
Abstract
RYR1 is the gene encoding the ryanodine receptor 1, a calcium release channel of the endo/sarcoplasmic reticulum. I4898T in RYR1 is one of the most common mutations that give rise to central core disease (CCD), with a variable phenotype ranging from mild to severe myopathy to lethal early-onset core-rod myopathy. Mice with the corresponding I4895T mutation in Ryr1 present mild myopathy when the mutation is heterozygous while I4895T homozygous is perinatal-lethal. Here we show that skeletal muscles of I4895T homozygous mice at birth present signs of stress of the endoplasmic reticulum (ER stress) and of the related unfolded protein response (UPR) with increased levels of the maladaptive mediators CHOP and ERO1. To gain information on the role of CHOP in the pathogenesis of RYR1I4895T-related myopathy, we generated compound Ryr1I4895T, Chop knock-out (-/-) mice. However, the genetic deletion of Chop, although it attenuates ER stress in the skeletal muscle of the newborns, does not rescue any phenotypic or functional features of Ryr1I4895T in mice: neither the perinatal-lethal phenotype nor the inability of Ryr1I4895T to respond to its agonist caffeine, but protects from ER stress-induced apoptosis. These findings suggest that genetic deletion of the ER stress response mediator CHOP is not sufficient to counteract the pathological Ryr1I4895T phenotype.
Similar content being viewed by others


Introduction
Three distinct genes encode three isoforms of the ryanodine receptor (RYR1, RYR2, and RYR3) which have different tissue distributions: RYR1 is enriched in the skeletal muscle, RYR2 in cardiac muscle, and RYR3 is expressed ubiquitously1,2,3. All three RYRs are calcium release channels, localized in the membrane of the endoplasmic reticulum (ER) and/or sarcoplasmic reticulum (SR). RYR1 is involved in the mechanism of excitation–contraction (E-C) coupling in skeletal muscle4. This implies that an action potential travels through the transverse (T)-tubule triggering the physical interaction between the dihydropyridine receptor (DHPR) and RYR1, which results in Ca2+ release from SR/ER, hence contraction of muscle fibers4.
Mutations in RYR1 can be either homozygous or heterozygous and can lead to distinct channel defects, i.e. leaky, E-C uncoupling, and loss of RYR1 channel. These mutations can give rise to many debilitating rare myopathies ranging from central core disease (CCD, OMIM n. 117,000), to multiminicore disease (MmD, OMIM n. 602,771), to nemaline rod myopathy (OMIM n. 161,800), and including life-threatening malignant hyperthermia (OMIM n.145,600)5,6,7,8.
I4898T mutation is one of the most common mutations in RYR1, and results in a variable phenotype ranging from mild to severe CCD to the lethal early-onset core-rod myopathy9. Mice knock-in for Ryr1I4895T, which corresponds to I4898T in humans, show a mild myopathic phenotype in the C57BL/6 background when the mutation is in heterozygosity while it is perinatal-lethal when the mutation is in homozygosity10. Functional studies of Ryr1I4895T suggest that this mutation falls into the category of an E-C uncoupling receptor, meaning RYR1 is not responsive to electrical or agonist stimulation11.
The mild myopathy in heterozygous mutant mice (Ryr1I4895T/+) is associated with progressive ER stress and UPR and increased levels of the mediators CHOP (CAATT enhancer-binding protein homologous protein) and ERO1-alpha (Endoplasmic reticulum oxidoreductin 1-alpha, henceforth ERO1). Importantly, treatment with the chemical chaperone 4-PBA rescues this pathological muscle phenotype while it attenuates ER stress, suggesting that ER stress/UPR might be an important pathogenic component of this RYR1-related myopathy10.
During UPR, CHOP and its downstream targets ERO1 and GADD34 might be mediators of the maladaptive branch of this response given their ability to generate reactive oxygen species (ROS) and to restart the protein synthesis respectively in condition of altered proteostasis thereby triggering apoptotic stimuli. Indeed, in some cases, deletion of the gene encoding CHOP preserves tissue function in case of chronic ER stress/maladaptive UPR12,13,14. In the field of genetic myopathies, ablation of CHOP rescued the muscle pathological phenotype of a mouse model with loss of function of SepN1, another gene whose mutations are associated with MmD15,16,17,18. Thus, to test whether CHOP is also an important contributor to the Ryr1I4895T muscle phenotype we engineered a mouse model lacking Chop and expressing Ryr1I4895T. Although skeletal muscles of Ryr1I4895T at birth show signs of ER stress with high levels of CHOP, supporting the rationale for the development of this mouse model, Chop deletion in Ryr1I4895T genetic background does not rescue the perinatal lethality of Ryr1I4895T or the channel function, but protects cells from ER-stress induced apoptosis.
