1-y}\) (3.X). Assumption of the fact, that all three regions correspond to half-Heusler phases with different types of defects, is justified by rather large amount of defective grains - too large to overlook in pXRD if they corresponded to impurity phases, (c.f. almost phase-pure XRD patterns in Supplementary Figure S1). To confirm that observed hither PS is representative for large areas of TmNiSb samples, we prepared also EDS maps with scale bar 100 \(\upmu\)m (see Supplementary Figure S2 and corresponding Table S2), which demonstrate that phase distribution is representative for large areas of the samples.
To the best of the authors knowledge, TmNiSb is the first half-Heusler compound in which PS is intrinsic for ternary composition. Similar formation energies for TmNiSb, TmNi\(_{1-x}\)Sb and Tm(NiSb)\(_{1-y}\) phases under enhanced pressure are the most likely reason for appearance of such peculiar microstructure. Three predicted polymorphic modifications of TmNiSb (cubic, hexagonal and the hypothetical orthorhombic) make obvious association with three types of observed stoichiometry, however no clear quantitative connection was found between those. Further ab-initio study seems necessary to clarify the driving force of this intriguing type of phase separation. PS was not observed for TmNiSb samples prepared at ambient pressures48,60, neither it was reported for Sc\(_{1-x}\)Tm\(_{x}\)NiSb solid solutions prepared by HPHT method59. In the latter case, reason for overlooking this feature was high magnification in EDS mapping (scale bar 1 \(\upmu\)m), allowing only observation of elemental distribution in single grain and possibly the neighboring impurity.
Areas marked as (4.X), (5.X), and (6.X) in Fig. 4 correspond to NiSb, pure Tm (or Tm\(_2\)O\(_2\)), and other nickel-rich precipitation, respectively. Small amount of Tm- and Ni-rich alloys are distributed randomly, while NiSb seem to be located mainly at the grain boundaries.
Electron back-scattered diffraction
To confirm proper interpretation of phase transition in TmNiSb synthesized at 1000 \(^{\circ }\)C, we performed for this sample crystallographic analysis with electron-backscattered diffraction (EBSD) technique. The method enables multiphase crystallographic orientation scans and provides detailed information on each individual component, not only as the phase distribution or grain size, but also the individual misorientations of grains as well as boundaries for each phase separately. In our paper, experimental Kikuchi patterns have been indexed for the MgAgAs-type, LiGaGe-type and TiNiSi-type crystal structures. For cubic and hexagonal phases, lattice parameters were taken from Rietveld analysis, while for orthorhombic phase, which was present in too small amount in the sample (if any) for the refinement, we employed the lattice parameters and atomic coordinates from ab-initio structural relaxation calculations: a = 6.783 Å, b = 4.392 Å, c = 7.498 Å , Tm (0.975; 1/4; 0.603), Ni (0.126; 1/4; 0.071), Sb (0.323; 1/4; 0.312), see below. Panel (a) of Fig. 5 shows overlay fit of phase map. Consistently with XRD analysis, majority (89.2 %) of the examined surface was ascribed to LiGaGe-type phase (green area), while 10.1 % and 0.7 % of the area were determined as the MgAgAs-type (orange) and TiNiSi-type (cyan) phases, respectively. The cubic phase formed a relatively large, irregular and well-separated grains. Presence of large grains of low temperature (MgAgAs-type) phase indicates on the fact, that structural transition was not complete in our sample. It is also necessary to underline than minuscule amount of TiNiSi-type phase detected still does not allow for unambigous confirmation of its real presence in sample studied. Around 2-3 % of the area were difficult to index during the preliminary analysis. The effect could be ascribed to minuscule amount of impurity phases, e.g. NiSb, or Tm\(_2\)O\(_3\), see “Scanning electron microscopy, energy dispersive X-ray spectroscopy” section. Random distribution of the impurities led to their automatic incorporation to grain boundaries or neighboring phases in the final analysis. Minuscule content of the contaminant is expected to have minor influence on the most important conclusions from EBSD.

(a) EBSD overlay phase map of TmNiSb specimen synthesized at 1000 \(^{\circ }\)C. Orange, green and cyan regions correspond to MgAgAs-type, LiGaGe-type and TiNiSi-type phases, respectively. (b) Inverse Pole Figure (IPF) map in [001] direction and with the IPF color key on the right side.
The middle and right panels of Fig. 5 shows the inverse pole figure (IPF). IPFs are used to present the grain orientation distribution, where each color is related to the different crystallographic orientation. As apparent from the figure, orientation of the grains of LiGaGe-type phase are independent from large MgAgAs-type areas. This feature is indicative of reconstructive phase transitions, i.e. transformations between structures that do not share group-subgroup relations. Transitions of that kind require breaking of near-neighbor bonds, strong change in coordination numbers and structural patterns, which is indeed observed for TmNiSb, c.f. Fig. 1. Similar situation was observed by EBSD for TiPtGe, which exhibits structural transition between MgAgAs-type and TiNiSi-type unit cells7. Speculatively, we assume that the structural path of TmNiSb under pressure is starting from low temperature polymorph in MgAgAs-type, then transfers through metastable LiGaGe structure and finishes up in orthorhombic TiNiSi-type. Higher temperatures, pressures or longer time of reactions technically unavailable in our equipment might be necessary for stabilization of TmNiSb in TiNiSi-type cell.
