
Abstract
The human gut microbiota has been shown to be significantly perturbed by antibiotic use, while recovering to the pre-treatment state several weeks after short antibiotic exposure. The effects of antibiotics on the gut microbiota have however been mainly documented in high-income settings with lower levels of antibiotic resistance as compared to lower and middle income countries (LMIC). This study aimed to examine the long-term consequences of repeated exposure to commonly use antibiotics on the fecal microbiota of residents living in a low income setting with high prevalence of antibiotic resistance. Fecal samples from household individuals (n = 63) participating in a rural cohort in northern Vietnam were collected monthly for a period of 6 months. Using 16S V4 rRNA gene region amplicon sequencing and linear mixed-effects models analysis, we observed only a minor and transient effect of antibiotics on the microbial richness (ß = − 31.3, 95%CI = − 55.3, − 7.3, p = 0.011), while the microbial diversity was even less affected (ß = − 0.298, 95%CI − 0.686, 0.090, p = 0.132). Principal Component Analyses (PCA) did not reveal separation of samples into distinct microbiota-based clusters by antibiotics use, suggesting the microbiota composition was not affected by the antibiotics commonly used in this population. Additionally, the fecal microbial diversity of the subjects in our study cohort was lower when compared to that of healthy Dutch adults (median 3.95 (IQR 3.72–4.13) vs median 3.69 (IQR3.31–4.11), p = 0.028, despite the higher dietary fiber content in the Vietnamese as compared to western diet. Our findings support the hypothesis that frequent antibiotic exposure may push the microbiota to a different steady state that is less diverse but more resilient to disruption by subsequent antibiotic use.
Similar content being viewed by others


The crucial importance of the human gut microbiota in modulation of host health, including prevention of infections, immune-mediated, metabolic and neurological diseases, has been well-established1,2.
After the initial maturation of the gut microbiota during the first years of life, it exists as a highly diverse and relatively stable ecosystem in healthy individuals3,4. Transient or long term alterations can however still occur in response to various external factors such as diet, living conditions and medical drug use5. In general, the human gut microbiota is rather resilient and recovers after drastic pulse perturbations, such as courses of antibiotics. Maintaining a resilient and robust microbiome is pivotal to minimize the period of microbial disruption upon antibiotic use which may put the host at increased risk of opportunistic infections6.
Microbiota resilience after antibiotic exposure has been shown to be highly dependent on diet, environmental context, past perturbations, and specific properties of the used antibiotics, as well as the individual-specific microbiome7,8. Regarding the latter, four main factors have been recognized to play an essential role in the resilience and recovery of the human gut microbiota: initial composition, capability to maintain its functional redundancy, abundance of (opportunistic) pathogens and the antibiotic resistance genes (ARGs) reservoir9,10. Indeed, when a simulated human gut microbiota was exposed to amoxicillin, it was shown that its biodiversity was extended by increasing abundance of pathogens carrying resistance genes11. A study on 12 healthy adults found that bacterial species harboring β-lactam (ARGs) modulated the resilience/recovery patterns by increasing the chances of survival and novel colonization after a broad-spectrum antibiotic treatment12.
β-lactam antibiotic therapy has been associated with profound alterations in the gut microbiota of both healthy individuals and patients, including a significant decrease in species richness, changes in abundance of specific bacterial taxa and a relative increase in antimicrobial resistance13. For instance, previous studies demonstrated that the abundance of Enterobacteriaceae was decreased after treatment with penicillin and cephalosporin antibiotics while the abundance of enterococci and Bacteroides spp was increased5,14. Moreover, the ensuing perturbations related to the use of β-lactam antibiotics is conductive for the selective outgrowth of opportunistic pathogens as illustrated by the increased risk of Clostridioides difficile infection15,16. A recent study illustrated that a course of penicillin or amoxicillin does not alter human gut microbiota of western obese individual17, whereas other studies found more profound yet temporary microbiota alterations upon amoxicillin treatment18. Previous culture-based studies reported that bacterial abundance returned to pre-antibiotics state 4 to 14 days after discontinuation of cephalosporin use19,20,21, but some studies observed an altered state that lasted longer than 42 days22,23.
