
Abstract
Pancreatic-β-cell-specifying transcription factor Nkx6.1, indispensable for embryonic development of the pancreatic epithelium and commitment to β-cell lineage, directly controls the expression of a glucose transporter (Glut2), pyruvate carboxylase (Pcx), and genes for insulin processing (endoplasmic reticulum oxidoreductase-1β, Ero1lb; zinc transporter-8, Slc30a8). The Nkx6.1 decline in aging diabetic Goto-Kakizaki rats contributes to β-cell trans-differentiation into δ-cells. Elucidating further Nkx6.1 roles, we studied Nkx6.1 ablation in rat INS-1E cells, prepared by CRISPR/Cas9 gene editing from single colonies. INS-1ENkx6.1–/– cells exhibited unchanged glucose-stimulated insulin secretion (GSIS), moderately decreased phosphorylating/non-phosphorylating respiration ratios at high glucose; unchanged but delayed ATP-elevation responses to glucose; delayed uptake of fluorescent glucose analog, but slightly improved cytosolic Ca2+-oscillations, induced by glucose; despite approximately halved Glut2, Pcx, Ero1lb, and Slc30a8 expression, and reduced nuclear receptors Nr4a1 and Nr4a3. Thus, ATP synthesis was time-compensated, despite the delayed GLUT2-mediated glucose uptake and crippled pyruvate-malate redox shuttle (owing to the PCX-deficiency) in INS-1ENkx6.1–/– cells. Nkx6.1 thus controls the expression of genes that are not essential for acute insulin secretion, the function of which can be compensated for. Considerations that Nkx6.1 deficiency is an ultimate determinant of β-cell pathology beyond cell trans-(de-)differentiation or β-cell identity are not supported by our results.
Similar content being viewed by others

Introduction
Recent advances in studies of the etiology of type 2 diabetes focus on a consensus, pointing out to the “altered β-cell identity” as a concept describing processes in type 2 diabetes development in humans1, in contrast to concepts2 of pancreatic β-cell de-differentiation and/or trans-differentiation, which were previously considered to be essential causes1,2,3,4,5,6,7,8,9,10. The maintained β-cell identity allows an altered transcriptome and metabolome, but only when the main minimum functional features of pancreatic β-cells are conserved. The precise number and nature of these minimum features is currently unknown.
Pancreatic β-cell dedifferentiation denotes a regression toward an earlier progenitor state with a stem-cell-like phenotype, whereas transdifferentiation means a change into other islets cell types, mostly to α-cells or δ-cells1,2,3,4,5,6,7,8,9,10. Progress in our understanding of the transcription factors controlling rodent embryonic development of the pancreatic epithelium and commitment to β-cell lineage identified several transcription factors as “β-cell-specifying”11,12, such as Nkx6.1, Nkx2.2, Pdx1, FoxA2, Six2 and Six3.
Nkx6.1 is a homeobox-containing transcription factor13, with increased expression in the trunk area of the pancreatic epithelium from mouse embryonic days 9.5 to 13 (E9.5 to E13), and it determines the later endocrine lineage12. Pancreatic progenitor cell (Pdx1+/Nkx6.1+), endocrine progenitor cell (Ngn3+/Nkx6.1+), and pancreatic β-cell lineage can be recognized, i.e. those expressing the insulin gene (Ins), (Ins+/Gcg–/Pdx1+/Nkx6.1+ after E13)14. Numerous relationships between transcription factors have been reported, while details of the differentiation into functional β-cells have been discovered4,5,10,11,12.
Another sorting was performed for mice, based on expression of the Flattop gene (Fltp) and Wnt/Pcp effector gene15. Thus the predominant β-cell fraction was Nkx6.1+Fltp+, which has high insulin, high oxidative phosphorylation (OXPHOS), i.e. the mitochondrial activity essential for glucose-stimulated insulin secretion (GSIS), and higher metabolism; while exhibiting a certain specific gene expression pattern. Another fraction (up to 10%) of so-called “hub” β-cells was identified in mouse pancreatic islets, which influences the remaining “follower’’ population of β-cells16. These “hub” β-cells have lower insulin granule content, reduced expression of Pdx1 and drastically low Nkx6.1 levels; but exhibit an essential orchestrating role during glucose-induced Ca2+ signaling16. These findings demonstrated a lack of exclusivity of Nkx6.1 for β-cells. The transcription factor Nkx6.1 could be recognized, together with Nkx2.2, Pdx1, FoxA2, Six2 and Six3, as β-cell-specifying factors, however with the exception of “hub” β-cells, which have almost no Nkx6.115.
Previous experiments with mice have suggested an essential role of Nkx6.1 for β-cells, claiming an indisputable Nkx6.1 requirement for β-cell homeostasis and GSIS17,18. GSIS was also found to be suppressed in INS-1 832/13 cells19. The overexpression of Nkx6.1 led to increased GSIS17. Nkx6.1 was reported to upregulate genes for insulin processing, such as endoplasmic reticulum (ER) oxidoreductase-1β (Ero1lb) and zinc transporter-8 (Slc30a8); glucose homeostasis genes, such as those encoding glucose transporter (Glut2), and pyruvate carboxylase (Pcx)18; likewise the enzyme of the last gluconeogenesis step, glucose-6-phosphatase catalytic subunit-2 (G6pc2)18. Also, some genes required for β-cell proliferation are directly induced by Nkx6.1, such as Aurora kinase A (Aurka, which degrades the cell cycle regulator p53)20; G1/S-specific cyclin-D2 (Ccnd2)18; G0/G1 switch regulatory protein-7 (c-Fos); and nuclear receptor subfamily-4 group A member-1 (Nr4a1) and 3 (Nr4a3)18. Transcription factors of β-cell development are also under the control of Nkx6.118, notably Rfx6, MafA, Mnx1, and Tle3.
The suppression of Nkx6.1 typically activates neurogenin 3 (Ngn3) in β-cells, hence stimulating their transdifferentiation into δ-cells15. This explains our observed Nkx6.1 decrease in aging diabetic Goto-Kakizaki rats in association with β-cell transdifferentiation into δ-cells21,22. Nkx6.1 also competes with Pax6 for the G1 element of the glucagon (Gcg) promoter23. Nkx6.1-deficient mice reportedly have a decreased pancreatic β-cell number24 and a defect in nutrient-induced β-cell expansion, associated with a low expression of glucagon-like peptide-1 (GLP-1) receptor (Glp1r)25.