Ab-initio calculations
To support our findings regarding changes in crystal structure of TmNiSb under pressure, we performed series of ab-initio calculations. First of all, we checked whether minimum in total energy for lattice parameters is close to the experiments results. The optimized parameters are a = 6.290 Å for HH compound, a = 4.394 Å, c = 7.178 Å for LiGaGe-type phase and a = 7.123 Å, b = 4.417 Å, and c = 7.534 Å for TiNiSi-type structures, respectively. The above results support our finding from Rietveld refinement of the HH and LiGaGe-type phase, see “Powder X-ray diffraction” section; slight overestimation (approx. 1 %) of theoretical lattice parameters with respect to the experiment data is typical for DFT methods.
As the second step of theoretical stability investigation, we calculated phonon dispersions relations for MgAgAs-type, ZrBeSi-type, LiGaGe-type, and TiNiSi-type structures to check thermodynamical stabilities; see left panel of Fig. 6. MgAgAs-type, LiGaGe-type, and TiNiSi-type polymorphs were found to exhibit no imaginary modes, indicating these phases are thermodynamically stable. On the other hand, the hexagonal phase in ZrBeSi-type structure exhibit imaginary modes in \(\Gamma\) and A points, which suggests, that the structure is thermodynamically unstable. We found Ni and Sb atoms are vibrating very strongly along c-axis according to the imaginary vibrational modes. As we mentioned above, Ni and Sb atoms constitute planar structure parallel to a-b plane, fixed in the c-axis direction in ZrBeSi-type. Thus, after relaxing atomic positions of Ni and Sb atoms in the c-axis direction with reducing symmetry to P3m in ZrBeSi-like structure, the imaginary modes disappeared. These atoms eventually formed distorted planar structure in LiGaGe-type, in which imaginary modes were not found. Hence, ab-initio calculations provide additional proof that high temperature high-pressure modification of TmNiSb is in LiGaGe-type structure. We found no further extraordinary features in phonon dispersions like rattling modes or avoided crossing.
The last step in theoretical verification was Gibbs energy calculations in different temperature and pressures (Fig. 6, right panel). The method can provide insight into mechanisms behind structural phase transition. In 0 GPa, MgAgAs-type half-Heusler phase is the most stable up to 1500 K, consistently with current state of experimental knowledge60. With increasing pressure, lower-symmetry polymorphic modifications of TmNiSb are becoming closer in energy to HH phase, and eventually in 15 GPa the LiGaGe-type phase is the most energetically stable in range 600–1200 K. Above that temperature, the TiNiSi-type modification attains the lowest energy. This result qualitatively corroborates general understanding of phase transition mechanism—from HH compound through metastable LiGaGe-type phase, up to hypothetical TiNiSi-type polymorph that plausibly can be observed at even higher temperatures and pressures. The interpretation is analogous to the case of TiPtGe7. Similar qualitative agreement between theory and experiment, where phase transition was calculated to occur in larger pressures than it was observed experimentally was reported for other HH phases, e.g. for CaAuBi and VCoSb37,38,42
Transport properties
Electrical resistivity (\(\rho\)) measurements are shown on Fig. 7a. Values of \(\rho\) are moderate, from 20 to 35 \(\upmu \Omega\)m near room temperature to 8-15 \(\upmu \Omega\)m at 950 K. The decrease of \(\rho\) values with temperature is associated with semiconducting-like charge transfer. In the intrinsic region, i.e. above 750 K, \(\rho\)(T) dependencies were fitted with Arrhenius formula:
where \(E_g\) corresponds to band gap and \(\sigma _0\) denotes semiconducting charge transfer coefficient. Both parameters resultant from least-squares fitting are collected in Table 2. The effective band gap increases with temperature of sintering from 170 meV for sample synthesized at room temperature to c.a. 260 meV for specimen synthesized at 500 \(^{\circ }\)C and 750 \(^{\circ }\)C. Based on the fact that structural disorder reduce \(E_g\) of HH compounds9,68,69 increase of \(E_g\) with \(T_{HPHT}\) might be tentatively associated with reduction of the disorder for samples prepared at higher temperatures. The observed \(E_g\) for specimens prepared at 500 \(^{\circ }\)C and 750 \(^{\circ }\)C could be considered closer to the intrinsic energy gap of TmNiSb, however it is still smaller than result of ab-initio calculations (\(E_g\) = 359 meV)70. Further precise studies at atomic arrangement in TmNiSb e.g. by synchrotron radiation diffraction and high resolution microscopy appear as interesting topic. Available literature on TmNiSb provides values of electrical resistivity near room temperature in 13–40 \(\upmu \Omega\)m range48,59,60. The discrepancies likely result from sample-specific disorder in crystal lattice and deviations from ideal stoichiometry, typical for HH compounds51,71.

Left panel: phonon dispersion relations for MgAgAs-type, TiNiSi-type, LiGaGe-type, and ZrBeSi-type polymorphic modifications of TmNiSb; Right panel: Temperature variations of Gibbs energy for constant pressures 0–15 GPa.

