
Abstract
A simple predictive biomarker for fatty liver disease is required for individuals with insulin resistance. Here, we developed a supervised machine learning-based classifier for fatty liver disease using fecal 16S rDNA sequencing data. Based on the Kangbuk Samsung Hospital cohort (n = 777), we generated a random forest classifier to predict fatty liver diseases in individuals with or without insulin resistance (n = 166 and n = 611, respectively). The model performance was evaluated based on metrics, including accuracy, area under receiver operating curve (AUROC), kappa, and F1-score. The developed classifier for fatty liver diseases performed better in individuals with insulin resistance (AUROC = 0.77). We further optimized the classifiers using genetic algorithm. The improved classifier for insulin resistance, consisting of ten microbial genera, presented an advanced classification (AUROC = 0.93), whereas the improved classifier for insulin-sensitive individuals failed to distinguish participants with fatty liver diseases from the healthy. The classifier for individuals with insulin resistance was comparable or superior to previous methods predicting fatty liver diseases (accuracy = 0.83, kappa = 0.50, F1-score = 0.89), such as the fatty liver index. We identified the ten genera as a core set from the human gut microbiome, which could be a diagnostic biomarker of fatty liver diseases for insulin resistant individuals. Collectively, these findings indicate that the machine learning classifier for fatty liver diseases in the presence of insulin resistance is comparable or superior to commonly used methods.
Similar content being viewed by others


Introduction
Fatty liver disease (FL), or hepatic steatosis, is diagnosed when a liver has at least 5% hepatocytes containing fat1. Depending on its etiology, it can be categorized into alcoholic fatty liver disease (AFLD) or nonalcoholic FL disease (NAFLD). The international prevalence of NAFLD was estimated to be 25.2%2. However, it was estimated to be 55.5% in participants with type 2 diabetes mellitus (T2DM)3. Insulin resistance (IR), a major pathological contributor to T2DM and metabolic syndrome, is a potential driver of NAFLD progression in nonalcoholic steatohepatitis (NASH)4,5. IR is a common pathological factor of NASH and T2DM. It may also be involved in hepatocellular carcinoma (HCC) development6.
Liver biopsy or magnetic resonance imaging/spectroscopy (MRI/MRS) is the gold standard for diagnosing FL7. However, it has certain disadvantages. For instance, liver biopsy is invasive, and the examination is spatially limited to the sample. MRI/MRS is inaccessible and expensive. The most popular method for FL diagnosis is ultrasonography (USG). However, it also has several limitations: (i) variability depending on the spots observed, (ii) low sensitivity in mild hepatic steatosis occupying < 30% of the liver8; and (iii) inconsistencies in diagnosis between interpreters. Therefore, an additional non-invasive diagnostic biomarker for FL is required urgently.
The symbiosis or dysbiosis between the human gut microbiome (HGM) and hosts is linked to the health or disease of the host, respectively9. According to recent reports, alterations in the gut microbiome could be a driver of obesity and IR10,11,12. It has also been shown that specific signatures of the HGM can serve as biomarkers of liver diseases, including NASH, liver fibrosis, and cirrhosis13,14,15. However, only a few studies have proposed the HGM as a biomarker for general FL rather than for advanced liver diseases. Most studies investigated either the correlation or causality of HGM with liver diseases only in a pathological cohort rather than a healthy one. Furthermore, those studies did not invest in participants with IR, who have a higher risk for advanced FL disease, in the healthy cohort. Therefore, our study established a potential gut microbiome to identify the FL of participants with IR in a healthy cohort using supervised machine learning (ML) methods for classification, such as random forest (RF), gradient boosting machine (GBM), extreme gradient boosting (XGB) algorithm, along with genetic algorithm (GA), a random-based algorithm inspired by natural selection in biology to obtain the optimized solution16.
Materials and methods
Human participants and data collection
Stool samples were collected from the study participants (n = 1,463) from the routine annual comprehensive physical examination of the Kangbuk Samsung Health (KSH) cohort. The study participants underwent extensive periodic PE between June and September 201417 and 213 participants were excluded from 1,463 participants because of missing data and poor detection. FL was diagnosed using abdominal USG with a 3.5-MHz transducer based on conventionally captured images by trained radiologists who were blinded to the study's predetermined parameters as previously described18,19. In the diagnosis, the inter-observer reliability value was Cohen’s kappa coefficient of 0.74, and the intra-observer reliability value was 0.9420.
The Institutional Review Board of Kangbuk Samsung Hospital authorized the study's protocol (2019-05-015). All participants signed a written informed consent form after being informed of possible outcomes and the nature of the study. In the study, we obeyed all applicable regulations of institutions and governments regarding human research ethics for participants, following the guidelines of the Declaration of Helsinki21.