Materials and methods
Animals
Ryr1I4895T mice in the C57BL/6 background were imported from the laboratory of Prof. Susan Hamilton (Baylor College, Houston, Texas) and crossed with Chop−/− mice in the C57BL/6 background already available in our laboratory. Genotyping at the Chop and Ryr1I4895T locus followed published procedures10,16. To determine the sex of the newborn pups, we employed a multiplex PCR to detect the male-specific sequence Sry together with IL3 in DNA extracted from pups at birth. Primers Sry F: TGGGACTGGTGACAATTGTC and R: GAGTACAGGTGTGCAGCTCT. Primers IL3 F: GGGACTC-CAAGCTTCAATCA and R: TGGAG-GAGGAAGAAAAGCAA (synthesized by Life Technologies/Gibco-BRL, Rockville, MD) as in19.
All experimental protocols were approved by the Mario Negri Institute licensing committee. Procedures involving animals and their care were conducted in conformity with the principle of ARRIVE 2.0 and the laws, regulations and policies governing the care and use of laboratory animals: Italian Governing Law (D. lgs 26/2014, authorization number 19/2008-A issued 6 March 2008 by Ministry of Health; 1043/2020-PR to E. Zito); Mario Negri Institutional Regulations and Policies providing internal authorization for people conducting animal experiments (Quality Management System Certificate—UNI EN ISO9001: 2008—registration number 6121); the NIH Guide for the Care and Use of Laboratory Animals (2011 edition); EU directives and guidelines (EEC Council Directive 2010/63/UE).
Real-time quantitative RT-PCR analysis
RNA was isolated from leg muscles of newborn pups and from mouse embryonic fibroblats (MEFs) using the RNeasy Mini Kit (Qiagen), reverse-transcribed and analyzed using the Applied Biosystems’ real-time PCR System and the ∆∆Ct method. Relative gene expression in cells was normalized to GAPDH mRNA levels. The primer sequences are described in16. Sequence of Ryr3 primers were Ryr3F: ACCAGCAGG AGCAAG TACG, Ryr3R: GGGGTCGTGTCAAAGTAGTCA.
MEFs
MEFs with RyR1IT or CHOP mutation were isolated on embryonic day (E.) 13.5 and studied as primary MEFs. MEFs were also immortalized after serial passages following transfection with SV-40 large T antigen and cultured in DMEM supplemented to 25 mM glucose, 10% FCS and non-essential amino acid. ERO1 alpha knock out MEFs were characterized in20.
Western blotting
Protein concentration was determined with a standard BCA assay (Pierce). Samples with the same protein concentration were mixed with non-reducing Laemmli buffer (62.5 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol and 0.01% bromophenol blue), supplemented with 100 mM DTT and heated for 5 min at 95 °C. Protein samples separated by reducing SDS-PAGE were transferred to Protran nitrocellulose membrane (GE10600002, Amersham Protran, pore size 0.45 μm) and probed with monoclonal mouse anti-Actin (MAB1501, Sigma Aldrich) polyclonal rabbit anti-ERO1 alpha21. The fluorescent secondary antibodies IRDye 680RD goat anti-mouse IgG (926–68,070, Li-Cor) and IRDye 800CW goat anti-rabbit IgG (926–32,211, Li-Cor) were used for protein detection. The fluorescent signal was acquired on a ChemiDoc MP Imaging System and quantified by Image Lab analysis software (Bio-Rad laboratories).
MTS assay
Three thousand cells were incubated in MTS [3-(4,5-dimethylthiazol-2-il)-5-(3-carboxymethoxyphenjl)-2-(4-sulfophenjl)-2H-tetrazolio] and PMS (phenazine methosulfate), as indicated in the CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay (Promega). For acquisition, we used TECAN infinite M200 with excitation wavelengths at 490 nm.
FACS analysis
DNA flow cytometric analyses were performed on 5 × 104 cells labeled with Annexin V-FITC and propidium iodide (Invitrogen) at the acquisition rate of 300 events per second, using a CytoFLEX LX flow cytometer (Beckman Coulter, Brea, CA, USA).
Chemicals
Thapsigargin (Tg, Santa Cruz Biotechnology Inc.) and Tunicamycin (Tun, Sigma Aldrich) were incubated on cells for 24 h at the concentrations indicated. Caffeine (Cf), ATP, 2-Aminoethoxydiphenyl borate (2-APB), ryanodine (Ry) and Ru360 were purchased from Sigma-Aldrich (Milan, Italy). Fluo-4-acetoxymethyl ester and Rhod 2-acetoxymethyl ester were purchased from Thermo Fisher Scientific (Milan, Italy). The cells were loaded with the two probes, then incubated for 10 min with 10 mM Cf or 1 mM ATP with or without 50 µM 2-APB (2-aminoethoxydiphenyl borate, an inhibitor of IP3R), 100 µM Ry (ryanodine, an inhibitor of RYR) or 10 µM Ru360 (an inhibitor of mitochondrial calcium uptake).