Temperature variations of (a) electrical resistivity, (b) Seebeck coefficient, and (c) power factor for TmNiSb samples sintered at different temperatures.
Results of thermopower measurements are presented in Fig.7b. Two trends are observed within the series: (1) values of maximum thermopower (\(S_{max}\)) increase with rising temperature of synthesis; (2) temperature at which \(S_{max}\) is attained, moves slightly towards lower values with rising temperature of preparation. Due to complex interplay of metallic impurities acting as carrier injectors, porous microstructure for samples sintered at 20 \(^{\circ }\)C and 250 \(^{\circ }\)C, possible modification of electronic structure of non-stoichiometric HH phases, we do not attempt to provide here more quantitative reasoning regarding tendencies in S. Literature values of thermopower near room temperature for TmNiSb are in range 60-80 \(\upmu\)V/K48,59,60, while \(S_{max}\) for TmNiSb samples measured in high temperature regime are 90 \(\upmu\)V/K at 600 K59 and 140 \(\upmu\)V/K at 550 K60. The discrepancies observed for different TmNiSb samples highlight influence of structural imperfections on electron transport of HH compounds.
Figure 7c displays temperature dependences of power factor. Maximum values are achieved in 600–700 K range and increase with rising \(T_{HPHT}\) up to 13 \(\upmu\)WK\(^{-2}\)cm\(^{-1}\) for sample synthesized at 750 \(^{\circ }\)C, which shows that higher temperature of sintering up to 750 \(^{\circ }\)C grants better thermoelectric performance. Obtained \(PF_{max}\) is similar to those obtained for the other rare-earth-bearing HH compounds72,73,74. It is worth noting that despite phase separation, PF of currently examined samples of TmNiSb shown slightly better thermoelectric performance than their arc-melted counterpart (PF = 11 \(\upmu\)WK\(^{-2}\)cm\(^{-1}\))49.
Approximate carrier concentration (n) and mobility (\(\mu _H\)) calculated from Hall in single band approximation at 300 K are gathered in Table 2. Values of n are moderate, 1.4-3.6 \(\times\) 10\(^{20}\)carriers cm\(^{-3}\), similarly to reported previously undoped sample of TmNiSb (\(n_H\) = 1.9 \(\times\) 10\(^{20}\) carriers cm\(^{-3}\))48 and lanthanide-based RENiSb phases49. Mobility attains maximum of 17 cm\(^2\) V\(^{-1}\) s\(^{-1}\) in the studied series. The related HH phase ErPdSb show significantly higher mobility (\(\mu _H\) = 165 cm\(^2\) V\(^{-1}\) s\(^{-1}\)). The discrepancy might result from different electronic structures of Ni- and Pd- based HH series and distinct microstructure.
Accepting rough approximation of parabolic band modelling for our system, we calculated average effective masses (\(m_{eff}\)) of TmNiSb samples near room temperature11. The results of the calculations together with the values of experimental transport parameters near 300 K are gathered in Table 2. Effective masses are rather low, not exceeding 2.5 \(m_e\) for TmNiSb pressed at 500 \(^{\circ }\)C. Similar value of \(m_{eff}\) for TmNiSb was obtained in samples synthesized by arc-melting and spark plasma sintering (2.61 \(m_e\))51. Last, but not least, we calculated weighted mobility (\(\mu _w\)) at 340 K from \(\rho\) and S data using formula proposed in Ref.75, see Table 2. The highest value of \(\mu _w\) obtained for sample sintered at 750 \(^{\circ }\)C is consistent with the highest value of \(PF_{max}\) observed for this material. The overall higher values of weighted mobility for specimen synthesized at 750 \(^{\circ }\)C with respect to all other materials, can be further rationalized by the values of Hall mobility and effective mass. In the original form, weighted mobility is \(\mu _w = \mu _0 m_{eff}^{3/2}\)76, where \(\mu _0\) denotes the intrinsic mobility, i.e. the upper limit of mobility in pure materials in the intrinsic regime of conductivity. It’s quantitative relation to \(\mu _H\) can be found e.g. in Ref.51. The difference between \(\mu _H\) and \(\mu _0\) is usually by factor of smaller than two. In our simplified consideration we might assume, that \(\mu _H \approx \mu _0\). Then the product of \(\mu _H m_{eff}^{3/2}\) gives values in range 11, 13, and 29 cm\(^2\) V\(^{-1}\) s\(^{-1}\) for samples synthesized in temperatures 20, 250, and 500 \(^{\circ }\)C. The for TmNiSb obtained at 750 \(^{\circ }\)C the product of \(\mu _H m_{eff}^{3/2}\) gives higher value of 57 cm\(^2\) V\(^{-1}\) s\(^{-1}\), due to good mobility and decent effective mass, which eventually justifies its highest \(\mu _w\) obtained with formula from Ref.75 and its highest PF.