To further investigate the role of Nkx6.1 in the function of β-cells, we prepared rat insulinoma INS-1E cells lacking Nkx6.1 (INS-1ENkx6.1–/– cells), using CRISPR/Cas9 gene editing. We initially intended to modify this pancreatic β-cell model into a presumable model of trans/de-differentiation (or altered β-cell identity). However, unlike in the previous reports18,19, we still found efficient GSIS, equal to the control cells, whereas ATP-elevations were delayed, but reached the same maximum, and ATP synthesis intensity (the latter determined from respiration parameters) was only slightly decreased. We conclude that INS-1ENkx6.1–/– cells are not dedifferentiated β-cells, but due to their phenotype, they instead resemble the “hub” β-cells.
Results
Unaffected GSIS in Nkx6.1-ablated INS-1E cells
Similarly to control INS-1Escrl/scrl cells (Fig. 1a), INS-1ENkx6.1–/– cells exhibited a relatively fast kinetics of insulin secretion stimulated by 20 mM glucose after 1-h pre-incubation in the KRH medium, containing 3 mM glucose (Fig. 1). These cells responded with a very low insulin secretion rate, when kept still at 3 mM glucose (not shown, but see Ref.26,27,31). When after 1-h pre-incubation, glucose was initially adjusted to 20 mM, a significant insulin release occurred in both control INS-1Escrl/scrl cells as well as two lines, a and b, of INS-1ENkx6.1–/–cells (Fig. 1a). For the lack of Nkx6.1 in these two cell lines originating from single cells see Fig. 1b and Supplementary Information Fig. S1. The control INS-1Escrl/scrl cells were prepared using the same CRISPR/Cas9 gene editing procedure followed by line selection from a single cell, however using a scrambled gRNA sequence. The rates of GSIS, obtained by linear regressions of data, were calculated to be 101% ± 20% and 99% ± 30% for lines a and b of INS-1ENkx6.1–/–cells, respectively. The amounts of insulin released at 60 min exhibited statistically insignificant differences. Control INS-1Escrl/scrl cells exhibited similar GSIS rates to parental (wt) INS-1E cells (not shown, see Refs.26,27,31).

(a) Insulin secretion kinetics is not affected by Nkx6.1 ablation—insulin secretion was assayed in triplicates for control INS-1Escrl/scrl cells (black) or lines (a) (yellow green) and (b) (green) of INS-1ENkx6.1–/– cells, when stimulated with 20 mM glucose adjustment after 1-h preincubation in the KRH medium, containing 3 mM glucose. Data were fit by linear regressions yielding GSIS rates of 101% ± 20% and 99% ± 30% for lines (a) and (b) relatively to INS-1Escrl/scrl cells. (b) Nkx6.1 protein expression.
Retained glucose-induced ATP elevations in Nkx6.1-ablated INS-1E cells
Since besides redox signaling26, GSIS depends strongly on OXPHOS, and hence on the elevated ATP synthesis resulting from the enhanced glucose catabolism26,27,28,29,30,31,32, we further estimated total cellular ATP levels, which increase in response to the elevated glucose. After 1-h pre-incubation with 3 mM glucose and subsequent adjustment of glucose to 20 mM, both INS-1ENkx6.1–/– line a and line b cells exhibited about a 10-min and 20-min delayed time course of the increase in total ATP levels, respectively, with almost unchanged amplitudes of maxima (Fig. 2; for wt parental cells, see also Ref.31).

Time course of ATP elevation after 3 to 20 mM glucose adjustment—relative ATP amounts assayed by bioluminescence after adjustment of glucose from 3 to 20 mM are plotted for parental INS-1E cells (wt, gray), INS-1Escrl/scrl cells (black) and line (a) (yellow green) and line (b) of INS-1ENkx6.1–/– cells (green ).
Insignificantly altered OXPHOS in Nkx6.1-ablated INS-1Ecells
Since the OXPHOS intensity can be semi-quantified from the respiratory parameters27,31, we further estimated the phosphorylating respiration rate (V3) (Fig. 3a), non-phosphorylating respiration rate (V4; established with oligomycin) and maximum respiration rate (Vmax; determined as maximum from the sequential titrations with aliquots of a mitochondrial uncoupler FCCP) (Fig. 3a–d). When calculated for average respiration rates for transitions from 3 to 20 mM glucose (number of cell samples N = 3–6; number of estimations n = 5–13), the parameters Rr = V3/V4 increased from 1.1 ± 0.2 to 1.6 ± 0.1 and from 1.2 ± 0.5 to 2.2 ± 0.2 for the two INS-1ENkx6.1–/– lines a and b, respectively, compared to the increase from 1.6 ± 0.3 to 2.1 ± 0.4 in INS-1Escrl/scrl cells. A slightly diminished Rr for line a, notably its lower rise, indicates a slightly decreased intensity of ATP synthesis (OXPHOS is zero when Rr = 1), and its reduced increase with low-to-high glucose transition. Similar data were obtained when calculated for each individual cell batch and averaged afterwards (Fig. 3b).

Phosphorylating respiration is not affected by Nkx6.1 ablation—panels show for wt (gray), INS-1Escrl/scrl (black), line a (green) and b (dark green) of INS-1ENkx6.1–/– cells at 3 mM and 20 mM glucose: (a) Rates of phosphorylating (endogenous) respiration (V3); (b) parameters Rr = V3/V4, i.e. ratios of V3 to non-phosphorylating respiration rates (V4), which reflect OXPHOS intensity; (c) parameters Ar = (V3 – V4) / FCCP, where the FCCP is the uncoupled rate in the presence of uncoupler FCCP (titrated to optimum concentration yielding a maximum rate); (d) Ratios of FCCP/V4. Number of cell passages N was 3–6 on which n of 5–13 respiration assays were performed. ANOVA: ***p < 0.001; **p < 0.05.