DNA purification and 16S rDNA gene sequencing
The Illumina MiSeq platform was used to sequence the fecal DNA samples, following the provided protocol (Illumina, San Diego, CA, USA)22. The DADA2 plugin of the QIIME 2 package (v.2020.8) was utilized in filtering out chimeras and low-quality sequences and to produce amplicon sequence variants (ASVs)23,24. The naïve Bayes classifier were trained, and the classifier was used to assign ASVs to microbial taxonomy against the SILVA 132 with a 99% operational taxonomy unit dataset. All 16S rDNA gene sequencing files are available in the Clinical & Omics Data Archive of the Korea National Institute of Health (accession number: R000635).
Development of ML classifier and evaluation
R Package “caret” v.6.0-86. was used for ML approach using three ML algorithms (RF, GBM, and XGB)25. The RF parameter options were set to default option in the ML approach. In the GBM models’ hyperparameter settings, 10, 20, 30, 40, and 50 were used as “n.trees”; one, two, three, and four as “interaction.depth”; 0.01 and 0.001 as “shrinkage”; three, five, seven, and nine as “n.minobsinnode.” For the hyperparameter setting in the XGB models, 10, 20, 30, 40, and 50 was used as “nrounds”; three, five, seven, and nine as “max_depth”; 0.01 and 0.2 as “eta”; 0.01 as “gamma”; 0.75 as “colsample_by_tree”; 1 as “min_child_weight”; and 0.5 as “subsample.” The microbiome dataset was randomly partitioned into training (80%) and test (20%) datasets using the createDataPartition function. The dataset was preprocessed using the zv, scale, and center methods of the training function. The Synthetic Minority Over-sampling Technique (SMOTE) function in the R package “smotefamily” (v.1.3.1) was used to handle the sample imbalance issue. The tenfold three-times repeated cross-validation were applied to the training dataset for ML-based development into the previously described classification model13. The sequential feature selection was conducted based on the Gini importance of the features. The performance of the developed classifiers was assessed using the area under the receiver operating curve (AUROC), representing their sensitivity and specificity in the training dataset. Using the test dataset, the classifiers were evaluated using AUROC, accuracy, F1-score, and kappa.
The optimal feature selection by GA
For a GA-based optimal feature selection, 300 individuals were randomly generated as the initial population to be sequentially evolved further by GA. The individuals carry a specific number of genera (described as “genes”) randomly selected from 87 gut microbial genera detected in the fecal samples. The selected genera were encoded as one, and the other genera were encoded as 0 in individuals to be evolved by GA. Each individual in the population was evaluated using a fitness score as follows:
where Sk is the AUROC score from the RF model in the k-th fold during M fold cross-validation; x is the number of genera selected by the RF; W is a penalty weight; and b ∈ {6, 7, 8, 9, 10} is the optimal number of biomarker genera. M was set to 3 and W to 10.
According to the fitness score, GA repeatedly searches for the best solution for classifying every generation. Firstly, in the initial population, GA selected the fittest individual with the highest fitness score in the initial population (first generation). To generate the population of the next generation, the fittest individual of the previous generation is kept, while the other individuals of the previous generation are influenced by crossovers and mutations, resulting in different individuals. Thus, we obtained 300 individuals in each generation. Then, these steps were iterated 100 times (generations) to get the best solution through the entire generation.
Package “DEAP” v.1.3.1 under python 3.7.1 was used for the GA simulations, revealing the optimal genera having the best fitness. The optimal genera served as the features of an optimal RF classification model. The crossover rate, mutation rate, and generations for GA simulations were set at 0.8, 0.003, and 300, respectively.
Data visualization and statistical analyses
All statistical analysis and visualizations were conducted using RStudio with R v.4.1. The normality of the overall data was analyzed using Shapiro’s test. Statistical significance was computed with either two-tailed Wilcoxon’s test or Kruskal–Wallis test upon the normality and distribution, and a p-value < 0.05 was deemed statistically significant. The R package “ggplot2” and “pROC” were used for data visualization.
Evaluation of model performance
To evaluate the model performance, we obtained a 2 × 2 confusion matrix from each classifier using the test dataset and calculated true positive, true negative, false positive, and false negative using predicted and observed classes. Then, we calculated and adopted four metrics for the model’s performance evaluation: accuracy26,27, F1-score27, kappa index28, and AUROC, plotted using true positive rate and false positive rate (FPR)29.
Results
Data processing in the healthy and FL cohort
Physical examination data, including high throughput 16S rDNA sequencing results from 1463 participants of KSH cohorts, were collected for the study. After removing missing and poorly detected values, 1,250 participants were included in the analysis. Subsequently, participants whose ASV number were < 5 000 were filtered out, leaving only 777 participants for the study (Fig. 1). It was revealed that 290 of the 777 participants had FL, while the remaining 487 did not. We regarded participants as insulin resistant or sensitive if their values for homeostatic model assessment of IR (HOMA-IR) were over the following cutoff: 1.8 for men and 2.2 for women, calculated as the critical threshold for T2DM development based on the KSH cohort internal investigation (data not shown). Among the 777 participants, 611 were classified into the insulin-sensitive (IS) group, while 166 were classified into the IR group based on the criteria for insulin resistance. The biological and physical characteristics of these groups are described in Table 1.