Transport properties of TmNiSb sample sintered at 1000 \(^{\circ }\)C were not studied due to coexistence of HH and ZrBeSi-type phases, which are likely to make interpretation difficult. High fragility of disc-shaped TmNiSb specimens synthesized by HPHT process made measurements of thermal conductivity (\(\kappa\)) at high temperatures difficult by standard laser-flash method. Separate project aimed at investigation of PS influence on \(\kappa\) will be arranged with related HH phases that show better mechanical properties after HPHT synthesis. Preliminarily, LuNiSb was chosen as the most stable compound from RENiSb series, where RE is rare earth metal (unpublished data).
Conlusions
Samples of f-electron half-Heusler compound TmNiSb were synthesized by combination of arc-melting and high-pressure high-temperature sintering in conditions: p = 5.5 GPa, T = 20-1000 \(^{\circ }\)C. The pressure-induced phase separation intrinsic for ternary composition was observed by division of HH compound into (1) stoichiometric TmNiSb, (2) nickel-deficient TmNi\(_{1-x}\)Sb, and (3) thulium-rich Tm(NiSb)\(_{1-y}\). For material prepared at 1000 \(^{\circ }\)C we observed structural transition, result of which was interpreted as hexagonal polymorphic modification with LiGaGe-type structure. Proper interpretation of phase transition is confirmed by refinements in XRD and EBSD experiments as well as phonon dispersion and Gibbs energy energy calculations. Both analogy to known HH counterparts and results of ab-initio analysis suggest possible existence of TiNiSi-type modification of TmNiSb at even higher temperatures and pressures that the currently examined.
Electrical resistivity in 350–950 K range measured for TmNiSb specimens with the phase separation was relatively low, from 5 to 35 \(\upmu \Omega\)m, and of non-degenerate type. Energy gaps were obtained by Arrhenius fit and led to results of 170–260 meV, where the largest \(E_g\) was obtained from specimens sintered at 750 \(^{\circ }\)C. Seebeck coefficient reached maximum of 145 \(\upmu\)V K\(^{-1}\) at 500 K for sample synthesized at 750 \(^{\circ }\)C, which resulted in the highest PF for this specimen among the series, PF = 13 \(\upmu\)W K\(^{-2}\)cm K\(^{-1}\) at 630 K. Good thermoelectric performance encourages further studied of TmNiSb other HH phases synthesized under pressure. We suggest, that in the future the phenomenon might be used for reduction of phonon transport in half-Heusler thermoelectrics, especially for those having intrinsically high thermal conductivity.
Methods
Samples were prepared in the first step by arc-melting the elemental constituent (purities Tm 99.9 at.%, Ni 99.999 at.%, Sb 99.999 at.%). For the sake of homogenization, the buttons were remelted and flipped several times. Small amount of Sb from 2 to 5% range was added in order to compensate for the evaporation losses. The obtained specimens were hand-grounded in agate mortar and consolidated at room temperature into a pellet 10 mm in diameter and 2 mm in height at room temperature and 100 MPa pressure. Then the pellet was placed in the special shape container with the graphite heater inside. After applying the 5.5 GPa pressure, the pellet was heated to assumed temperature with the heating rate 16.7 \(^{\circ }\)C/s. When the specified temperature was reached, the pellet was sintered for 60 s. Then the temperature was lowered to room temperature for 60 s.
Powder X-ray diffraction (XRD) was performed with Xpert Pro PANalytical diffractometer employing Cu K\(\alpha\) radiation. XRD patterns were analyzed by Rietveld method incorporated in FullProf software77. Density was measured with Archimedes method. Morphology and chemical composition as well as crystallographic orientation and grain boundary type were investigated by using a Field Emission Scanning Electron Microscope (FE-SEM) FEI NovaNano SEM 230 equipped with an energy dispersive X-ray spectrometer (EDS) EDAX Genesis XM4 and a high resolution EBSD camera DigiView V (EDAX Inc. OIM AnalysisTM). SEM images and EDS spectra/maps were performed at 5 and 20 kV acceleration voltage, respectively. Moreover, SEM images were recorded in a beam deceleration mode in order to enhance visibility of morphological features. Since, the surface of the TmNiSb sample sintered at 1000 \(^{\circ }\)C was too rough and defected to achieve high quality images of Kikuchi patterns (EBSD), the sample was polished prior to scanning. In the first step, the sample was subjected to mechanical polishing using SiC paper from 500 to 4000 grain and SiO\(_2\) suspension with particle size equal to 0.05 \(\upmu\)m (G &G Surface Technology, Italy) to a total removal the surface deformation layers. After that, the sample was cleaned for several times in an ethanol ultrasonic bath and drying using an infrared lamp. Finally, the polished sample was analysed in the scanning electron microscope (SEM) equipped with a TSL OIM analysis software. The Kikuchi patterns were generated at 30 kV acceleration voltage and 32 nA beam current. To produce a crystallographic orientation map, the electron beam was scanned over a selected surface area and the resulting Kikuchi patterns were indexed and analysed automatically (i.e., the Kikuchi bands were detected by means of the software). An image quality (IQ) parameter and a confidence index (CI) were recorded for each Kikuchi pattern. Based on the analysis of the recorded CI value, a multi-phase analysis was realised.