Next, we calculated the parameters Ar = (V3 – V4)/FCCP, where the FCCP rate is equal to Vmax. The parameter Ar indicates the maximum capacity of the respiratory chain, which is used for ATP synthesis. For transitions from 3 to 20 mM glucose, calculations using average rates yielded increases in parameters Ar from 0.14 ± 0.05 to 0.19 ± 0.09, and from 0.1 ± 0.06 to 0.44 ± 0.05 for lines a and b, respectively; compared to the increase from 0.13 ± 0.09 to 0.36 ± 0.17 for INS-1Escrl/scrl cells. Individual calculations of Ar gave similar data (Fig. 3c). Note that all cell lines, parental, and INS-1Escrl/scrl INS-1ENkx6.1–/– lines a and b exhibited nearly identical ratios of Vmax to the non-phosphorylating respiration rate V4 (Fig. 3d), which is substantiated by the existing H+ leak. We conclude that INS-1ENkx6.1–/– lines a and b exhibit bioenergetics parameters that are compatible with insignificantly altered ATP synthesis and reflect its increase upon the transition between 3 and 20 mM glucose.
Delayed 6-NBD deoxyglucose uptake in Nkx6.1-ablated INS-1E cells
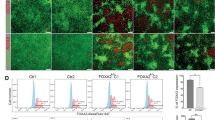
In agreement with the previously reported Nkx6.1-mediated control of the gene for a glucose transporter (Glut2)18, we observed decreased and/or delayed transport of fluorescent glucose analog 6-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-6-deoxyglucose (6-NBDG, present at 100 μM) into INS-1ENkx6.1–/– cells, relative to control INS-1Escrl/scrl cells (Fig. 4). J6-NBDG rates of increases in integral fluorescence intensities in regions of interest (ROI, equal to individual cells, Fig. 4a,b) were faster in control and parental cells in contrast to slower rates in INS-1ENkx6.1–/– lines a and b (Fig. 4c–f), when the inhibited transport rates by the specific GLUT1,2 inhibitor BAY-876 were subtracted. 100 μM BAY-876 blocked J6-NBDG rates by ~ 95%, while 25 mM glucose caused a ~ 50% inhibition of J6-NBDG rates. Therefore, we can conclude that the recorded J6-NBDG rates predominantly reflected the GLUT2 transport activity and were delayed by 40% in INS-1ENkx6.1–/– lines a and b (Fig. 4g) in which Glut2 transcript was approximately halved (Fig. 4h).

Glucose uptake is delayed Nkx6.1-ablated INS1E cells containing around half GLUT2, but intact KATP channel—(b–g) 6-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-6-deoxyglucose (6-NBDG) uptake into cells assessed in a confocal microscope; (h) relative transcripts rates of GLUT2; (i) Responses of Tl+ fluxes to glucose as surrogate for K+ fluxes, to which a KATP is a major contributor. Panels (a,b) shows confocal images at time zero (a), when wt INS-1E cells are dark and the just added 100 μM 6-NBDG causes a fluorescence background around cells; and 20 min after (b), when cells are filled with 6-NBDG. (c) Compares time-courses averaged from individual time courses, displayed on panels (d–f). Basal rates were already subtracted, i.e. those when the GLUT2 inhibitor (100 μM BAY-876) blocked the 6-NBDG uptake by up to ~ 95%. (g) Shows statistics of initial rates of the 6-NBDG uptake. (h) Shows relative Glut2 transcripts amounts. (i) Indicates no closure of KATP channels in the absence of glucose (Tl+ influx rate is normalized to 100%) and equal glucose-stimulated closure of KATP channels independent of the Nkx6.1 ablation.
Retained function of ATP-sensitive K+ channel in Nkx6.1-ablated INS-1E cells
INS-1ENkx6.1–/– cells, preincubated for 1-h in KRH with 3 mM glucose, exhibited a similar rate of the Tl+ influx (Tl+ as a surrogate for K+), which was completely blocked by glibenclamide (Fig. 4i). After setting glucose to 20 mM, the resulting Tl+ influx almost fell to that of glibenclamide in both INS-1ENkx6.1–/– and INS-1Escrl/scrl cells. These results demonstrate the unaffected closure of the ATP-sensitive K+ channel (KATP) by the Nkx6.1 deletion.
Unchanged cytosolic Ca2+ oscillations during GSIS in Nkx6.1-ablated INS-1E cells
Since so-called “hub” β-cells, lacking Nkx6.1, were reported to likely possess an enhanced Ca2+-signaling 16, we evaluated oscillations in cytosolic Ca2+ at increasing glucose between 3 and 20 mM, monitored in cells with an expressed slow variant of the GCaMP6 fluorescence probe (Supplementary Information Fig. S2) by confocal microscopy (Fig. 5a). Fluorescence was collected from ROI within each single cell. However, the intensity and overall character of cytosolic Ca2+-oscillations was dependent on the ROI of choice, since some cells did not exhibit Ca2+-oscillations. Even when we did not account for an unknown noise content in the signal, cytosolic Ca2+-oscillations [Ca2+]c(t) exhibited very similar dependences of peak intensity histograms on glucose concentration in the responsive cells of INS-1ENkx6.1–/– and INS-1Escrl/scrl cell lines as well as parental wt cells (Fig. 5b–e). However, there were distinctions in [Ca2+]c(t) histograms at 3 mM glucose, at which much higher peak intensities were encountered in both INS-1ENkx6.1–/– cell lines a and b, so the histogram was significantly shifted to the right towards higher peak intensity values (Fig. 5b). Such a phenotype of excitability is natural for actual hub β-cells.

Ca2+ oscillations are slightly improved at 3 and 20 mM glucose in Nkx6.1-ablated INS-1E cells as surveyed by GCaMP6 fluorescence probe—(a) shows typical oscillations in cytosolic Ca2+, monitored by confocal microscopy of GCaMP6 fluorescence probe in each single cell: color-coding indicates Ca2+-oscillations after glucose adjustment to distinct concentrations as indicated. In the end, 5 μM nimodipine was added. (b–e) Show the corresponding histograms of intensities of Ca2+-oscillations, binned in deciles for INS-1Escrl/scrl cells (left, black), grouped data of lines (a) and (b) of INS-1ENkx6.1–/– cells (middle, green) and wt cells (right, gray). Around 1000 Ca2+-oscillation peaks were taken into the statistics for each condition.
With increasing glucose concentration to 5 mM and 9 mM, [Ca2+]c(t) spikes of up to ~ 33% of the maximum intensity were the most frequent and even higher intensities were abundant (Fig. 5c,d). However, histograms for wt cells, INS-1Escrl/scrl cells as well as INS-1ENkx6.1–/– cells had the same characteristics. A slight improvement in [Ca2+]c(t) oscillations was again encountered for INS-1ENkx6.1–/– cells at 20 mM glucose, at which INS-1Escrl/scrl cells had almost 40% of the lowest decile intensity, whereas INS-1ENkx6.1–/– cells only had 30% and peak of higher intensity peaks were more frequently present (Fig. 6e). In all cells, 5 μM nimodipine blocked [Ca2+]c oscillations (Fig. 5a, red part of the trace).