Schematic of the analysis pipeline. Participants (n = 777) from the KSH cohort were included in the final analysis and were divided into four subgroups, ISNF (n = 449), ISFL (n = 162), IRNF (n = 38), and IRFL (n = 128). ASV: amplicon sequence variants; HOMA-IR: homeostatic model assessment of insulin resistance index; IR: insulin resistant; KSH cohort: Kangbuk Samsung Hospital cohort.
Subject demographics
Among the 777 participants in the study, IS and IR included 611 and 166 individuals, respectively. Men accounted for 55.97% of IS and 84.94% of the IR group. The IS group had significantly lower values than those of the IR group for age, body mass index (BMI), waist circumference, heart rate, HOMA-IR, glucose, insulin, HbA1c, albumin, aspartate aminotransferase, alanine transaminase, triglycerides (TG), low-density lipoprotein cholesterol (LDL-C), and both diastolic and systolic blood pressure (BP). The IS group had lower total cholesterol than the IR group, but the difference was not statistically significant. Moreover, participants of IS group had higher high-density lipoprotein cholesterol (HDL-C) levels than those of the IR group with statistical significance (Table 1).
Microbiome comparison and classification between the groups with or without FL
Alpha diversity, particularly Shannon’s entropy, was used to compare the diversity of the gut microbiome in participants of the non-fatty liver control group (NF) and FL groups. For Shannon’s entropy, representing biodiversity integrated with community richness and evenness, NF (median = 6.677; interquartile range [IQR] = 6.179–7.103) had a significantly higher value than that of FL (median = 6.475; IQR = 5.961–6.941; p = 0.0017; Fig. 2a). Consistent with previous reports, FL (median = 15.090, IQR = 12.859–18.057) had a significantly lower value of Faith’s phylogenetic diversity (PD: biodiversity based on phylogeny) than that of NF (median = 15.848, IQR = 13.648–18.594) (p = 0.004; Fig. 2b). Additionally, FL (median = 0.910; IQR = 0.885–0.928) had a lower value of Pielou’s evenness (a measure of biodiversity and species richness) than that of NF (median = 0.918; IQR = 0.899–0.931; p = 0.00045; Fig. 2c). Subsequently, we performed principal coordinate analysis (PCoA) to obtain representative relationships between the NF and FL groups. However, the two groups had no observable distant clusters (Fig. 2d).

Comparison of the alpha and beta diversity of gut microbiome between fatty liver disease (FL) and nonfatty liver control (NF) groups. (A–C) Alpha diversity of NF and FL was measured using Shannon’s entropy (A), Faith’s phylogenetic diversity (PD) (B), and Pielou’s evenness (C). Boxes represent the IQR, whereas the upper whiskers represent the range from minimum (upper quartile − 1.5IQR) to maximum (lower quartile + 1.5IQR), and black dots represent outliers excluded in the range. (D) Beta diversity among participants in NF and FL was measured using the Principal Coordinates Analysis (PCoA). (E–F) The predictive power (AUROC) of the RF prediction model featuring different discriminative gut microbial genera in the training dataset (E) and test dataset (F). Statistical significance was analyzed using the Kruskal–Wallis test. *p < 0.05, **p < 0.01. FL: participants with fatty liver disease; IQR: interquartile range; ML: machine learning; NF: nonfatty liver control group; RF: random forest.
As NF and FL showed differential alpha diversity but not beta diversity, we generated a classification model with informative gut microbial features. We can perform Gini importance-based core informative feature selection based on the algorithm. The predictive power of the models from the training dataset with two, four, eight, 12, 16, 24, and 32 features were 0.53, 0.59, 0.60, 0.59, 0.62, and 0.64 of AUROC, respectively (Fig. 2e). The FL prediction using the model featuring 32 gut microbial features displayed 0.65 (0.56–0.73) of AUROC in the test dataset (Fig. 2f). This implies an inefficient classification based on a set of most informative features between the NF and FL groups.