The phonon frequencies and corresponding thermodynamic properties were calculated under the harmonic approximation by the finite displacement method78,79 and supercell as implemented in the PHONOPY code80. To calculate the total energy of the supercells including displacements, we employed the plane-wave projector augmented wave (PAW) method81 in the framework of DFT within the generalized gradient approximation (GGA) functional in the Perdew–Burke–Ernzerhof (PBE) parameterization82, as implemented in the Quantum ESPRESSO code83. We examined the initial structures of MgAgAs-type, ZrBeSi-type, LiGaGe-type, and TiNiSi-type of TmNiSb. A 0.13 eV smearing width of the Methfessel–Paxton scheme84 was used for all crystal structures except for MgAgAs-type TmNiSb. Force constants were obtained from the cells whose sizes corresponded to the 2\(\times\)2\(\times\)2, 2\(\times\)2\(\times\)1, 2\(\times\)2\(\times\)1, and 2\(\times\)2\(\times\)1 supercells of the primitive MgAgAs-type, ZrBeSi-type, LiGaGe-type, and TiNiSi-type unit cells, respectively. An atomic displacement of 0.01 Å was used for all supercells. The phonon band paths were determined by using SeeK-path85. We used the quasi-harmonic approximation that transforms thermodynamic parameters from the function of volume (V) to the function of pressure (P)86. The Gibbs free energy (G) at the finite P and T was obtained as G(P, T) = min\(_V\)[U\(_{\text{el}}\) +U\(_{\text{ph}}\) + PV - TS\(_{\text{vib}}\)], where U\(_{\text{el}}\), U\(_{\text{ph}}\), and S\(_{\text{vib}}\) are the electronic internal energy at 0 K, the phonon energy, and vibrational entropy, respectively.
Hall effect was investigated in PPMS system produced by Quantum Design using perpendicular 4-point method with horizontal rotator. The electrical contacts were prepared with 50 \(\upmu\)m silver wires and Ag epoxy. Electrical resistivity and Seebeck coefficient in the high-temperature range were measured on commercial Linseis LSR-3 device. To ensure repeatability, for each sample several measurements were made with a different value of the gradient, i.e. 30, 40 and 50 K; in the article results with 50 K gradient are shown. Experimental uncertainties were 3% and 5% for \(\rho\) and S, respectively.
Data availability
The data set relevant for the current study is available from the corresponding author on reasonable request.
References
Bos, J.-W.G. & Downie, R. A. Half-Heusler thermoelectrics: A complex class of materials. J. Phys. Condens. Matter 26, 433201 (2014).
Rogl, G. et al. Mechanical properties of half-Heusler alloys. Acta Mater. 107, 178–195 (2016).
Synoradzki, K., Ciesielski, K. & Kaczorowski, D. Magnetocaloric effect in antiferromagnetic half-Heusler alloy DyNiSb. Acta Phys. Pol. A 133, 691–693 (2018).
Pavlosiuk, O., Kaczorowski, D., Fabreges, X., Gukasov, A. & Wiśniewski, P. Antiferromagnetism and superconductivity in the half-Heusler semimetal HoPdBi. Sci. Rep. 6, 1–9 (2016).
Pietri, R., Andraka, B., Kaczorowski, D., Leithe-Jasper, A. & Rogl, P. Magnetoresistance and low-temperature specific heat of the Yb compounds YbRhSn, YbPdBi, and YbPtSn. Phys. Rev. B 61, 12169–12173 (2000).
de Groot, R. A., Mueller, F. M., Engen, P. G. & Buschow, K. H. J. New class of materials: Half-metallic ferromagnets. Phys. Rev. Lett. 50, 2024–2027 (1983).
Ackerbauer, S.-V. et al. Structural transformation with “negative volume expansion’’: Chemical bonding and physical behavior of TiGePt. Chem. Eur. J. 18, 6272–6283 (2012).
Chadov, S. et al. Tunable multifunctional topological insulators in ternary Heusler compounds. Nat. Mater. 9, 541–545 (2010).
Larson, P., Mahanti, S. & Kanatzidis, M. Structural stability of Ni-containing half-Heusler compounds. Phys. Rev. B 62, 12754 (2000).
Kim, K. S. et al. Direct observation of inherent atomic-scale defect disorders responsible for high-performance Ti\(_{1-x}\)Hf\(_x\)NiSn\(_{1-y}\)Sb\(_y\) half-Heusler thermoelectric alloys. Adv. Mater. 29, 1702091 (2017).
Xie, H. et al. Beneficial contribution of alloy disorder to electron and phonon transport in half-Heusler thermoelectric materials. Adv. Funct. Mater. 23, 5123–5130 (2013).
Xie, H. et al. The intrinsic disorder related alloy scattering in ZrNiSn half-Heusler thermoelectric materials. Sci. Rep. 4, 6888 (2014).
Harmening, T., Eckert, H. & Pöttgen, R. Defects in half-Heusler type antimonides ScTSb (T = Ni, Pd, Pt). Solid State Sci. 11, 900–906 (2009).
Synoradzki, K. et al. Thermal and electronic transport properties of the half-Heusler phase ScNiSb. Materials 12, 1723 (2019).
Ciesielski, K. et al. Structural, thermodynamic and magnetotransport properties of half-Heusler compound HoPtSb. J. Alloys Compd. 829, 154467 (2020).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst. 44, 1272–1276 (2011).