Measurement of cytosolic and mitochondrial Ca2+
MEFs grew in 35 mm tissue culture dishes with an uncoated coverslip, treated for 20 min with 4 µM Fluo 4-AM (a fluorescent probe for the detection of cytosolic calcium) or 10 µM Rhod 2-AM (a fluorescent probe for the detection of mitochondrial calcium), then exposed for another 10 min to the IP3R or RyR agonists. The cells were finally washed three times in phosphate buffer saline (PBS; 136 mM NaCl, 10 mM Na2HPO4, 1.5 mM KH2PO4, 3 mM KCl; pH 7.4) and fluorescence images were collected with a BX-51 microscope (Olympus, Milan, Italy), equipped with a SPOT-RT camera unit (Diagnostic Instruments, Delta Sistemi, Rome, Italy) using an Olympus LCAch 40 × /0.55 objective lens. The excitation and emission wavelengths were set at 488 and 515 nm for Fluo 4, and at 540 and 590 nm for Rhod 2 with a 5-nm slit width for both emission and excitation. Images were collected with exposure times of 100–400 ms, acquired digitally and processed for fluorescence determination at the single cell level, with ImageJ software. Mean fluorescence was determined by averaging the fluorescence values of at least 50 cells/treatment condition/experiment.
Statistics
Data are the mean ± SEM, and were analyzed with Prism 7 (Graphpad). Statistical significance was established using the unpaired t-test for two-group analysis and one-way ANOVA multiple comparison tests for three or more groups. One asterisk indicates p < 0.05, two for p < 0.01, three for p < 0.001 and four for p < 0.0001.
Results
Compound homozygous Ryr1 I4895T/I4895T, Chop −/− mice die at birth
To study the importance of CHOP on the phenotypical consequence of Ryr1I4895T we crossed Ryr1WT/I4895T mice with Chop−/− mice. The compound homozygous Ryr1I4895T/I4895T, Chop−/− was obtained by crossing Ryr1WT/I4895T, Chop+/− trans heterozygote mice. Crosses between these trans heterozygotes yielded the compound homozygous Ryr1I4895T/I4895T, Chop−/− as well as Ryr1I4895T/I4895T and Ryr1I4895T/I4895T, Chop+/−. Mice were genotyped at birth with previously established procedures10,16 and females were distinguished from males by a PCR-based genotyping to detect the male-specific sequence (Sry). However, on a total of one hundred thirty-five pups, all Ryr1I4895T/I4895T pups also with mixed Chop genotypes were recovered at a lower frequency than that predicted by Mendelian transmission of the mutant alleles. The yield was better for the females with Ryr1I4895T/I4895T genotype and mixed Chop deletion which was very close to that predicted by Mendelian transmission in Ryr1I4895T/I4895T, Chop−/− (Fig. 1A).

Low recovery rate and gross morphology of Ryr1I4895T/I4895T, Chop−/− newborn pups. (A) The expected and observed distribution of males and females, as percentages of genotypes on day one among a progeny of one hundred thirty-five (sixty-eight males and sixty-seven females) newborns pups from a cross between Ryr1WT/I4895T, Chop+/− trans-heterozygote female and male mice. (B) Representative gross lateral view of newborn pups (the vertical black lines indicate that the views of the pups were trimmed from different snapshots from which the original background was erased and the views of the different pups copied on the same background) of the indicated genotypes. Ryr1I4895T/I4895T, Ryr1I4895T/I4895T, Chop+/− Ryr1I4895T/I4895T, Chop−/− have a curved posture. (C) Bar graphs depicting the mean weight of newborn mice at birth.
All the recovered pups with Ryr1I4895T/I4895T genotypes, with or without any Chop deletion, died soon after birth while siblings of the other mixed genotypes survived (Fig. 1A). The pups with Ryr1I4895T/I4895T genotypes and with or without any Chop deletion were readily identifiable at birth among other siblings given their curved posture and lack of any response to stimuli. Soon after birth, these mice became cyanotic and died, probably unable to breathe as previously reported11 (Fig. 1B). Mice with Ryr1I4895T/I4895T genotypes had lower birth weight but no appreciable difference in weight were seen among mice with Ryr1I4895T/I4895T genotypes and mixed Chop deletion (Fig. 1C). These findings unequivocally suggest that CHOP deletion does not rescue the perinatal lethality of Ryr1I4895T/I4895T mice.