Classification between NF and FL using reported gut microbial features
Recent reports have proposed a novel microbiome-based diagnostic tool for liver cirrhosis, the most advanced FL13. We built an RF classifier using gut microbial markers to test whether the markers could distinguish between FL and NF in our data. Differential abundance of gut microbial genera features, including Acidaminococcus spp., Alistipes spp., Bacteroides spp., Dorea spp., Enterobacter spp., Escherichia-Shigella spp., Eubacterium spp., Faecalibacterium spp., Klebsiella spp., Ruminococcus (gnavus group) spp., Streptococcus spp., and Veillonella spp., were selectively observed between FL and NF (Supplementary Fig. 1a). Among these features, Acidaminococcus spp. (p = 8.9e−05), Alistipes spp. (p = 7.5e−07), Faecalibacterium spp. (p = 0.011), and Ruminococcus spp. (gnavus group; p = 0.0018) had significantly different abundances in NF and FL, consistent with previous studies. Furthermore, we evaluated the sensitivity and selectivity of a set of these features using the AUROC. However, the predictive power of each model with different number of features was insufficient to distinguish between NF and FL (0.60 AUROC for a 12-feature model, 0.60 AUROC for an 8-feature model, 0.56 AUROC for a 6-feature model, and 0.56 AUROC for 4-feature model; Supplementary Fig. 11).
Microbiome comparison and classification between IRNF and IRFL
NAFLD is considered the hepatic component of IR. Therefore, it is critical to distinguish FL from NF in participants with IR. The participants in the IR groups were divided into the following based on the presence of FL: NF featuring IR (IRNF) and FL featuring IR (IRFL), to find the most informative microbial features differentiating FL from NF in the participants with IR. Then, we observed the differential biodiversity of the two microbiomes. IRNF (median = 6.808, IQR = 6.289–7.183) had a significantly higher value of the index than that of IRFL (median = 6.403, IQR = 5.988–6.805; p = 0.032) in terms of Shannon’s entropy (Fig. 3a). Additionally, IRFL (median = 14.906, 12.876–17.804) had significantly lower Faith’s PD than that of IRNF (median = 16.293, IQR = 14.311–20.455; p = 0.032; Fig. 3b). In terms of other alpha diversity indices, IRFL (median = 0.902, IQR = 0.878–0.921) had lower Pielou’s evenness than that of IRNF (median = 0.915, IQR = 0.905–0.930; p = 0.021; Fig. 3c). However, PCoA and uniform manifold approximation and projection (UMAP) of the total microbiome of both groups showed no difference in clusters between IRFL and IRNF (Fig. 3d, e, Supplementary Fig. 2a and b).

Comparing the alpha and beta diversity of the gut microbiome of IRNF and IRFL. (A–C) Alpha diversities of IRNF and IRFL were measured using Shannon’s entropy (A), Faith’s phylogenetic diversity (PD) (B), and Pielou’s evenness (C). Boxes represent the IQR. The upper whiskers represent the range from minimum (upper quartile − 1.5IQR) to maximum (lower quartile + 1.5IQR), and black dots represent outliers excluded in the range. (D–E) Beta diversity among participants in IRNF and IRFL was measured using PCoA (D) and UMAP analyses (E). (F) The model’s predictive power featuring different number of gut microbial genera constructed using RF, GBM, and XGB algorithms in the test dataset. The statistical significances were analyzed using Wilcoxon’s test. *p < 0.05, **p < 0.01. IRFL, fatty liver participants featuring insulin resistance; IQR: interquartile range; IRNF: nonfatty liver control group featuring insulin resistance.
To classify IRFL and IRNF from their gut microbiome, we constructed ML models using three ML algorithms for classification, RF, GBM, and XGB. Among the three ML models featuring different numbers of gut microbial genera, the RF model demonstrated the most reliable prediction in the test dataset (AUROC 0.77), while the AUROCs of classification for the other two ML models, GBM and XGB, were 0.62 and 0.63, respectively (Fig. 3f). Next, we built the models in the same manner, but individually for each gender, to see if any gender had better predictive results. In the training dataset, the RF model for females displayed AUROC values of 0.81, 0.96, 0.88, and 0.73 for the models using six-, eight-, twelve-feature, and entire gut microbiome, respectively (Supplementary Fig. 2c). The predictive power of the eight-feature model showed 0.67 AUROC. Surprisingly, the aforementioned outcome in the training dataset was superior to the results from the RF model for male, presenting AUROC values of 0.63 (six-feature model), 0.76 (eight-feature model), 0.69 (twelve-feature model), and 0.58 (entire gut microbiome-based model) (Supplementary Fig. 2d). During model validation using the male test dataset, the RF model had an AUROC of 0.76, the GBM model had an AUROC of 0.62, and the XGB model had an AUROC of 0.77 (Supplementary Fig. 2e). Together, it was determined that the models using the RF algorithm are appropriate for further research.
Then, we built RF models and assessed their efficacy in predicting FL in the IR groups after applying the SMOTE algorithm to the dataset to minimize the present class imbalance (30% IRNF: 70% IRFL). The model’s predictive power was 0.87 AUROC in the training dataset, but it only displayed 0.72 AUROC in the test dataset (Supplementary Table S1).