mtDNA copy number is not affected by Nkx6.1 ablation—the copy number evaluated as described in “Methods” is compared for parental INS-1E cells (wt, gray; number of passages N was 6), INS-1Escrl/scrl cells (black N was 4) and line a (green, green points) and line b (green, blue points) of INS-1ENkx6.1–/– cells (N was 3 for each).
Unaltered expression of OXPHOS-related proteins in Nkx6.1-ablated INS-1Ecells
The moderately decreased OXPHOS in INS-1ENkx6.1–/– cells was also in accordance with findings of the unchanged copy number of mitochondrial DNA (mtDNA) (Fig. 6) and higher transcript levels of PGC1α protein (Fig. 7). The latter suggests the possibility of enhanced expression of relevant proteins under conditions, in which the expression of mtDNA-encoded subunits of respiratory chain complexes and ATP-synthase is still not saturated, hence there is no need for higher mtDNA replication.

Change in selected transcripts upon Nkx6.1 ablation—a volcano plot shows increased (right) and decreased (left) transcripts of selected genes as indicated on a log2 scale, while the z-axis shows the probability of such changes (statistical significance of the assay evaluated by Student’s T-tests). Grouped data for lines a and b of INS-1ENkx6.1–/– cell are compared to INS-1Escrl/scrl cells.
Pattern of transcripts for selected transcription factors and GSIS-relevant genes in Nkx6.1-ablated INS-1Ecells
Figure 7 shows a Volcano plot of changes in the transcripts of selected transcription factors and GSIS-relevant genes. Data for lines a and b were not significantly different (see examples in Fig. S3), hence the respective data were grouped together. Besides the undetectable Nkx6.1 mRNA, there is also a significantly enhanced expression of Nrf1, Mfn2, and Pgc1a (and slightly for Ssb), i.e. important factors of mitochondrial biogenesis. The transcript of the Kcnj11 gene, which encodes the KATP channel subunit KIR6.2, was also elevated; unlike Abcc8 encoding SUR1; whereas the transcription of the CaL-encoding gene Cacna1d was reduced in in INS-1ENkx6.1–/– cells. The most significant decrease in transcription was found for Pcx, which encodes pyruvate carboxylase (Figs. S3a, 7), as well as for Glut2, which encodes the glucose transporter GLUT2 (Figs. 4h, 7), the Slc30a8 gene of the zinc transporter-8, and genes encoding the nuclear receptor subfamily 4 group A member 1 and 3, i.e. Nr4a1 (Fig. S3c) and Nr4a3. The most reduced expression was found for endoplasmic reticulum oxidoreductase-1β (Ero1lb), which was downregulated to a high probability of 26% (Figs. S3b, 7). As for the lactate dehydrogenase Ldha, a so-called disallowed gene in pancreatic β-cells, it remained at very low expression that was unaffected by the Nkx6.1 knockdown (Fig. 7). These data confirmed the pattern of previously established genes18 regulated by Nkx6.1.
Discussion
We reinvestigated on the status of Nkx6.1 as an important transcription factor for embryonic β-cell development12,13,14 and for nutrient-induced β-cell expansion25. Our main conclusion is that the protein machinery required for the acute triggering mechanism of GSIS is not dependent on Nkx6.1. We demonstrated that the metabolic plasticity of INS-1ENkx6.1–/– cells is relatively wide, so that GSIS is largely maintained, due to the maintained OXPHOS. Only transients from a lower to higher intensity of ATP synthesis induced by higher glucose intake are delayed due to the Nkx6.1 deficiency and the resulting deficiency of GLUT2. This result contrasts with previous measurements18,19, which, however, did not assess the kinetics of the insulin release. Similarly as with the deficiency of DAPIT (a membrane FO-ATP-synthase subunit)31, this should also be partly explained by the fact that the redox signaling26 by NOX4-derived H2O2 cannot be suppressed by the Nkx6.1 deficiency, as reflected by the preserved normal closure of KATP channels upon the addition of glucose. Despite elevations in ATP after the initial glucose intake being delayed by ~ 10 min in INS-1ENkx6.1–/– cells vs. control INS-1Escrl/scrl cells, the time compensated OXPHOS was sufficient for sustained GSIS.
The Nkx6.1 ablation was confirmed to reduce the GLUT2 expression18, resulting in a delayed glucose uptake. In accordance with the reduced Glut2, we demonstrated that the kinetics of glucose uptake exhibits a distinct slowdown. This was reflected by concomitantly delayed ATP elevations, induced by glucose, that however later reached sufficiently high levels that reflected high OXPHOS levels. Thus OXPHOS was time-compensated. On the other hand, a somewhat premature Ca2+-influx triggering at 3 mM glucose (a property of hub β-cells) and unaffected KATP channel closure-responses keep orchestrated insulin granule vesicles exocytosis and hence lead to the preserved insulin secretion kinetics. Since the time integral of the latter is rather directly proportional to the released number of insulin granule vehicles, the total amount of insulin released over 60 min as well as the kinetics of such release should not be influenced very much.
The Pcx deficiency18, i.e. decrease in pyruvate carboxylase, might be manifested as a partial blockage of the the pyruvate-malate shuttle, i.e. one of the three pyruvate redox shuttles26 (Fig. S4). However, compensation, by the other two shuttles might be sufficient, but disadvantageous perhaps during the early onset of type 2 diabetes. One may speculate for INS-1ENkx6.1–/– cells with a deficiency in the pyruvate-malate shuttle, that there is a faster NADH release to the cytosol27 and hypothetically a fractionally faster respiration (for a given pyruvate or glucose intake). This is relative to control cells, where the shuttle function disables the production of NADH by the malate dehydrogenase. Instead, transport and the cytosolic enzyme reaction sequence enables the export of redox equivalents of such NADH to the cytosol, which also decreases superoxide release into the matrix27. Thus speculatively, the Pcx deficiency might lead to higher superoxide release into the matrix and cells could be more vulnerable to oxidative stress.