The majority of studies were conducted in high-income settings or settings with a low prevalence of antibiotic resistance. Studies on impact of antibiotics on microbiota conducted in low- and middle-income countries (LMICs) with a high consumption of antibiotics and high prevalence of antibiotic resistance are underrepresented24. The effects of antibiotics on the microbiota composition in these settings may differ given the different initial microbiota composition resulting from different lifestyles, dietary habits and history of antibiotic exposures. The fecal microbiota composition of rural populations in LMICs like Vietnam, is expected to be more diverse than in high income countries25 at least in part due to the lower fat and higher fibre-content of the diet26,27 as compared to the Western diets which are typically enriched in saturated fats and simple carbohydrates28,29. We therefore conducted this study (i) to explore the long-term impact of antibiotics on diversity of the human gut microbiota in a resource limited setting and (ii) to evaluate whether repeated exposure to commonly used antibiotics induces the human gut microbiota with high prevalence of antibiotic resistance to be resilient to further antibiotic effects.
Study design and methods
Study cohort, study design and fecal sample collection
This study was conducted within the context of an existing prospective household-based community cohort in Ha Nam in northern Vietnam, which was established to quantify the burden of influenza and to gain insights into the influenza virus transmission in a tropical setting. A full description of the demographics and lifestyle of the Ha Nam cohort has been published previously30.
To examine the impact of antibiotic use on shifts in the gut microbiota of residents of this community, we conducted a longitudinal study over a period of six months from November 2014 to June 2015. The study design encompassed weekly interviews to collect information on health issues and consumption of antibiotic use in the preceding week and monthly fecal samples for microbial profiling. The interviews included questions to indicate the indications for which antibiotics were used in the preceding week (including having respiratory symptoms (cough, breathless, sore throat)).
From 80 households (HHs), we selected 11 HHs in which at least one household member used antibiotics during the six months follow-up. In total this resulted in 41 participants of whom 32 used at least one course of antibiotics.
In addition, we randomly selected a control group from 11 HHs (22 participants) where none of the household members used antibiotics during the study period. We used randomizer (www.randomizer.org) for the selection.
To investigate the effects of antibiotic use, samples were divided into one of three categories: samples collected within two weeks after last antibiotic use (E2), samples collected within two to four weeks after last antibiotic exposure (E4), and samples collected without any antibiotic exposure in the preceding four weeks (Fig. 1).

Study design and classification of samples into categories based on the number of days of exposure to antibiotics prior to sampling. The blue boxes indicate HHs, subjects and samples are part of the assessment. The light grey boxes indicate HHs of the study cohort and part of participants whose samples did not be included in the assessment.
To compare the microbial diversity between our cohort and a cohort with less antibiotic use, we included a random selection of healthy Dutch adults (≥ 18 years) who participated in the Carriage Of Multiresistant Bacteria After Travel (COMBAT) Study. This study was conducted to evaluate the travel-associated risk factors on the acquisition, persistence and transmission of antimicrobial resistance (AMR) in gut microbiota of healthy people. The detailed information of demographic characterization of the cohort has been reported previously31. For the purpose of the present study, we only included fecal samples collected at baseline (before travel) of 106 subjects with no history of antibiotic use during the past year. Differences in Shannon diversity and observed species richness in fecal samples of Dutch and Vietnamese individuals were examined by linear regression analyses with adjustment for age and sex as potential confounding factors.
We used the term “microbial resistance to pulse perturbations” to indicate the resistance of the microbiota to change upon exposure to antibiotic use. Microbial resilience, on the other hand, reflects the recovery speed upon a shift in the microbiota. In this study, to examine the microbial resistance to pulse perturbations, we stratified Vietnamese individuals who used antibiotics according to their microbial resistance. For this purpose, we calculated the Aitchison distance between the samples of each subject collected before and immediately after antibiotic use. Subjects were classified as having a low microbial resistance when the Aitchison distance was above the median, whereas subjects with an Aitchison distance below the median were classified as having a high microbial resistance to pulse perturbations changes.
We also examined if the number of different antimicrobial resistance genes (ARGs) encoding extended-spectrum beta-lactamases, carbapenemases and plasmid-mediated colistin resistance in fecal samples was associated with the microbial community structure.
DNA extraction, 16S –rRNA amplicon sequencing and determination of antimicrobial resistance genes
Fecal samples were transported within 4 h after defecation to the National Institute of Hygiene and Epidemiology (NIHE) in cold conditions (4–8 °C). Within 3 h after transport, samples were stored at − 80 °C until DNA extraction. Microbial DNA was manually extracted from frozen feces according to protocol Q of the International Human Microbiome Standards consortium32. Briefly, fecal samples were firstly subjected to mechanical lysis by repeated Bead-Beating (RBB) followed by column-based purification.