Skeletal muscle of Ryr1 I4895T/I4895T mice shows signs of ER stress
Quantitative real-time PCR on cDNA from the leg muscles of the few Ryr1I4895T/I4895T pups recovered indicated increased levels of CHOP, ERO1, GADD34, XBP1 spliced, ATF4 and BIP, suggesting ER stress and UPR in their muscle while the levels of these ER stress/UPR markers were low and comparable to those of WT in the skeletal muscle of Ryr1I4895T/WT. These findings suggest ER stress and UPR in the skeletal muscle of newborn homozigous Ryr1I4895T/I4895T but not in heterozigous Ryr1I4895T/WT pups. However, while CHOP levels were half in the skeletal mice of Ryr1I4895T/I4895T, Chop+/− and abolished, as expected, in Ryr1I4895T/I4895T, Chop−/−, and GADD34 followed the same pattern, the other ER stress/UPR markers ERO1 and BIP did not follow the CHOP patterns, i.e.; ERO1 and BIP were not downregulated in Ryr1I4895T/I4895T, Chop+/− but only in Ryr1I4895T/I4895T, Chop−/− and SEPN1 was even upregulated in mice with the latter genotype (Fig. 2). These findings support the possibility that Chop deletion might lower ongoing ER stress/ER stress response in the skeletal muscle of Ryr1I4895T/I4895T pups.

Analysis of ER stress and UPR in skeletal muscle with mixed Ryr1I4895T CHOP background. Semi-quantitative real-time RT-PCR analysis of ER stress/ER stress response markers (together with Sepn1 and Ryr3) from mRNA from leg muscles of newborn pups of the genotypes indicated (N = 3).
Chop deletion does not protect Ryr1 I4895T/I4895T from the consequences of severe ER stress
The difficulty of recovering enough live Ryr1I4895T/I4895T pups to isolate skeletal myotubes limited our studies on RYR1 activity in skeletal muscle-derived cell cultures. Early reports indicated RYR1 expression in cells different from those of the skeletal muscle, such as the fibroblasts22, and indicated early expression of Ryr1 transcript already at E (embryo day) 9.511. We then looked at MEFs, which are extracted from embryos on E13.5, for functional studies on Ryr1I4895T/I4895T. There was no detrimental effect of the I4895T mutation at this embryonal stage, and in fact Ryr1I4895T/I4895T embryos were recovered at the predicted yield of the Mendelian transmission of the mutant alleles (Fig. 3A).

Effect of CHOP deletion on ER stress levels and the viability of Ryr1I4895T/I4895T cells. (A) The expected and observed distribution of twenty embryos on E13.5 as a percentage of the different genotypes obtained from a cross between Ryr1WT/I4895T, Chop+/− trans-heterozygote female and male mice. (B) Semi-quantitative real-time RT-PCR of ER stress/ER stress response markers (and Ryr3) from mRNA prepared from MEFs of the indicated genotypes (N = 4). (C) Bar graphs presenting the growth rate (MTS) in 24 h of an equal number of cells with the indicated genotypes (N = 5). (D) Survival of MEFs that received no treatment or were treated with the indicated concentrations of thapsigargin (Tg) and tunicamycin (Tun) for 24 h. Survival is expressed as the relative amount of MTS signal reduced by thapsigargin- and tunicamycin-exposed cells compared with unexposed cells (arbitrarily set to 100%) (N = 5). (E) Apoptosis analysis. Cells treated with or without tunicamycin (1μg/mL) were analyzed for Annexin-V and propidium iodide (PI) by flow cytometry and divided into necrotic, late apoptotic, early apoptotic and live cells.
WT, Chop−/−, Ryr1I4895T/I4895T and Ryr1I4895T/I4895T, Chop−/− MEFs were extracted from embryos, genotyped and cultured (Fig. Supplementary 1A). Quantitative real-time PCR on cDNAs from MEFs indicated the presence of Ryr1 transcript in MEFs, although sixty times less than in the diaphragm (data not shown). We tested also Ryr3, the ubiquitously expressed Ryr isoform, which resulted upregulated in Ryr1I4895T/I4895T muscle but not in MEFs (Fig. 2 and Fig. 3B). Therefore, we used MEFs as a surrogate cell system to study whether Chop deletion protects Ryr1I4895T/I4895T cells from the consequences of ER stress and influences RYR1 activity.
Primary Ryr1I4895T/I4895T MEFs, and SV40-immortalized Ryr1I4895T/I4895T MEFs had increased CHOP, ERO1, GADD34, ATF4, XBP1 spliced levels, as already seen in the skeletal muscle of mice with the same genotype, but BIP did not increase significantly in these MEFs. Importantly and in line with what was seen in skeletal muscle, ERO1, GADD34, ATF4, XBP1 spliced levels were significantly reduced in Ryr1I4895T/I4895T, Chop−/− (Fig. 3B).