Classification between IRNF and IRFL by using GA-optimized classifier (IRFL-GARF classifier)
GA is a heuristic algorithm that determines the global optimum based on natural selection30,31,32. It can be used to select model features such that the model demonstrates the best prediction. We used GA to create an ML classifier with better prediction performance using the RF algorithm. We developed an RF classifier presenting higher accuracy in distinguishing IRFL from IRNF, based on the features selected by GA. The RF classifier optimized by GA was termed “IRFL-GARF classifier,” with the potential gut microbial biomarkers33. Using the fitness score, the classifier can repeatedly search for the best solution for classifying IRFL and IRNF every generation.
In the development of the IRFL-GARF classifier, we first generated 300 individuals to be evolved further as the initial population (Fig. 4). Then, the fittest individual was selected following evaluation based on the fitness scores of every individual. With the fittest individual, the next generation is produced with crossover and mutation (Supplementary Fig. 3).

The overview of biomarker genera mining using GA. From the randomly generated initial 300 individuals consisting of genera, the classification model was optimized using GA methods, including crossover and mutation. The model with the highest fitness score is selected for each generation and further sequentially optimized in the next generation. The final model was evaluated using the average AUROC of the tenfold CV model. Further model validation was conducted using test data for the corresponding biomarker subset and accuracy, an F1-score, a kappa, and an AUROC.
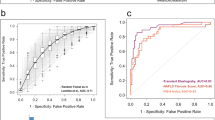
Consequently, the GA reported ten optimal features (equivalently, genera) for an optimal RF model: Christensenellaceae (R-7 group) spp., Lachnospiraceae (UCG-004) spp., Fusicatenibacter spp., Butyricimonas spp., Weissella spp., Ruminococcaceae (UCG-004) spp., Erysipelatoclostridium spp., UBA1819 spp., Allisonella spp., and Collinsella spp. The classifier model’s predictive power was 0.93 in the test dataset (95% confidence interval: 0.83–1.00; Fig. 5a). Between gut microbial features in the classifier model, Butyricimonas spp. (mean of IRNF: 0.111%; IRFL: 0.070%), Christensenellaceae (R-7 group) spp. (IRNF: 0.736%; IRFL: 0.202%), Collinsella spp. (IRNF: 0.052%; IRFL: 0.021%), Erysipelatoclostridium spp. (IRNF: 0.153%; IRFL: 0.020%), and UBA1819 spp. (IRNF: 0.104%; IRFL: 0.014%) displayed higher relative abundances in IRNF than in IRFL. In contrast, Allisonella spp. (IRNF: 0.008%; IRFL: 0.049%), Fusicatenibacter spp. (IRNF: 0.216%; IRFL: 0.305%), Lachnospiraceae (UCG-004) spp. (IRNF: 0.219%; IRFL: 0.358%), Ruminococcaceae (UCG-004) spp. (IRNF: 0.020%; IRFL: 0.026%), and Weissella spp. (IRNF: 0.089%; IRFL: 0.117%) were more abundant in IRFL than in IRNF. Notably, Butyricimonas spp. (p = 0.0094), Christensenellaceae (R-7 group) spp. (p = 0.00056), and Ruminococcaceae (UCG-004) spp. (p = 0.026) had significantly different relative abundances between the two groups (Fig. 5b). The visualization of fold change in ten GA-selected features in the rate per hundred showed that Christensenellaceae (R-7 group) spp. (fold change of log2 [log2FC]: − 1.006), Weissella spp. (log2FC: − 0.168), UBA1819 spp. (log2FC: − 1.967), Collinsella spp. (log2FC: − 0.185), and Erysipelatoclostridium spp. (log2FC: − 0.032) had lower relative abundances in IRFL. In contrast, Lachnospiraceae (UCG-004) spp. (log2FC: 0.536), Fusicatenibacter spp. (log2FC: 0.823), Butyricimonas spp. (log2FC: 0.070), Allisonella spp. (log2FC: 0.408), and Ruminococcaceae (UCG-004) spp. (log2FC: 1.229) were more abundant in both groups (Fig. 5c). Then, we performed UMAP projection to dimensionally reduce the dataset, presenting an IRNF clustering. Christensenellaceae spp. (R-7 group) were highly distributed in the green circle, where most IRNFs were distributed, whereas the Lachnospiraceae (UCG-004) group was highly distributed in the purple circle, where most of the dots represent IRFL (Fig. 5d–f).

Prediction of FL in the presence of insulin resistance using GA-optimized classifier. (A) The predictive power (AUROC) derived from the test dataset using GA-optimized RF classifier with ten features. (B) Violin plots displaying relative abundances of core informative features in IRNF and IRFL. (C) Average relative abundances of discriminative features in the 10-feature prediction model in IRNF and IRFL. (D–F) UMAP analysis and heatmap of Christensenellaceae (R-7 group) spp. (E) and Lachnospiraceae (UCG-004) spp. onto UMAP. *p < 0.05, **p < 0.01, and ***p < 0.001 (Wilcoxon’s test). IRFL: fatty liver participants featuring insulin resistance; IRNF: nonfatty liver control group featuring insulin resistance; UMAP: uniform manifold approximation and projection.