We performed amplicon libraries preparation and sequencing as described previously33. In brief, we amplified the V4 region of the 16S rRNA gene from each DNA sample in triplicate using the 515 f./806r primer pair34. PCR amplicons of the triplicate reactions were pooled; next products were purified using AMPure XP purification (Agencourt, Massachusetts, USA) according to the manufacturer’s instructions and eluted in 25 µl 1 × low TE (10 mM Tris–HCl, 0.1 mM EDTA, pH 8.0). Subsequently, we quantified DNA concentration by Quant-iT PicoGreen dsDNA reagent kit (Invitrogen, New York, USA) using a Victor3 Multilabel Counter (Perkin Elmer, Waltham, USA). The purified amplicons were mixed in equimolar concentrations to ensure equal representation of each sample and, were sequenced on an Illumina MiSeq instrument using the V3 reagent kit (2 × 250 cycles).
Bioinformatics
After demultiplexing raw reads, we visualized the quality profiles of forward and revered reads by uploading the Fastq files into R using package DADA2 (v1.16.0)35. We set maximum number of expected errors (E_max) at 0.5 to remove low-quality reads (with expected errors (E) > E_max) from demultiplexed reads. We then remained good-quality regions of reads for further steps by truncation at positions 200 of forward and 140 of reverse reads. Samples with total read counts below 12,000 were not further analyzed. Merged reads were used to generate amplicon sequence variants (ASV) using DADA2 (V1.16.0)35. Identified ASVs were aligned to construct a phylogeny with FastTree236. Taxonomy was assigned using the RDP v16 (Ribosomal Database Project—SILVA 1.38 v2) with the set formatted for DADA2 package37.
ASVs that were present in less than 5% of all samples as well as reads with a total relative abundance < 0.01% were removed from downstream analysis. At ASV level of taxonomic aggregation, we calculated the alpha-diversity (observed richness and Shannon index) and beta-diversity (Aitchison and Bray–Curtis distances) as measures of within- and between-sample microbial diversity, respectively. To examine the change in alpha-diversity from baseline (M1) to the subsequent time points (M2, M3, M4, M5, M6), we next calculated the delta of the alpha diversity (\(d\Delta\)) using the formula:
Statistical analysis
All statistical and computational analysis and visualizations were performed in R v4.1.1 (2021–08-10) using the following packages: reshape2 (v.1.4.4), purrr (v.0.3.4), readr (v.1.3.1), tidyr (v.1.1.2), tibble (v.3.0.4), tidyverse (v.1.3.0)38, RColorBrewer (v.1.1–2), ggthemes (v.4.2.0), circlize (v.0.4.10), ComplexHeatmap (v.2.0.0)39, ggplot2 (v.3.3.2)38, lme4 (v.1.1–27.1), glmmTMB (v.1.1.2.3), ggstatsplot (v.0.9.0)40,dplyr (v.1.0.2)38, phyloseq (v.1.28.0), microViz (0.7.10.9004)41.
Linear mixed effects models were used to test for the effects of antibiotic use, age, and time points on alpha diversity (IBM SPSS Statistics, v 27.0, IBM, Armonk (NY), USA). The model included exposure to antibiotics, age and time point of sample collection as fixed effects and participant ID as a random effect to control for repeated measures. The variance inflation factors (VIF) were calculated prior to model fitting to assess correlated predictor parameters, none were excluded on the basis of collinearity. In addition, the residuals were plotted and checked for homoscedasticity. We next used generalized linear mixed effects models using a negative binomial distribution to test the effect of antibiotic use on the relative abundance of individual bacterial genera (glmmTMB package, R). For each genus, we fitted the model Genus (Counts) ~ Exp_ABx_3codes + Age + Time_point + Age *Exp_ABx_3codes + offset (log (Total_counts)) + (1|ParticipantID). In the model, the variance ‘Exp_ABx_3codes’ is a logical values including three categories that was grouped by number of days since the last antibiotic use prior to sampling as described in the study design.
To examine differentially abundant genus-level taxa between Dutch and Vietnamese individuals, we conducted Analysis of Compositions of Microbiomes with Bias Correction (ANCOM-BC). Differentially abundant genera (p < 0.05 upon Bonferroni correction) are presented42.
Ethical approval
The research was approved by the Oxford University Tropical Research Ethics Committee (OxTREC, 49–14), the National Institute of Hygiene and Epidemiology, Vietnam (NIHE) institutional review board and National Hospital for Tropical Diseases, Vietnam.
All methods were performed in accordance with the relevant guidelines and regulations.
We confirm that informed consent was obtained from all participants and/or their legal guardians.
Results
Antibiotic use in the Ha Nam cohort (November 2014–June 2015)
To analysis the impact of antibiotic use on the microbiota in the Ha Nam cohort. This observational study investigated gut microbiota responses among 63 individuals belonging to 11 HHs that used at least one course of antibiotics (n = 32) or 11 HHs that did not use antibiotics (n = 31) during the study period. The median age was 28 years (IQR = 8.5–45.2) for those that consumed antibiotics (including 6 subjects aged < 6 years) versus 41 years (IQR = 17.5–52.5) for those who did not (no subjects aged < 6 years).