Schwall, M. & Balke, B. Phase separation as a key to a thermoelectric high efficiency. Phys. Chem. Chem. Phys. 15, 1868–1872 (2013).
Rausch, E., Balke, B., Ouardi, S. & Felser, C. Enhanced thermoelectric performance in the p-type half-Heusler (Ti/Zr/Hf)CoSb\(_{0.8}\)Sn\(_{0.2}\) system via phase separation. Phys. Chem. Chem. Phys. 16, 25258–25262 (2014).
Rausch, E. et al. Fine tuning of thermoelectric performance in phase-separated half-Heusler compounds. J. Mater. Chem. C 3, 10409–10414 (2015).
Krez, J. et al. Optimization of the carrier concentration in phase-separated half-Heusler compounds. J. Mater. Chem. A 2, 13513–13518 (2014).
Rausch, E. et al. Short and long range order of half-Heusler phases in (Ti, Zr, Hf)CoSb thermoelectric compounds. Acta Mater. 115, 308–313 (2016).
Page, A., Van der Ven, A., Poudeu, P. F. P. & Uher, C. Origins of phase separation in thermoelectric (Ti, Zr, Hf)NiSn half-Heusler alloys from first principles. J. Mater. Chem. A 4, 13949–13956 (2016).
Schwall, M. & Balke, B. On the phase separation in n-type thermoelectric half-Heusler materials. Materials 11, 649 (2018).
Krez, J. et al. Long-term stability of phase-separated half-Heusler compounds. Phys. Chem. Chem. Phys. 17, 29854–29858 (2015).
Berche, A., Tedenac, J. & Jund, P. Phase separation in the half-Heusler thermoelectric materials (Hf, Ti, Zr)NiSn. Scr. Mater. 139, 122–125 (2017).
Mena, J. M., Schoberth, H. G., Gruhn, T. & Emmerich, H. Nanophase separation in cosb-based half-Heusler thermoelectrics: A multiscale simulation study. Phys. Status Solidi A 213, 706–715 (2016).
Fecher, G. H., Rausch, E., Balke, B., Weidenkaff, A. & Felser, C. Half-Heusler materials as model systems for phase-separated thermoelectrics. Phys. Status Solidi A 213, 716–731 (2016).
Gelbstein, Y., Davidow, J., Girard, S. N., Chung, D. Y. & Kanatzidis, M. Controlling metallurgical phase separation reactions of the Ge\(_{0.87}\)Pb\(_{0.13}\)Te alloy for high thermoelectric performance. Adv. Energy Mater. 3, 815–820 (2013).
Gelbstein, Y. Phase morphology effects on the thermoelectric properties of Pb\(_{0.25}\)Sn\(_{0.25}\)Ge\(_{0.5}\)Te. Acta Mater. 61, 1499–1507 (2013).
Samanta, M. & Biswas, K. Low thermal conductivity and high thermoelectric performance in (GeTe)\(_{1-2x}\)(GeSe)\(_x\)(GeS)\(_x\): competition between solid solution and phase separation. J. Am. Chem. Soc. 139, 9382–9391 (2017).
Girard, S. N. et al. Analysis of phase separation in high performance PbTe–PbS thermoelectric materials. Adv. Funct. Mater. 23, 747–757 (2013).
Wu, H. et al. Broad temperature plateau for thermoelectric figure of merit \(ZT>\) 2 in phase-separated PbTe\(_{0.7}\)S\(_{0.3}\). Nat. Commun. 5, 1–9 (2014).
Hsu, K. F. et al. Cubic AgPbmSbTe\(_{2+m}\): Bulk thermoelectric materials with high figure of merit. Science 303, 818–821 (2004).
Barabash, S., Ozolins, V. & Wolverton, C. First-principles theory of competing order types, phase separation, and phonon spectra in thermoelectric AgPb\(_m\)SbTe\(_{m+2}\) alloys. Phys. Rev. Lett. 101, 155704 (2008).
Liu, J. et al. Achieving high thermoelectric performance with Pb and Zn codoped polycrystalline SnSe via phase separation and nanostructuring strategies. Nano Energy 53, 683–689 (2018).
Mishraa, R. et al. Ternary antimonides YbTSb (T= Ni, Pd, Pt, Cu, Ag, Au)-synthesis, structure, homogeneity ranges, and 121Sb Mössbauer spectroscopy. Z. Naturforsch. B Chem. Sci. 57, 1215–1223 (2002).
Noda, Y., Shimada, M. & Koizumi, M. Synthesis of high-pressure phases of vanadium-cobalt-antimony (VCoSb) and vanadium-iron-antimony (VFeSb) with a dinickel-indium (Ni\(_2\)In)(B\(_{82}\)) type structure. Inorg. Chem. 18, 3244–3246 (1979).
Kong, B., Chen, X.-R., Yu, J.-X. & Cai, C.-L. Structural, elastic properties and pressure-induced phase transition of ‘half-Heusler’ alloy CoVSb. J. Alloys Compd. 509, 2611–2616 (2011).