Also, we developed a GA-optimized classifier for IS (a classification between IS participants without FL, ISNF, and IS participants with FL, ISFL). The model featured eight gut microbial genera, namely, Eubacterium spp. (coprostanoligenes group), Alistipes spp., Bifidobacterium spp., Erysipelotrichaceae spp. (UCG-003), Lachnoclostridium spp., Parabacteroides spp., Ruminococcus spp. (torques group), and Subdoligranulum spp. However, the model’s predictive power was insufficient to classify ISNF and ISFL (0.52 of an AUROC; Supplementary Fig. 4a and b).
Model evaluation
To assess the GA-optimized model’s performance in IR, the model’s predictive power was compared with previously and broadly used non-invasive indexing scores calculated from clinical data for predicting FL, including FL index (FLI)34, NAFLD liver fat score (NAFLD-LFS)35, hepatic steatosis index (HSI)36, and Framingham steatosis index (FSI)37. For comparison, each score was calculated for each IR analyzed for the study and used for FL prediction with a partitioned test dataset. Our classifier displayed 0.93 AUROC, as the FLI, NAFLD-LFS, HSI, and FSI values were 0.82, 0.62, 0.80, and 0.82, respectively (Fig. 6a). The prediction accuracies of the GA-optimized classifier, FLI, NAFLD-LFS, HSI, and FSI were 0.83, 0.57, 0.60, 0.67, and 0.84, respectively (Fig. 6b). Additionally, the FL prediction by our classifier presented a kappa of 0.50, while the kappa of FLI, NAFLD-LFS, HSI, and FSI were 0.24, 0.17, 0.33, and 0.53, respectively (Fig. 6c). Finally, our classifier displayed 0.89 F1-score, which was similar to the FSI (0.90), whereas FLI, NAFLD-LFS, and HSI displayed 0.63, 0.63, and 0.72 of F1-scores, respectively (Fig. 6d). As shown above, among all measuring methods for predicting the power of predictors, our classifier gave the highest diagnostic accuracy compared with other predictors. This result implied that our gut microbiome-based classifier could be used with the abovementioned established predictors.

Evaluating prediction using GA-optimized classifier differentiating IRFL from IRNF. Bar plots comparing the predictive power derived from the test dataset using the GA-optimized classifier with other predictors by (A) AUROC, (B) accuracy, (C) kappa, and (D) F1 score. IRFL: fatty liver participants featuring insulin resistance; IRNF: nonfatty liver control group featuring insulin resistance.
Discussion
In this study, we performed an RF classification model using the KSH cohort comprising 777 healthy individuals, and we applied GA to improve the predictive efficiency of the RF-generated classifier (an AUROC of 0.77) (Fig. 3f). Based on the IRFL-GARF, we proposed ten genera as biomarkers for identifying FL in IR individuals, including Christensenellaceae (R-7 group) spp., Weissella spp., UBA1819 spp., Collinsella spp., Erysipelatoclostridium spp., Lachnospiraceae (UCG-004) spp., Fusicatenibacter spp., Butyricimonas spp., Allisonella spp., and Ruminococcaceae (UCG-004) spp. The IRFL-GARF classifier containing ten microbial genera had an AUROC, accuracy, kappa, and F1-score of 0.93, 0.83, 0.50, and 0.89, respectively, which are comparable or even superior to common diagnostic indices for FL diseases, such as FLI (AUROC of 0.82), HSI (AUROC of 0.80), NAFLD-LFS (AUROC of 0.60), and FSI (AUROC of 0.82). This demonstrates the potential of a diagnostic marker for FL disease in insulin-resistant participants (Fig. 6a–d).
GA is an adaptive metaheuristic search algorithm that identifies the global optimum based on the principle of natural selection in evolution. GA does not evaluate solutions individually but evaluates a group of solutions simultaneously and explores the space of possible solutions. Furthermore, GA has the advantage of being less likely to fall into a local minimum and does not require assumptions about the interaction between features.
The ten genera could be developed into a non-invasive biomarker for FL disease in insulin-resistant participants, who could have a higher chance of developing advanced chronic liver diseases. To the best of our knowledge, this is the first study that used GA to successfully classify FL diseases, encouraging GA application in future studies. The development of a non-invasive, inexpensive, and accurate method for diagnosing FL disease is required. Several recent studies have proposed the gut microbiome as a potential biomarker for advanced chronic liver diseases13,14,15,38; however, only a few studies have been conducted in generally healthy populations to identify individuals at higher risk of developing advanced chronic liver diseases based on the gut microbiome. For instance, it would be important to identify generally healthy participants showing IR without symptoms with a higher risk of advanced FL diseases.