Ryr1I4895T/I4895T MEFs had reduced cell viability compared to WT, which it was better in Ryr1I4895T/I4895T Chop−/− MEFs, suggesting that there is a positive correlation between reduced ER stress and improved viability in the latter (Fig. 3C). To understand whether lack of CHOP protected Ryr1I4895T/I4895T against the consequences of severe ER stress, MEFs underwent treatments with different doses of the two ER stress inducer drugs tunicamycin and thapsigargin and their viability was analyzed. Ryr1I4895T/I4895T, Chop−/− MEFs were not protected from the detrimental effect on the viability (as detected by a MTS assay) of the exposure to these two drugs (Fig. 3D). However, the analysis of an apoptotic readout, i.e., the staining Annexin V and propidium iodide, on cells treated for twelve hours with tunicamycin indicated a higher rate of apoptosis in Ryr1I4895T/I4895T which was much lower in Ryr1I4895T/I4895T, Chop−/− MEFs, suggesting that CHOP deletion preserved Ryr1I4895T/I4895T cells from tunicamycin-induced apoptosis (Fig. 3E).
These observations indicate that Chop deletion does not protect the overall viability of Ryr1I4895T/I4895T MEFs against the consequences of severe ER stress but protects them from ER stress-induced apoptosis.
RYR1 I4895T activity is not rescued following CHOP deletion
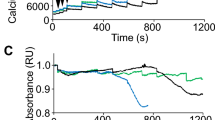
WT, as well as Chop−/− MEFs, were responsive to the RYR agonist caffeine (Cf)23, as suggested by the increases in calcium levels in the cytosol and mitochondria, pointing to a functional RYR in MEFs (Fig. 4A). Consistently with this, the calcium levels were blunted in both organelles by a high concentration of the alkaloid ryanodine (Ry), a RYR antagonist23. Mitochondrial calcium accumulation was impaired by Ru360, an inhibitor of the mitochondrial calcium uniporter24 (Fig. 4A). In contrast, caffeine did not raise calcium levels in cytosol and mitochondria of Ryr1I4895T/I4895T and Ryr1I4895T/I4895T, Chop−/− MEFs, suggesting that both MEFs express a non-functional caffeine-insensitive RYR (Fig. 4A).

Calcium handling in Ryr1I4895T/I4895T and in Ryr1I4895T/I4895T, Chop−/− cells. (A) Bar graphs depicting the effects of caffeine (Cf, an agonist of the RYR), Ryanodine (Ry, an inhibitor of the RYR) and Ru360 (an inhibitor of mitochondrial calcium uptake) on calcium levels in cytosol (detected by the Fluo 4 probe) and in mitochondria (detected by the Rhod 2 probe) in the MEFs of the indicated genotypes. (B) Bar graphs depicting the effects of ATP (an agonist of IP3r), 2-APB (an inhibitor of IP3r), Ry and Ru360 on calcium levels in cytosol (detected by the Fluo 4 probe) and in mitochondria (detected by the Rhod 2 probe) in the MEFs of the indicated genotypes. For clarity, the value of untreated cells is duplicated and presented in panels (A) and (B). Results are the means ± SD calculated from three separate experiments (ANOVA followed by Dunnett's test).
However, ATP, an IP3 receptor (r) agonist25, raised calcium levels in cytosol and mitochondria through a mechanism sensitive to 2-APB, an IP3r antagonist, in all four genotype-divergent MEFs26 (Fig. 4B). Interestingly, Ry partially reduced the ATP-dependent increase in cytosolic calcium and suppressed the mitochondrial calcium accumulation in WT and Chop−/− MEFs, implying the involvement of the RYR in mitochondrial calcium accumulation after IP3r stimulation, as already seen in other RYR expressing cells27. However, while Ry had no detectable effects in Ryr1I4895T/I4895T and Ryr1I4895T/I4895T, Chop−/− MEFs, the combination of ATP with Ry raised cytosolic and mitochondrial calcium in Ryr1I4895T/I4895T and Ryr1I4895T/I4895T, Chop−/− (Fig. 4B).
Thus, in Ryr1I4895T/I4895T MEFs and in their counterpart without CHOP, calcium mobilization was compensated by the involvement of IP3r. Impressed by28, which reported a hyperactive effect of the loss of the CHOP mediator ERO1 on the cardiac-selective isoform of RyR, RyR2, we evaluated RyR-dependent calcium transients to caffeine in ERO1-alpha knock-out MEFs. However, ERO1 loss in these cells and in basal conditions did not change neither cytosolic or mitochondrial calcium transients (Fig. Sup. 1B and C).