Also, despite INS-1ENkx6.1–/– cells exhibiting reduced GLUT2 expression, possible expected resulting growth defects are likely to be compensated. This would proceed simply by allowing a longer development of the related phenomena, such as the glucose-stimulated expression of genes important for β-cells. Moreover, the observed enhanced expression of Nrf1, Mfn2, and Pgc1a (slightly for Ssb), which are the important factors of mitochondrial biogenesis, reflects the adaptability of INS-1E cells which lack an important regulatory transcription factor such as the Nkx6.1. Thus the enhanced mitochondrial biogenesis can balance any deteriorating effects of the Nkx6.1 deficiency. This is reflected by the unchanged mt DNA copy number in INS-1ENkx6.1–/– cells.
Despite reduction in the transcription of the CaL-encoding gene Cacna1d, Ca2+-oscillations [Ca2+]c(t) exhibited the same peak intensity histograms at 5 and 9 mM glucose and slightly more intensive Ca2+-spikes at 20 mM glucose were found for INS-1ENkx6.1–/–, as compared to INS-1Escrl/scrl cells. However, these subtle differences do not entitle us to conclude that INS-1ENkx6.1–/– phenocopies hub β-cells.
As previously reported for mice with Nkx6.1 ablation18, genes important for the biogenesis of insulin, such as endoplasmic reticulum oxidoreductase-1β (Ero1lb) and zinc transporter-8 (Slc30a8) were also significantly reduced in INS-1ENkx6.1–/– cells. This could lead to somewhat lower total insulin storage. However, in cultured cells the concomitantly slower insulin biogenesis did not prevent the acute insulin release stimulated by glucose. Most likely this is because the amount of insulin released during GSIS in vivo accounts for only a few percent of the total stored insulin. When the triggering machinery secretes an equal amount of insulin, then only a higher relative percentage of the stored insulin granule vesicles is released. Likewise, chronically after a sufficient time given that insulin granules are still sufficiently produced in cultured INS-1ENkx6.1–/– cells. Importantly, the expression of the disallowed Ldha gene was kept to a minimum.
Our findings contribute to the discussion on what a β-cell is. More and more attempts to define healthy and functional pancreatic β-cell are leading to a consensus understanding that there is a spectrum of β-cell subtypes1,10, which is relatively dynamic and results not only from the pancreatic islet embryonic development, but also from biogenesis maintenance in the adult period. Unfortunately, distinct islet morphological organization and biogenesis in rodents vs. humans makes the research on rodent models less relevant for translation into human medicine. Nevertheless, easier possibilities of gene editing could justify murine studies, as we present in this work.
Bearing in mind that pancreatic islets also contain a heterologous population of β-cells also under physiological or pre-diabetic conditions, we need some definition33 of a “minimum β-cell”, which requirements should be met and what aspects are not lost, for the transformed cell to not be considered as dedifferentiated or having lost its β-cell identity. In the past, a cell was considered to be a β-cell when its transcriptome involves mRNAs for certain key β-cell transcription factors2,17,34,35, including the expression of MafA, Neurod1, Foxo1, Pdx1 and notably Nkx6.1; in addition to properly tuned Ins expression and “correct” machinery for GSIS and insulin secretion induced by other major stimuli, e.g. by branched-chain keto acids and fatty acids. Notably, fetal β-cells also express36 also Nkx2.2 and Isl1. Our data of unchanged MafA and Pdx1 not only demonstrated the independence of these factors from Nkx6.1, but confirmed that these should belong to the list, probably being indispensable for β-cells. In contrast, in addition to the hub β-cells, we demonstrate that Nkx6.1 is dispensable for acute GSIS and should not be used as a β-cell marker.
The advent of single cell transcriptomics and mass cytometry also allowed a new definition of four sub-types of human β-cells, with the predominant β1 population and less glucose-responsive β2, β3 and β4 cells37. These subtypes exhibit different expression37 of ganglioside synthase ST8SIA1 and cell surface glycoprotein CD9. The human transcriptome contains exclusively or highly expressed PAX4, PDX1, MAFA, MAFB, DLK1, SIX2/3, ID1, IAPP, UCN3, and OLIG1938,39, plus crucial SIX2 and SIX340,41,42.
The change in β-cell identity caused by dedifferentiation involves declines in specific proteins, including insulin, and decreasing expression of transcription factors, which were originally enriched in β-cells, together with restoring the expression of so-called β-cell disallowed genes, such as lactate dehydrogenase (LDH) and monocarboxylate transporter MCT111,12,13,14,15,16. Upon dedifferentiation, expression is also elevated for transcription factors of progenitor cells that act during the original stem cell differentiation into β-cells. Thus, the expression of Neurog3, Nanog, Pax4 and Sox9 is enhanced2. Note that Neurog3 expression induces precursors of five endocrine (islet) cell types. Transcription factors specific for other islet cell types are repressed upon dedifferentiation, but restored upon trans-differentiation into the α-cells (Arx)17 or δ-cells (homeodomain transcription factor Hhex hematopoietically expressed homeobox)43, respectively. All these changes lead to the concomitant impairment of GSIS as a unique β-cell function, which is also caused by the reduced insulin gene (Ins) expression1,2,3,4,5,6,7,8,9,10.
Islets from type-2 diabetic patients contained a threefold elevated fraction of β-cells incapable of secreting any of the major pancreatic hormones and deficient in insulin, but expressing synaptophysin and aldehyde dehydrogenase 1 isoform A3 (ALDH1A3)44. Also, an increased fraction of chromogranin-A-positive cells was found in diabetic islets45,46. Nevertheless, other studies excluded the existence of a major β-cell de-differentiation in humans. Instead an “altered identity” was suggested as a term that better described the processes in type 2 diabetes development in humans1. For example, insulin-glucagon bihormonal cells exist as a 3 to 4% fraction in islets of type 2 diabetic subjects, elevated up to 16% by incretin treatment47. Also, an abnormal Nkx6.1 expression was found in type 2 diabetic subjects48. Moreover, there is a phenomenon of β-cell degranulation exists, referring to the depletion of insulin granules initiated by a metabolic stress with an unchanged transcriptome (at least transiently upon a retarded response)44.