Hoffmann, R.-D. & Pöttgen, R. Alb\(_2\)-related intermetallic compounds-a comprehensive view based on group-subgroup relations. Z. Kristallogr. Cryst. Mater. 216, 127–145 (2001).
Hartjes, K. & Jeitschko, W. Crystal structures and magnetic properties of the lanthanoid nickel antimonides LnNiSb (Ln = La, Nd, Sm, Gd, Tm, Lu). J. Alloys Compd. 226, 81–86 (1995).
Xie, L. S., Schoop, L. M., Medvedev, S. A., Felser, C. & Cava, R. Pressure-induced structural phase transition in the half-Heusler compound CaAuBi. Solid State Sci. 30, 6–10 (2014).
Shrivastava, D. & Sanyal, S. P. Structural phase transition, electronic and lattice dynamical properties of half-Heusler compound CaAuBi. J. Alloys Compd. 745, 240–246 (2018).
Merlo, F., Pani, M. & Fornasini, M. \(RMX\) compounds formed by alkaline earths, europium and ytterbium—I. Ternary phases with \(M\) = Cu, Ag, Au; \(X\) = Sb, Bi. J. Less Common Met. 166, 319–327 (1990).
Zhu, H. et al. Discovery of TaFeSb-based half-Heuslers with high thermoelectric performance. Nat. Commun. 10, 270 (2019).
Fu, C. et al. Realizing high figure of merit in heavy-band p-type half-heusler thermoelectric materials. Nat. Commun. 6, 1–7 (2015).
Zhu, H. et al. Discovery of ZrCoBi based half Heuslers with high thermoelectric conversion efficiency. Nat. Commun. 9, 2497 (2018).
Young, D. P., Khalifah, P., Cava, R. J. & Ramirez, A. P. Thermoelectric properties of pure and doped FeMSb (M = V, Nb). J. Appl. Phys. 87, 317–321 (2000).
Sportouch, S. et al. Observed properties and electronic structure of RNiSb compounds (R = Ho, Er, Tm, Yb and Y). Potential thermoelectric materials. MRS Proc. 545, 421 (1998).
Ciesielski, K. et al. High-temperature power factor of half-Heusler phases RENiSb (RE = Sc, Dy, Ho, Er, Tm, Lu). J. Alloys Compd. 816, 152596 (2020).
Hohl, H. et al. A new class of materials with promising thermoelectric properties: MNiSn (M = Ti, Zr, Hf). MRS Proc. 478, 109 (1997).
Ciesielski, K. et al. Thermoelectric performance of the half-Heusler phases RNiSb (R= Sc, Dy, Er, Tm, Lu): High mobility ratio between majority and minority charge carriers. Phys. Rev. Appl. 14, 054046 (2020).
Dwight, A. The crystal structure of DyNiSb, DyPtSb and related compounds. Proc. Rare Earth Conf. 11, 642–650 (1974).
Pecharskii, V., Pankevich, Y. & Bodak, O. Crystal structures of the compounds RNiSB with various rare earth elements. Sov. Phys. Crystallogr. 28, 97–98 (1983).
Zhu, P. et al. A new method of synthesis for thermoelectric materials: HPHT. Solid State Commun. 123, 43–47 (2002).
Guo, X. et al. Thermoelectric transport properties and crystal growth of BiSbTe\(_3\) bulk materials produced by a unique high-pressure synthesis. CrystEngComm 15, 7236–7242 (2013).
Sun, H. et al. Effect of HPHT processing on the structure, and thermoelectric properties of Co\(_4\)Sb\(_{12}\) co-doped with Te and Sn. J. Mater. Chem. A 3, 4637–4641 (2015).
Zhang, Y. et al. Effect of high pressure on thermoelectric performance and electronic structure of SnSe via HPHT. J. Alloys Compd. 667, 123–129 (2016).
Sun, B. et al. Effect of high-temperature and high-pressure processing on the structure and thermoelectric properties of clathrate Ba\(_8\)Ga\(_{16}\)Ge\(_{30}\). J. Phys. Chem. C 120, 10104–10110 (2016).
Wolańska, I. et al. Enhanced thermoelectric power factor of half-Heusler solid solution sc\(_{1-x}\)tm\(_x\)nisb prepared by high-pressure high-temperature sintering method. Mater. Chem. Phys. 227, 29–35 (2019).
Synoradzki, K., Ciesielski, K., Kępiński, L. & Kaczorowski, D. Power factor enhancement in a composite based on the half-Heusler antimonide TmNiSb. J. Appl. Phys. 123, 235101 (2018).
Ciesielski, K., Synoradzki, K., Wolańska, I., Stuglik, P. & Kaczorowski, D. High-temperature thermoelectric properties of half-heusler phases er\(_{1-x}\)ho\(_{x}\)nisb. Mater. Today Proc. 8, 562–566 (2019).
Gnida, D., Ciesielski, K. & Kaczorowski, D. Origin of the negative temperature coefficient of resistivity in the half-Heusler antimonides LuNiSb and YPdSb. Phys. Rev. B 103, 174206 (2021).
Synoradzki, K., Ciesielski, K., Kępiński, L. & Kaczorowski, D. Thermoelectric properties of (DyNiSn)\(_{1-x}\)(DyNiSb)\(_{x}\) composite. Physica B 536, 659–663 (2018).