Among the 32 individuals who used antibiotics during the study period, 20 individuals consumed two or more courses, which resulted in a total consumption of 69 courses of oral antibiotics (Figure S1). First and second generation cephalosporins (n = 52) were most frequently consumed, followed by penicillin (n = 15), trimethoprim (n = 1) and lincosamide antibiotic (n = 1).
The symptoms for which participants reported to have used antibiotics were symptoms of the upper respiratory tract (fever accompanied by cough) for 50 courses, urinary tract combined with upper respiratory tract symptoms for 15 courses and urinary tract combined with stomach complaints for 4 courses. In none of these cases individuals reported to have symptoms of diarrhoea or other intestinal tract symptoms. Therefore, it is unlikely that confounding by indication (i.e., enteric infections rather the antibiotics itself affecting the microbiome) affected our findings.
Transient effect of antibiotics on microbiota and low diversity at baseline
A total of 373 samples were collected from 63 individuals at month 1 (n = 60), month 2 (n = 62), month 3 (n = 63), month 4 (n = 63), month 5 (n = 63) and month 6 (n = 62) of follow-up.
After filtering of low-quality reads the remaining reads with a median sequencing depth of 81,488 reads per sample (IQR 71,719–93,257) were clustered into 5,367 amplicon sequence variants (ASVs). Upon removal of rare ASVs, defined as ASVs present in less than 5% (n = 19) of the samples and ASVs with an overall abundance of less than 0.01%, a total of 460 ASVs were retained.
We first investigated the richness and diversity of the microbiota. Overall, significant differences during the study period were not observed for both richness and biodiversity (Fig. 2). Although we did not observe a difference in alpha diversity metrics over time at the population-level, we did observe strong perturbations among individuals. At time-points when individuals had consumed antibiotics, the alpha diversity often was reduced as compared to baseline (M1). Differences then gradually narrowed at the subsequent time points, when those individuals did not use antibiotics. The diversity of microbiota from individuals who did not use antibiotics was more stable over 6 months (Fig. 3a, 3b).

Comparison of alpha diversity (observed richness and Shannon diversity) between follow-up time-points in the study cohort.

Within-subject shifts in microbial richness and diversity throughout the 6-months follow-up. The heatmaps depict the changes in microbial richness (a) and Shannon diversity (b) within each study subject between the first month (baseline) and the following months during the study period. Circles and triangles indicate that subjects used antibiotics within four weeks of the later time point (circles for cephalosprorins and triangles for penicillin/amoxicillin). The red lines under the participant ID indicate individuals under 6 years of age. The colour legends (Delta) indicate the difference of alpha diversity from baseline.
To account for the longitudinal design of our study and for potential confounding by age and carriage of extended-spectrum beta lactamases (ESBL) and carbapenemases, we next assessed the effects of antibiotics on the alpha diversity by linear mixed-effects models. Neither the follow-up time-point at which the fecal samples were collected, nor age, significantly affected the observed richness and Shannon index (all p > 0.05) (Table 1). Also, the fecal carriage of enzymes encoding ESBLs (n = 235) and carbapenemases (n = 22) did not affect these alpha diversity indices (Table 1). While antibiotic use was associated with a significant decrease in microbial richness, this effect was only observed when antibiotics were used within two weeks prior to fecal sampling (ß = − 31.3, 95%CI = − 55.3, − 7.3, p = 0.011) and had already disappeared when antibiotics were consumed 2–4 weeks prior to fecal sampling (ß = − 1.3, 95%CI = − 34.4, 31.7, p = 0.937). Also, the microbial diversity, as measured by the Shannon index, was reduced in samples collected within two weeks after antibiotic use, although this was not statistically significant (ß = − 0.298, 95%CI − 0.686, 0.090, p = 0.132). These findings suggest that in the current population the use of antibiotics has only an effect on the microbial richness for a short period, while the microbial diversity is even less affected. In contrast the reduction of microbial diversity remained significantly lower than that in the initial composition after 42 days of treatment with beta–lactam antibiotics, as previously described11,12.