Conclusions
We confirmed that the transcription factor Nkx6.1 controls several metabolic genes, notably glucose transporter (Glut2) and pyruvate carboxylase (Pcx) genes and genes important for insulin biogenesis (Ero1lb and Slc30a8). However, when their control is dysregulated by a deficiency in Nkx6.1 that also leads to their decreased expression, OXPHOS is virtually unaffected so that the GSIS triggering is still ensured by the NOX4-mediated redox (H2O2) signaling albeit at a more slowly increasing ATP levels. Thus, the metabolic plasticity is demonstrated in cultured INS-1ENkx6.1–/– cells, as well as a moderately altered transcriptome. Consequently, certain Nkx6.1-regulated genes, previously considered to be essential for insulin secretion, can be compensated for overall integral function. Our data suggest a caution in considering Nkx6.1 deficiency to be an ultimate determinant of β-cell pathology other than stemming from cell trans-(de-)differentiation and changes in the β-cell identity. In conclusion, the definition of the β-cell identity is compatible with the Nkx6.1 deficiency, similarly as in the hub-β-cells.
Methods
CRISPR/Cas9-edited Nkx6.1 ablation in INS-1E cells
Model pancreatic β-cells, rat insulinoma INS-1E cells, were purchased from AddexBio (San Diego, CA; cat. No. C0018009). Cells were cultivated with 11 mM glucose in RPMI 1640 medium with l-glutamine, supplemented with 10 mM HEPES, 1 mM pyruvate, 5% (v/v) fetal calf serum, 50 μmol/l mercaptoethanol, 50 IU/ml penicillin, and 50 μg/ml streptomycin21,22,26,27,31. These parental (wt) cells were assayed after 5–15 passages.
CRISPR/Cas9 gene editing was performed as follows: gRNA was designed using CRISPOR, a web-based tool for genome editing experiments (http://crispor.tefor.net, version 4.6, accessed on Nov. 2018). Two different INS-1ENkx6.1–/– cell lines a and b were produced as single cell colonies, using the sgRNA sequences 5′CACCGTTGGGCGCACACAACCCGGG3′ and 5′CACCGCCAGAAGATGGGCGTCCGAC. SgRNA sequences were subcloned into the LentiCRISPR (pXPR_001) vector (kindly provided by Jakub Rohlena, Institute of Biotechnology of the Czech Academy of Sciences, Prague-West, Czech Republic) and transfected into INS-1E cells with Lipofectamine 3000 (ThermoFisher). Cells were treated with 1 μg/mL puromycin for 10 days and then cultured from single cells as separate colonies for at least three weeks to generate cell lines with deleted Nkx6.1 (INS-1ENkx6.1–/– cells, lines a and b were selected for further assays). The Nkx6.1 deletion was verified by western blot (Supplemental Information, Fig. S1). A nonsense sgRNA sequence (5′CGCACTACCAGAGCTAACTCAGATAGTACT3′) was used to repeat the above procedure in parallel as a negative control (scrambled, Scrl; INS-1EScrl cells, were pooled from 50 single cell lines).
Prior to each experiment, cells were preincubated for 60 min (two washes of 30 min each) with 3 mM glucose, in the Krebs–Ringer HEPES (KRH) buffer (135 mM NaCl, 3.6 mM KCl, 10 mM HEPES, 0.5 mM MgCl2, 1.5 mM CaCl2, 0.5 NaH2PO4, pH 7.4).
Insulin secretion kinetics
Cells were seeded at 0.2 106 cells/well in poly-l-lysine-coated 12-well plates one day before the transient transfection and three days before the experiment. Insulin release was assayed using a Rat Insulin ELISA kit (U-E type, Shibayagi Co., Shibukawa, Japan) in a KRH buffer after 1-h of preincubation prior to glucose addition26,27,31.
Elevations in total cell ATP
The quantification of ATP was performed using an ATP Assay bioluminescence kit HSII (Roche). After reaching the desired conditions, cells were mixed with boiling lysis buffer (100 mM Tris, 4 mM EDTA, pH 7.75) and further boiled for another 2 min. Samples were centrifuged at 10,000×g for 1 min. Diluted supernatants were mixed with luciferase reagent and a Synergy HT luminometer was used to read the bioluminescence27,31. To confirm that the assay procedure does not interfere with the ATP concentration determination, internal ATP standards were added to the samples during the initial experiments.
High resolution respirometry
Routinely, an oxygraph 2k (Oroboros Instruments GmbH, Innsbruck, Austria) was used for experiments checking the respiration of INS-1E cells as described elsewhere27,31.
Monitoring of uptake of fluorecent glucose analog into INS-1E cells
6-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-6-deoxyglucose (6-NBDG; 100 μM) (ThermoFisher, Life Technologies Waltham, MA, USA) was used for monitoring glucose uptake into INS-1E cells. Cells were seeded onto the coverslips 48 h before the experiment and prior to confocal imaging they were incubated for 1 h in the KRH buffer with 3 mM mM glucose. 6-NBDG was added at the beginning (with no other agent or together with glucose to adjust it to 25 mM or with 100 μM BAY-876, which is both a GLUT1, but also GLUT2 inhibitor). Time-lapsed confocal monitoring was performed using a Leica TCS SP8 confocal microscope (Leica, Mannheim, Germany) with excitation at 480 and emission at 540 nm. A 2D spatial integral of the fluorescence intensity was derived from the series of confocal images within the individual cell ROIs.
Tl+ uptake assay as surrogate for K+ uptake into INS-1E cells
Assays of Tl+ influx rates were performed using the FluxOR potassium ion channel assay (ThermoFisher)26. Since plasma membrane depolarization is caused by an inhibition of KATP channels, the cells were first preloaded with “stimulus buffer” containing Tl+. Upon binding cytosolic Tl+, the de-esterified FluxOR dye strongly increases fluorescence at its peak emission of 525 nm, which was monitored in a Shimadzu RF 5301 PC spectrofluorometer.
GCaMP6 assay for Ca2+ oscillations in INS-1E cells
The vectors pGP-CMV-GCaMP6f (Plasmid #40753) and pGP-CMV-GCaMP6s (Plasmid #40755) were purchased from Addgene49, which express the fast-responding and slow-responding but more sensitive GCaMP6 fluorescent Ca2+indicator, respectively. GCaMP6 represents a synthetic fusion of green fluorescent protein (GFP), calmodulin and M13, a peptide sequence from myosin light-chain kinase50. Cells were transfected using Lipofectamine 2000 (ThermoFisher) for 48 h. A Leica TCS SP8 confocal microscope was employed for the time-lapsed recording of integral fluorescence intensity within the individual cells ROI with excitation at 480 and emission at 510 nm. For every second, integral fluorescence intensities F[Ca]c(ti) were collected from the image of each responding single cell in widefield and data were plotted as a time course. Typically, 10 min periods (trails) were set and in each period the glucose level was adjusted, starting from 3 mM, then typically 5, 7, 9, 11, and 20 mM glucose followed and at the end 30 mM KCl was added. Numerical 1st-derivatives were calculated to ascertain peaks of oscilations, so that a peak at time ti occurred when the derivative of F[Ca]c(ti) was zero at time ti. Only those zero derivatives were selected when preceded by the positive derivative and followed by the negative derivative values, thus excluding the negative–positive derivative pairs around the zero derivative, which represent functional minima. Peaks of oscillations in each period were sorted by intensities into ranges scaled by 1/10 of maximum intensity (deciles) and histograms were plotted for each period.