In this study, alpha diversity, which reflects the gut microbiome structure concerning its richness39, decreased significantly in the FL participants. In contrast, the PCoA plots, representing beta diversity of the gut microbiome, failed to generate two distinct groups, inconsistent with previous studies13,38. Estimating alpha and beta diversities implies that reduced richness was sufficient to show differences in alpha diversity between the groups. However, it occurred in only a few genera (components). Although it is insufficient to determine whether the altered genera drove FL or vice versa, several studies have reported that the family Christensenellaceae correlates with BMI40, and the Ruminococcaceae (R-7 group) genera correlate with blood TG, very-low-density lipoprotein- and HDL-particles levels41. Another recent human study observed a strong correlation between Collinsella spp. and NASH and cirrhosis42. Few studies have indicated that Butyricimonas spp. is altered in AFLD and HCC43,44, implying that Butyricimonas spp. might contribute to FL disease or hepatic inflammation.
There are a few limitations to our study. Firstly, our research was based on a Korean hospital cohort with not quite large patients; thus, our results could be racially and geographically biased. However, we tried to prove the feasibility of the discovered ten genera as a biomarker to identify FL disease among patients with IR from the supporting studies. Secondly, it was implied that predicting FL using gut microbiome-based ML in the female IR groups could be more reliable rather than in the male group. However, greater sample size is needed for further validation. Additionally, to our knowledge, there was no independent external validation cohort available; thus, we expect that further validation studies population will address these limitations. Although our study is primarily correlative, our data strongly support the value of future work exploring the causal role of the ten genera in liver diseases.
Conclusively, these findings indicate that the ML classifier combined with GA for FL in the presence of IR is comparable or superior to commonly used methods. The ten genera we discovered are useful as a non-invasive biomarker for FL among patients with IR.
Data and material availability
All 16S rDNA gene sequencing files are available in the Clinical & Omics Data Archive of the Korea National Institute of Health (http://coda.nih.go.kr; accession number: R000635). The source code for the GA simulation is available on GitHub (https://github.com/labnams/FLIRGAMB).
References
Nassir, F., Rector, R. S., Hammoud, G. M. & Ibdah, J. A. Pathogenesis and prevention of hepatic steatosis. Gastroenterol. Hepatol. 11, 167–175 (2015).
Younossi, Z. M. et al. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016).
Younossi, Z. M. et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 71, 793–801 (2019).
Chitturi, S. et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology 35, 373–379. https://doi.org/10.1053/jhep.2002.30692 (2002).
Birkenfeld, A. L. & Shulman, G. I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 59, 713–723. https://doi.org/10.1002/hep.26672 (2014).
Byrne, C. D. & Targher, G. NAFLD: A multisystem disease. J. Hepatol. 62, S47-64. https://doi.org/10.1016/j.jhep.2014.12.012 (2015).
Iwasaki, M. et al. Noninvasive evaluation of graft steatosis in living donor liver transplantation. Transplantation 78, 1501–1505 (2004).
Palmentieri, B. et al. The role of bright liver echo pattern on ultrasound B-mode examination in the diagnosis of liver steatosis. Dig. Liver Dis. 38, 485–489 (2006).
Kho, Z. Y. & Lal, S. K. The human gut microbiome - a potential controller of wellness and disease. Front. Microbiol. 9, 1835. https://doi.org/10.3389/fmicb.2018.01835 (2018).
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006).
Caricilli, A. M. & Saad, M. J. The role of gut microbiota on insulin resistance. Nutrients 5, 829–851 (2013).
Koh, A. et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell 175, 947-961.e917 (2018).
Oh, T. G. et al. A universal gut-microbiome-derived signature predicts cirrhosis. Cell Metab. 32, 878-888.e876 (2020).
Lee, G. et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat. Commun. 11, 1–13 (2020).
Loomba, R. et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 25, 1054-1062.e1055 (2017).
Vandewater, L., Brusic, V., Wilson, W., Macaulay, L. & Zhang, P. An adaptive genetic algorithm for selection of blood-based biomarkers for prediction of Alzheimer’s disease progression. BMC Bioinform. https://doi.org/10.1186/1471-2105-16-S18-S1 (2015).
Chang, Y. et al. Nonheavy drinking and worsening of noninvasive fibrosis markers in nonalcoholic fatty liver disease: A cohort study. Hepatology 69, 64–75 (2019).
Mathiesen, U. et al. Increased liver echogenicity at ultrasound examination reflects degree of steatosis but not of fibrosis in asymptomatic patients with mild/moderate abnormalities of liver transaminases. Dig. Liver Dis. 34, 516–522 (2002).
Hwang, Y. C., Ahn, H. Y. & Park, C. Y. Association between nonalcoholic fatty liver disease and future deterioration of metabolic health: A cohort study. Obesity 27, 1360–1366 (2019).