Altogether, the above pharmacological studies indicate a non-responsive RyR to the agonist caffeine in Ryr1I4895T/I4895T MEFs whose activity is not rescued by CHOP deletion.
Discussion
RYR1-related myopathy is a class of rare muscular diseases due to heterozygous or homozygous mutations in RYR1, which encodes the Ca2+ channel RYR1. These mutations can result in a leaky, E-C-uncoupling or complete loss of the channel. There is still no treatment for RYR1-RM. Progress has been made for some RYR1 mutations: Rycals are a novel class of small molecules which restore the calstabin1 binding to RyR1 channel, stabilizing the closed state of the channel and resulting in a potential valid treatment for RYR1-RM with a leaky channel29. However, there is still a critical need for studies on the pathogenesis of RYR1-RM to identify treatments for the other mutations such as those that give rise to E-C-uncoupling.
The significant degree of clinical (i.e., muscle weakness, breathing problems, scoliosis) and histopathological (i.e., minicores) overlap between SEPN1-RM and RYR1-RM might reflect molecular defects common to these two genetically distinct pathologies. Although with two distinct functions SEPN1 and RYR1 are both localized in the ER/SR membrane and involved in calcium homeostasis of the ER/SR. Selenoprotein N1 is a type II selenocysteine-containing transmembrane protein of the ER which works as a calcium sensor, activating the Ca2+ pump SERCA and protecting it against the excessive oxidation imposed by the ER stress-induced oxidase ERO1 alpha (henceforth ERO1)17,30,31. RYR1 is a redox-sensitive Ca2+ channel of the ER/SR, that triggers Ca2+ efflux from this cellular compartment, leading to excitation–contraction (E-C) coupling32. Primary myotubes from patients with either SEPN1 null or RYR1 mutations present oxidative stress which can be rescued by the reductant N-acetylcysteine (NAC)33. This result led to the first therapeutic trials with NAC in SEPN1-RM and RYR1-RM (ClinicalTrials.gov NCT02505087 and NCT02362425). However, the RYR1 trial with NAC failed to achieve its primary endpoint, while the SEPN1 results are still under analysis34. Thus, new molecular determinants are still needed common to the pathogenesis of these two diseases, which can justify the overlapping symptoms.
Skeletal muscle is exposed to physiological triggers of ER stress, such as hypoxia, reactive oxygen species (ROS) triggered during muscle contraction, and unbalanced Ca2+. Thus, skeletal muscle might experience ER stress despite not being a highly secretory tissue (i.e., with a high load of proteins to fold). Furthermore, the response to ER stress, referred to as the Unfolded protein response (UPR), is involved in the physiological function of the skeletal muscle, providing one more proof of the existence of ER stress and its response (UPR) in this tissue32,35. Although UPR is first of all a homeostatic response, in some circumstances, still to be completely elucidated, it can be maladaptive, and in fact deletion of one of its mediators, the gene encoding CHOP, preserves tissue function in cases of a pathological ER stress response12,13,14. Importantly, in the case that ER stress and the consequent UPR were pathogenic in skeletal muscle, new molecules that target specific branches of these pathways are available for pharmacological intervention36.
Although SEPN1 regulates SERCA and thus ER calcium uptake, while RYR1 regulates calcium release from ER, both SEPN1 and RYR1 mutants show ER calcium defects, which might imply a common underlying mechanism triggering ER stress and maladaptive UPR, giving rise to the overlapping pathological phenotype. In this regard, we found that the ablation of CHOP rescued the muscle phenotype of SEPN1 KO mice while it reduced ERO1 levels in skeletal muscle, suggesting that the CHOP/ERO1 axis might be the main contributor to SEPN1-RM16,30,37.
The I4898T mutation is one of the most common RYR1 mutations in humans. Analysis of myotubes from mice carrying Ryr1I4895T showed that the receptor lacks voltage- and 4-CMC-induced Ca2+ release, suggesting that it belongs to the class of mutations generating an uncoupling receptor. Skeletal muscles of Ryr1I4895T/WT mice point to altered Ca2+ handling, increased ROS, ER stress/UPR, with the upregulation also of CHOP and ERO1, associated with defective muscle force10. Importantly, the ER stress inhibitor and chemical chaperone 4-PBA rescues the pathological muscle phenotype of Ryr1I4895T/WT mice raising the possibility that ER stress is an important driver also for this myopathy.
On the basis of this rationale, we generated mice with genetic deletion of Chop in Ryr1I4895T genetic background to study whether ablation of CHOP and thus lack of a maladaptive component of UPR might protect from the pathological consequences of Ryr1I4895T.