Synoradzki, K., Ciesielski, K., Kępiński, L. & Kaczorowski, D. Effect of secondary LuNiSn phase on thermoelectric properties of half-Heusler alloy LuNiSb. Mater. Today Proc. 8, 567–572 (2019).
Winiarski, M. J., Bilińska, K., Ciesielski, K. & Kaczorowski, D. Thermoelectric performance of p-type half-Heusler alloys ScMSb (M = Ni, Pd, Pt) by ab initio calculations. J. Alloys Compd. 762, 901–905 (2018).
Ciesielski, K., Wolańska, I., Synoradzki, K., Szymański, D. & Kaczorowski, D. Mobility ratio as a probe for guiding discovery of thermoelectric materials: The case of half-Heusler phase ScNiSb\(_{1-x}\)Te\(_x\). Phys. Rev. Appl. 15, 044047 (2021).
Winiarski, M. J. & Bilińska, K. High thermoelectric power factors of p-type half-Heusler alloys YNiSb, LuNiSb, YPdSb, and LuPdSb. Intermetallics 108, 55–60 (2019).
Öğüt, S. & Rabe, K. M. Band gap and stability in the ternary intermetallic compounds NiSnM (M = Ti, Zr, Hf): A first-principles study. Phys. Rev. B 51, 10443–10453 (1995).
Kirievsky, K., Fuks, D. & Gelbstein, Y. Composition conserving defects and their influence on the electronic properties of thermoelectric TiNiSn. Phys. Chem. Chem. Phys. 22, 8035–8047 (2020).
Rykavets, Z., Ryzhak, D., Romaka, L., Shvachko, S. & Romaka, V. Experimental and first-principle study of TmNiSb half-Heusler alloy. In Litteris et Artibus 1, 262–264 (2017).
Tang, Y. et al. Impact of ni content on the thermoelectric properties of half-Heusler TiNiSn. Energy Environ. Sci. 11, 311–320 (2018).
Kawano, K., Kurosaki, K., Sekimoto, T., Muta, H. & Yamanaka, S. Effect of Sn doping on the thermoelectric properties of ErNiSb-based p-type half-Heusler compound. Appl. Phys. Lett. 91, 062115 (2007).
Li, S., Zhao, H., Li, D., Jin, S. & Gu, L. Synthesis and thermoelectric properties of half-Heusler alloy YNiBi. J. Appl. Phys. 117, 205101 (2015).
Gofryk, K. et al. Magnetic, transport, and thermal properties of the half-Heusler compounds ErPdSb and YPdSb. Phys. Rev. B 75, 224426 (2007).
Snyder, G. J. et al. Weighted mobility. Adv. Mater. 32, 2001537 (2020).
Wang, H., Pei, Y., LaLonde, A. D. & Snyder, G. J. Material design considerations based on thermoelectric quality factor. In Thermoelectric Nanomaterials 3–32 (Springer, 2013).
Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Physica B 192, 55–69 (1993).
Parlinski, K., Li, Z. & Kawazoe, Y. First-principles determination of the soft mode in cubic ZrO\(_2\) direct space approach. Phys. Rev. Lett. 78, 4063–4066 (1997).
Kresse, G., Furthmüller, J. & Hafner, J. Ab initio force constant approach to phonon dispersion relations of diamond and graphite. Europhys. Lett. 32, 729–734 (1995).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Giannozzi, P. et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Methfessel, M. & Paxton, A. T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 40, 3616–3621 (1989).
Hinuma, Y., Pizzi, G., Kumagai, Y., Oba, F. & Tanaka, I. Band structure diagram paths based on crystallography. Comput. Mater. Sci. 128, 140–184 (2017).
Togo, A., Chaput, L., Tanaka, I. & Hug, G. First-principles phonon calculations of thermal expansion in Ti\(_3\)SiC\(_2\), Ti\(_3\)AlC\(_2\), and Ti\(_3\)GeC\(_2\). Phys. Rev. B 81, 174301 (2010).
Acknowledgements
The authors gratefully thank Dr. Paweł Głuchowski for his help with sintering of the materials. This work was supported by the National Science Centre (Poland) under research Grants No. 2015/18/A/ST3/00057 and 2021/40/Q/ST5/00066. For the purpose of Open Access, the author has applied a CC-BY public copyright licence to any Author Accepted Manuscript (AAM) version arising from this submission. Open Access of the publication was supported by the AGH Excellence Initiative - Research University program (IDUB AGH, Action 9).
Author information
Authors and Affiliations
Contributions
K.C. and P.O. performed the syntheses and pXRD experiments, D.S. carried out SEM-EDS imaging, K.B. and D.S. conducted EBSD imaging, K.S. and K.C. performed transport properties measurement, K.T. and K.K. carried out ab-initio calculations, and D.K. was responsible for project supervision. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ciesielski, K., Synoradzki, K., Szymański, D. et al. Half-Heusler phase TmNiSb under pressure: intrinsic phase separation, thermoelectric performance and structural transition. Sci Rep 13, 1592 (2023). https://doi.org/10.1038/s41598-023-28110-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-28110-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