Kim, C.-W. et al. Sleep duration and quality in relation to non-alcoholic fatty liver disease in middle-aged workers and their spouses. J. Hepatol. 59, 351–357 (2013).
Shephard, D. A. The 1975 declaration of Helsinki and consent. Can. Med. Assoc. J. 115, 1191–1192 (1976).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120 (2013).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 28, 1–26 (2008).
Metz, C. E. Seminars in Nuclear Medicine 283–298 (Elsevier, 1978).
Chicco, D. & Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genomics 21, 1–13 (2020).
Chicco, D., Warrens, M. J. & Jurman, G. The Matthews correlation coefficient (MCC) is more informative than Cohen’s Kappa and Brier score in binary classification assessment. IEEE Access 9, 78368–78381 (2021).
Hanley, J. A. & McNeil, B. J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143, 29–36 (1982).
Sumida, B. H., Houston, A. I., McNamara, J. M. & Hamilton, W. D. Genetic algorithms and evolution. J. Theor. Biol. 147, 59–84 (1990).
Katoch, S., Chauhan, S. S. & Kumar, V. A review on genetic algorithm: Past, present, and future. Multimed. Tools Appl. 80, 8091–8126 (2021).
Trevino, V. & Falciani, F. GALGO: An R package for multivariate variable selection using genetic algorithms. Bioinformatics 22, 1154–1156 (2006).
Zhang, P. et al. Selection of microbial biomarkers with genetic algorithm and principal component analysis. BMC Bioinform. 20, 413. https://doi.org/10.1186/s12859-019-3001-4 (2019).
Bedogni, G. et al. The fatty liver index: A simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 6, 1–7 (2006).
Kotronen, A. et al. Prediction of non-alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology 137, 865–872 (2009).
Lee, J. H. et al. Hepatic steatosis index: A simple screening tool reflecting nonalcoholic fatty liver disease. Dig. Liver Dis. 42, 503–508 (2010).
Long, M. T. et al. Development and validation of the Framingham steatosis index to identify persons with hepatic steatosis. Clin. Gastroenterol. Hepatol. 14, 1172-1180.e1172 (2016).
Caussy, C. et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 10, 1–9 (2019).
Jovel, J. et al. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front. Microbiol. 7, 459 (2016).
Goodrich, J. K. et al. Human genetics shape the gut microbiome. Cell 159, 789–799 (2014).
Vojinovic, D. et al. Relationship between gut microbiota and circulating metabolites in population-based cohorts. Nat. Commun. 10, 1–7 (2019).
Astbury, S. et al. Lower gut microbiome diversity and higher abundance of proinflammatory genus Collinsella are associated with biopsy-proven nonalcoholic steatohepatitis. Gut Microbes 11, 569–580 (2020).
Piñero, F. et al. A different gut microbiome linked to inflammation found in cirrhotic patients with and without hepatocellular carcinoma. Ann. Hepatol. 18, 480–487 (2019).
Llopis, M. et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 65, 830–839 (2016).
Acknowledgements
We are very grateful to all participants of the Kangbuk Samsung Hospital cohort who participated in this study. We would like to thank all lab members of Prof. Nam, Park, and Ryu for their constructive discussions. This study was supported by the SKKU-KSH Future Convergence Research Program (to C.-Y.P. and D.R.). D.R. was supported by grants from the National Research Foundation of Korea (NRF; 2020R1A2C2010964 and 2021R1A5A8029876 funded by the Ministry of Science and ICT). C.-Y.P. was supported by grants from the NRF (2014R1A2A2A04006291, funded by the Ministry of ICT). S.N. was supported by the Gachon University Research Fund of 2019 (GCU-2019-0323). H.Y. was supported by grants from the NRF (2020R1I1A1A01073155, funded by the Ministry of Education).
Author information
Authors and Affiliations
Contributions
B.E.K., A.P., J.A., S.N., C.-Y.P., and D.R. conceived and designed the projects. B.E.K, S.N., C.-Y.P., and D.R. wrote the manuscript with technical support and conceptual advice from A.P., H.Y., Y.J., T.G.O., S.M.J., Y.J., and J.A. Machine learning including data processing were performed by B.E.K., A.P., S.N., C.-Y.P., and D.R. with technical support from T.G.O., S.M.J., Y.J., and J.A. Fecal sample preparation and 16S rDNA gene sequencing were done by H.‐L.K., and H.‐N.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests, except for S.M.J. and Y.J., who are employed by HEM Pharma Inc.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, B.E., Park, A., Yang, H. et al. Machine learning-derived gut microbiome signature predicts fatty liver disease in the presence of insulin resistance. Sci Rep 12, 21842 (2022). https://doi.org/10.1038/s41598-022-26102-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-26102-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