Ryr1I4895T/I4895T newborn mice with mixed Chop genotypes were recovered at a lower frequency than that predicted by Mendelian transmission of the mutant alleles, which might reflect missed animals on day one rather than embryonal lethality of the mutation. Indeed, the rare newborns with Ryr1I4895T/I4895T mutation and mixed Chop genotypes were recovered only when we assisted to the delivery and they died soon after. Unfortunately, the lack of CHOP, although it attenuates ERO1, GADD34 and the chaperone BIP levels in skeletal muscle of Ryr1I4895T mice, suggesting reduced ER stress, does not recover RYR1 activity or rescue the perinatal lethality of Ryr1I4895T mice. This suggests that the failure in the recovery of any RYR1 activity frustrates the rescue of the perinatal lethality despite the reduced ER stress.
This study highlights the untoward consequences of the lack of a mediator of the maladaptive branch of UPR in the missed rescue of Ryr1I4895T muscle phenotype. Thus, this study excludes the ER stress/maladaptive branch of UPR, or at least its mediator CHOP, as the primary cause of this RYR1-RM. Further studies will aim to clarify whether CHOP deletion in a genetic background with a more modest increase in Ryr1I4895T, as in Ryr1I4895T/WT, triggers any increase/change in RYR1 activity and rescues the related less severe muscle pathological phenotype.
Data availability
All data generated or analysed during this study are included in this published article.
References
Zorzato, F. et al. Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 265, 2244–2256 (1990).
Otsu, K. et al. Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J. Biol. Chem. 265, 13472–13483 (1990).
Giannini, G., Conti, A., Mammarella, S., Scrobogna, M. & Sorrentino, V. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J. Cell. Biol. 128, 893–904. https://doi.org/10.1083/jcb.128.5.893 (1995).
Sandow, A. Excitation-contraction coupling in muscular response. Yale. J. Biol. Med. 25, 176–201 (1952).
Zhang, Y. et al. A mutation in the human ryanodine receptor gene associated with central core disease. Nat. Genet. 5, 46–50. https://doi.org/10.1038/ng0993-46 (1993).
Ferreiro, A. et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann. Neurol. 51, 750–759. https://doi.org/10.1002/ana.10231 (2002).
MacLennan, D. H. et al. Ryanodine receptor gene is a candidate for predisposition to malignant hyperthermia. Nature 343, 559–561. https://doi.org/10.1038/343559a0 (1990).
Sewry, C. A., Laitila, J. M. & Wallgren-Pettersson, C. Nemaline myopathies: A current view. J. Muscle Res. Cell Motil. 40, 111–126. https://doi.org/10.1007/s10974-019-09519-9 (2019).
Lynch, P. J. et al. A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc. Natl. Acad. Sci. U. S. A. 96, 4164–4169. https://doi.org/10.1073/pnas.96.7.4164 (1999).
Lee, C. S. et al. A chemical chaperone improves muscle function in mice with a RyR1 mutation. Nat. Commun. 8, 14659. https://doi.org/10.1038/ncomms14659 (2017).
Zvaritch, E. et al. An Ryr1I4895T mutation abolishes Ca2+ release channel function and delays development in homozygous offspring of a mutant mouse line. Proc. Natl. Acad. Sci. U. S. A. 104, 18537–18542. https://doi.org/10.1073/pnas.0709312104 (2007).
Pennuto, M. et al. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-marie-tooth 1B mice. Neuron 57, 393–405. https://doi.org/10.1016/j.neuron.2007.12.021 (2008).
Walter, P. & Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 334, 1081–1086. https://doi.org/10.1126/science.1209038 (2011).
Song, B., Scheuner, D., Ron, D., Pennathur, S. & Kaufman, R. J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Invest. 118, 3378–3389. https://doi.org/10.1172/JCI34587 (2008).
Varone, E. et al. SELENON (SEPN1) protects skeletal muscle from saturated fatty acid-induced ER stress and insulin resistance. Redox Biol. 24, 101176. https://doi.org/10.1016/j.redox.2019.101176 (2019).
Pozzer, D. et al. A maladaptive ER stress response triggers dysfunction in highly active muscles of mice with SELENON loss. Redox Biol. 20, 354–366. https://doi.org/10.1016/j.redox.2018.10.017 (2018).
Chernorudskiy, A. et al. Selenoprotein N is an endoplasmic reticulum calcium sensor that links luminal calcium levels to a redox activity. Proc. Natl. Acad. Sci. U. S. A. 117, 21288–21298. https://doi.org/10.1073/pnas.2003847117 (2020).
Pozzer, D. et al. Endoplasmic reticulum oxidative stress triggers Tgf-beta-dependent muscle dysfunction by accelerating ascorbic acid turnover. Sci. Rep. 7, 40993. https://doi.org/10.1038/srep40993 (2017).