Mitochondrial DNA copy number
The mtDNA from INS-1E cells was isolated using DNA lysis buffer (10 mM Tris–Cl, 100 mM NaCl, 10 mM EDTA, 0.5% SDS pH 8.0) followed by incubation in proteinkinase K, protein salting in 6 M NaCl and the precipitation of DNA by isopropanol. This was followed by SYBR Green qPCR amplification with primers annealing on the Slco2a1 nuclear gene (which encodes solute carrier organic anion transporter family member 2A1) and the Nd5 mitochondrial gene (bp 11,092 to 11,191 according to Genebank sequences from The National Center for Biotechnology Information, USA)31. For primers see Ref.31. The ratio between Nd5 amplicon and half of the nuclear amplicon amounts was taken as the mtDNA copy number per cell.
RT-PCR
Total RNA was extracted from cells by acid guanidinium thiocyanate-phenol-chloroform extraction using Trizol-like buffer (0.4 M ammonium thiocyanatate, 0.8 M guanidine thiocyanate, sodium acetate 0.1 M, glycerol 5% (v/v), phenol 38% (v/v), pH 4.4). The reverse transcription was performed with a cDNA GrandScript reverse transcription kit (Tataa Biocenter, Prague, Czech Republic), using 500 ng of RNA previously quantified with a NanoDrop 2000 (ThermoFisher). The real-time polymerase chain reaction was performed with Fast Evagreen qPCR Master Mix (Biotium, Fremont, CA, USA). The PCR amplification was carried out with initial denaturation at 95 °C for 20 s, followed by 45 cycles of 95 °C for 5 s and 57 °C for 10 s and 72 °C for 20 s, followed by 5 min of terminal amplification at 72 °C, using a CFX96 cycler (Bio-Rad). The primers used were as indicated in Table 1. All of them led to a single amplicon product when the described qRT-PCR procedure was performed. Data were calculated by the 2−∆∆CT method (where CT is cycle threshold and ∆∆CT is CT for the gene of interest minus CT of the internal control), having selected housekeeping genes, indicated in Table 1, as an internal control for each experimental condition.
Statistical analysis
Results are presented as mean ± standard deviation (SD) for N biological replicates or total number of estimates (n). Graphs were plotted and statistical analyses were performed using SigmaPlot 6.0 and SigmaStat 3.1 (Systat-Software, San Jose, CA) and ANOVA followed by the Tukey test on the pre-validated data; or, alternatively using Prism (GraphPad Software, San Diego, CA) and an unpaired Student’s T-test (when comparing two groups) or one-way non-parametric ANOVA (Tukey test). Statistical significance was set at ***p < 0.001; **p < 0.05; *p < 0.1.
Data availability
The data sets supporting this study are available from the corresponding author, P.J., on request.
References
Moin, A. S. M. & Butler, A. E. Alterations in beta cell identity in type 1 and type 2 diabetes. Curr. Diab. Rep. 19, 83 (2019).
Hunter, C. S. & Stein, R. W. Evidence for loss in identity, de-differentiation, and trans-differentiation of islet β-cells in type 2 diabetes. Front. Genet. 8, 35 (2017).
Lawlor, N. & Stitzel, M. L. (Epi)genomic heterogeneity of pancreatic islet function and failure in type 2 diabetes. Mol. Metab. 27S, S15–S24 (2019).
Zhu, Y., Liu, Q., Zhou, Z. & Ikeda, Y. PDX1, Neurogenin-3, and MAFA: Critical transcription regulators for beta cell development and regeneration. Stem Cell Res. Ther. 8, 240 (2017).
Wang, Y. J. & Kaestner, K. H. Single-cell RNA-Seq of the pancreatic islets—a promise not yet fulfilled?. Cell Metab. 29, 539–544 (2019).
Ibarra Urizar, A. et al. Beta-cell dysfunction induced by non-cytotoxic concentrations of Interleukin-1β is associated with changes in expression of beta-cell maturity genes and associated histone modifications. Mol. Cell Endocrinol. 496, 110524 (2019).
Christensen, A. A. & Gannon, M. The beta cell in type 2 diabetes. Curr. Diab. Rep. 19, 81 (2019).
De Jesus, D. F. & Kulkarni, R. N. “Omics” and “epi-omics” underlying the β-cell adaptation to insulin resistance. Mol. Metab. 27S, S42–S48 (2019).
Nasteska, D., Viloria, K., Everett, L. & Hodson, D. J. Informing β-cell regeneration strategies using studies of heterogeneity. Mol. Metab. 27S, S49–S59 (2019).
Dominguez-Gutierrez, G., Xin, Y. & Gromada, J. Heterogeneity of human pancreatic β-cells. Mol Metab. 27S, S7–S14 (2019).
Aigha, I. I. & Abdelalim, E. M. NKX6.1 transcription factor: A crucial regulator of pancreatic β cell development, identity, and proliferation. Stem Cell Res. Ther. 11, 459 (2020).
Al-Khawaga, S. et al. Pathways governing development of stem cell-derived pancreatic beta cells: Lessons from embryogenesis. Biol. Rev. Camb. Philos. Soc. 93, 364–389 (2018).
Inoue, H. et al. Isolation, characterization, and chromosomal mapping of the human Nkx6.1 gene (NKX6A), a new pancreatic islet homeobox gene. Genomics 40, 367–370 (1997).
Villasenor, A., Chong, D. C. & Cleaver, O. Biphasic Ngn3 expression in the developing pancreas. Dev. Dyn. 237, 3270–3279 (2008).
Bader, E. et al. Identification of proliferative and mature beta-cells in the islets of Langerhans. Nature 535, 430–434 (2016).
Johnston, N. R. et al. Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab. 24, 389–401 (2016).
Schaffer, A. E. et al. Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic Beta cell identity. PLoS Genet. 9, e1003274 (2013).
Taylor, B. L., Liu, F. F. & Sander, M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 4, 1262–1275 (2013).
Schisler, J. C. et al. The Nkx6.1 homeodomain transcription factor suppresses glucagon expression and regulates glucose-stimulated insulin secretion in islet beta cells. Proc. Natl. Acad. Sci. USA 102, 7297–7302 (2005).
Donelan, W. et al. Distinct regulation of hepatic nuclear factor 1alpha by NKX6.1 in pancreatic beta cells. J. Biol. Chem. 285, 12181–12189 (2010).
Alán, L. et al. Delta cell hyperplasia in adult Goto-Kakizaki (GK/MolTac) diabetic rats. J. Diabetes Res. 2015, 385395–385416 (2015).
Špaček, T. et al. Nkx6.1 decline accompanies mitochondrial DNA reduction but subtle nucleoid size decrease in pancreatic islet β-cells of diabetic Goto Kakizaki rats. Sci. Rep. 7, 15674 (2017).
Gauthier, B. R. et al. The beta-cell specific transcription factor Nkx6.1 inhibits glucagon gene transcription by interfering with Pax6. Biochem. J. 403, 593–601 (2007).
Sander, M. et al. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 127, 5533–5540 (2000).
Taylor, B. L., Benthuysen, J. & Sander, M. Postnatal beta-cell proliferation and mass expansion is dependent on the transcription factor Nkx6.1. Diabetes 64, 897–903 (2015).
Plecitá-Hlavatá, L. et al. Glucose-stimulated insulin secretion fundamentally requires H2O2 signaling by NADPH oxidase 4. Diabetes 69, 1341–1354 (2020).
Plecitá-Hlavatá, L. et al. Mitochondrial superoxide production decreases on glucose-stimulated insulin secretion in pancreatic β cells due to decreasing mitochondrial matrix NADH/NAD + ratio. Antioxid. Redox Signal. 33, 789–815 (2020).
Ježek, P., Holendová, B., Jabůrek, M., Dlasková, A. & Plecitá-Hlavatá, L. Contribution of mitochondria to insulin secretion by various secretagogues. Antioxid. Redox Signal. 36, 920–952 (2022).
Ježek, P. et al. The pancreatic β-Cell: The perfect redox system. Antioxidants 10, 197 (2021).
Ježek, P., Jabůrek, M. & Plecitá-Hlavatá, L. Contribution of oxidative stress and impaired biogenesis of pancreatic β-cells to type 2 diabetes. Antioxid. Redox Signal. 31, 722–751 (2019).
Leguina-Ruzzi, A. A. et al. Glucose-induced expression of DAPIT in pancreatic β-cells. Biomolecules 10, 1026 (2020).
Rorsman, P. & Ascroft, F. M. Pancreatic β-cell electrical activity and insulin secretion: Of mice and men. Physiol. Rev. 98, 117–214 (2018).
Kaestner, K. H. et al. What is a β cell?—Chapter I in the Human Islet Research Network (HIRN) review series. Mol. Metab. 53, 101323 (2021).
Guo, S. et al. Inactivation of specific beta cell transcription factors in type 2 diabetes. J. Clin. Investig. 123, 3305–3316 (2013).
Gao, T. et al. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab. 19, 259–271 (2014).
Jennings, R. E. et al. Development of the human pancreas from foregut to endocrine commitment. Diabetes 62, 3514–3522 (2013).
Dorrell, C. et al. Human islets contain four distinct subtypes of beta cells. Nat. Commun. 7, 11756 (2016).
Xin, Y. et al. RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 24, 608–615 (2016).
Tritschler, S., Theis, F. J., Lickert, H. & Bottcher, A. Systematic singlecell analysis provides new insights into heterogeneity and plasticity of the pancreas. Mol. Metab. 6, 974–990 (2017).
Lawlor, N. et al. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res. 27, 208–222 (2017).
Segerstolpe, A. et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 24, 593–607 (2016).
Muraro, M. J. et al. A single-cell transcriptome atlas of the human pancreas. Cell Syst. 3, 385–394 (2016).
Zhang, J., McKenna, L. B., Bogue, C. W. & Kaestner, K. H. The diabetes gene Hhex maintains δ-cell differentiation and islet function. Genes Dev. 28, 829–834 (2014).
Cinti, F. et al. Evidence of beta-cell dedifferentiation in human type 2 diabetes. J. Clin. Endocrinol. Metab. 101, 1044–1054 (2016).
Butler, A. E. et al. Beta-cell deficit in obese type 2 diabetes, a minor role of beta-cell dedifferentiation and degranulation. J. Clin. Endocrinol. Metab. 101, 523–532 (2016).
Md Moin, A. S. et al. Increased frequency of hormone negative and polyhormonal endocrine cells in lean individuals with type 2 diabetes. J. Clin. Endocrinol. Metab. 101, 3628–3636 (2016).
Butler, A. E. et al. Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagonproducing neuroendocrine tumors. Diabetes 62, 2595–2604 (2013).
Liu, T. et al. Increased frequency of beta cells with abnormal NKX6.1 expression in type 2 diabetes but not in subjects with higher risk for type 2 diabetes. BMC Endocr. Disord. 21, 47 (2021).
Nakai, J., Ohkura, M. & Imoto, K. A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat. Biotechnol. 19, 137–141 (2001).
Chen, T. W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013).
Acknowledgements
The project was supported by the Grant Agency of the Czech Republic, grant No. 20-00408S to P.J.; and by the project National Institute for Research of Metabolic and Cardiovascular Diseases (Programme EXCELES, ID Project No. LX22NPO5104)—Funded by the European Union—Next Generation EU. The excellent technical assistance of Jana Vaicová and Jitka Smiková is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
V.P. and H.E. designed and conducted all experiments, but auxiliary western blots made by J.Š. V.P. and J.Š. designed and V.P. constructed INS-1ENkx6.1–/– cells using CRISPR/Cas9 gene editing. P.J. designed hypotheses and experiments, analyzed and plotted data, performed statistical analysis and wrote the article. Therefore, P.J. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pavluch, V., Engstová, H., Špačková, J. et al. Deficiency of transcription factor Nkx6.1 does not prevent insulin secretion in INS-1E cells. Sci Rep 13, 683 (2023). https://doi.org/10.1038/s41598-023-27985-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-27985-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