Lambert, J. F. et al. Quick sex determination of mouse fetuses. J. Neurosci. Meth. 95, 127–132. https://doi.org/10.1016/s0165-0270(99)00157-0 (2000).
Chin, K. T. et al. The sarcoplasmic reticulum luminal thiol oxidase ERO1 regulates cardiomyocyte excitation-coupled calcium release and response to hemodynamic load. FASEB J. 25, 2583–2591. https://doi.org/10.1096/fj.11-184622 (2011).
Zito, E., Chin, K. T., Blais, J., Harding, H. P. & Ron, D. ERO1-beta, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J. Cell Biol. 188, 821–832. https://doi.org/10.1083/jcb.200911086 (2010).
Zhou, H. et al. Epigenetic allele silencing unveils recessive RYR1 mutations in core myopathies. Am. J. Hum. Genet. 79, 859–868. https://doi.org/10.1086/508500 (2006).
Meissner, G. The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 149, 1065–1089. https://doi.org/10.1085/jgp.201711878 (2017).
Kirichok, Y., Krapivinsky, G. & Clapham, D. E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. https://doi.org/10.1038/nature02246 (2004).
Berridge, M. J. Inositol trisphosphate and calcium signalling. Nature 361, 315–325. https://doi.org/10.1038/361315a0 (1993).
Berridge, M. J. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96, 1261–1296. https://doi.org/10.1152/physrev.00006.2016 (2016).
Guidarelli, A. et al. Functional organization of the endoplasmic reticulum dictates the susceptibility of target cells to arsenite-induced mitochondrial superoxide formation, mitochondrial dysfunction and apoptosis. Food Chem. Toxicol. 156, 112523. https://doi.org/10.1016/j.fct.2021.112523 (2021).
Hamilton, S. et al. Ero1alpha-dependent ERp44 dissociation from RyR2 contributes to cardiac arrhythmia. Circ. Res. 130, 711–724. https://doi.org/10.1161/CIRCRESAHA.121.320531 (2022).
Kushnir, A. et al. Intracellular calcium leak as a therapeutic target for RYR1-related myopathies. Acta Neuropathol. 139, 1089–1104. https://doi.org/10.1007/s00401-020-02150-w (2020).
Marino, M. et al. SEPN1, an endoplasmic reticulum-localized selenoprotein linked to skeletal muscle pathology, counteracts hyper-oxidation by means of redox-regulating SERCA2 pump activity. Hum. Mol. Genet. 24, 1843–1855. https://doi.org/10.1093/hmg/ddu602 (2015).
Zito, E. & Ferreiro, A. Calcium and redox liaison: A key role of selenoprotein N in skeletal muscle. Cells 10, 1116. https://doi.org/10.3390/cells10051116 (2021).
Boncompagni, S., Pozzer, D., Viscomi, C., Ferreiro, A. & Zito, E. Physical and functional cross talk between endo-sarcoplasmic reticulum and mitochondria in skeletal muscle. Antioxid. Redox. Signal 32, 873–883. https://doi.org/10.1089/ars.2019.7934 (2020).
Dowling, J. J. et al. Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain 135, 1115–1127. https://doi.org/10.1093/brain/aws036 (2012).
Todd, J. J. et al. Randomized controlled trial of N-acetylcysteine therapy for RYR1-related myopathies. Neurology 94, e1434–e1444. https://doi.org/10.1212/WNL.0000000000008872 (2020).
Wu, J. et al. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1α/ATF6α complex. Cell Metab. 13, 160–169. https://doi.org/10.1016/j.cmet.2011.01.003 (2011).
Zito, E. Targeting ER stress/ER stress response in myopathies. Redox. Biol. 26, 101232. https://doi.org/10.1016/j.redox.2019.101232 (2019).
Zito, E. ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 83, 299–304. https://doi.org/10.1016/j.freeradbiomed.2015.01.011 (2015).
Acknowledgements
Prof. Susan Hamilton (Baylor College, Houston, Texas) is acknowledged for kindly providing Ryr1I4895T mice. Dr. Chiara Grasselli is acknowledged for her help with the FACS analysis. This study was supported by Institutional and the RYR-1 foundation funding (2019).
Author information
Authors and Affiliations
Contributions
S.G. and A.C.M. ran the experiments, A.G. and O.C. critically analysed the experiments and provided reagents, V.S. and E.Z. acquired funding, E.Z. designed and oversaw the experiments, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Germani, S., Marchetti, A.C., Guidarelli, A. et al. Loss-of-rescue of Ryr1I4895T-related pathology by the genetic inhibition of the ER stress response mediator CHOP. Sci Rep 12, 20632 (2022). https://doi.org/10.1038/s41598-022-25198-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-25198-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