Microbiota of Vietnamese as compared to Dutch adults
As this modest effect of antibiotic use is in contrast to previous studies43,44, we hypothesized that the microbiota diversity of individuals in this cohort might have already been reduced by extensive past antibiotic use to an extent that the effect of further perturbations would be limited. To test this hypothesis, we compared the microbial richness and diversity of the adults in the Ha Nam cohort (n = 63) to that of a cohort of healthy Dutch adults (n = 106). Several previous studies have shown that rural communities in LMICs have a higher microbial richness and diversity compared to individuals in Western countries, mainly as a result of a diet that is rich in fibers and low in animal fat and protein. Indeed, most Vietnamese adults take grains (96.4%) and vegetables (93.9%) every day45. Meanwhile, less than 10% of the Dutch take the recommended amount of food originate from plant46. Moreover, on average, each Vietnamese person consumes 134 g meat per day47, much lower than the Dutch’s (450 g meat and dairy products, daily)46. However, the microbiota of the Ha Nam cohort participants showed a statistically significantly lower microbial diversity at baseline with Shannon median 3.69 (IQR 3.31–4.11) when compared to the Dutch individuals with median 3.95 (IQR 3.72–4.13), (p = 0.028). This supports the hypothesis of a reduction in microbial diversity as a consequence of frequent antibiotic exposure (Fig. 4).

Microbial richness (a) and diversity (b) in Vietnamese (n = 57) and Dutch healthy adults (n = 106) who did not use antibiotics for at least one year prior to sampling. P-values are based upon linear regression analysis with adjustment for age and sex.
We next examined whether the microbial resistance to antibiotic-induced pulse perturbations among participants in the Ha Nam cohort was associated with the similarity of their microbiota to that of Dutch individuals. Ordination analyses showed that participants with a more resistant microbiota did neither cluster apart from individuals with a less resistant microbiota nor was the microbiota of these participants more (dis)similar to that of Dutch individuals (Figs. 5, S3 and S4).

Comparison of within-subject microbiota dissimilarity (Aitchison distance) between samples at the adjacent time point before and after exposure to antibiotics (exposed) versus within-subject microbiota dissimilarity between adjacent samples without intermittent antibiotic exposure.
Analysis of the differentially abundant taxa at genus level showed that many bacterial genera, including Prevotella, Blautia, and Holdemanella, of which specific species/strains have previously been identified as Volatile and/or Associated Negatively with Industrialized Societies of Humans (VANISH)48, were depleted in Dutch as compared to Vietnamese individuals. Next to these vanishing microbes, also members of the Enterobacteriaceae family including, Klebsiella and Escherichia-Shigella were enriched in the Vietnamese individuals. On the other hand, Akkermansia and Alistipes, genera of which members have been classified as BloSSUM (Bloom or Selected in Societies with Urbanization/Modernization) microbial taxa, were enriched in the Dutch individuals (Figure S5).
Impact of antibiotic use on microbial community structure and composition
To examine the impact of antibiotic use on the microbial community structure, we calculated the dissimilarity between samples using the Aitchison distance metric. As expected, Aitchison distances between samples from the same individuals were significantly lower than the distance between samples from different individuals (p < 0.001, Wilcoxon test) (Supplementary Figure S2). However, Principal Component Analyses (PCA) did neither reveal separation of samples into distinct microbiota-based clusters by time points nor by antibiotics use (Supplementary Fig. S6).
We examined the impact of antibiotic use on the stability of the microbial community structure among subjects who used antibiotics at any given time during the study period. To this end, we compared the within-subject Aitchison distance of samples collected at the adjacent time points before and immediately after exposure to antibiotics to the within-subject Aitchison distance of the same individuals but between adjacent samples without intermediate exposure to antibiotics. These analyses revealed that the stability in microbial community structure was not affected by antibiotic use (Fig. 6). Visualization of the longitudinal changes in the microbial community structure of a random selection (for visual purposes) of participants that did or did not use antibiotics also corroborated the findings that the stability in the microbial community structure was not affected by antibiotic use (Fig. S7).

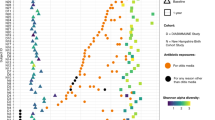
Principal Component Analyses (PCA) comparing the resistance to change of the microbiota of Vietnamese subjects who used antibiotics (both adults and infants, n = 39) to the microbiota of Dutch individuals (n = 106) without antibiotic exposure. Samples of Vietnamese subjects were collected in the month preceding the antibiotic use and are coloured according to the resistance to antibiotics defined as the Aitchison distance between the depicted pre-antibiotic sample and the sample collected after antibiotic use (not depicted in this plot), with a large dissimilarity (within-subject Aitchison distance above the median) depicted as green triangles and low dissimilarity as orange triangles.
We next examined whether exposure to antibiotics was associated with changes in the relative abundance of bacterial genera by generalized linear mixed effects models using a negative binomial distribution. We used exposure to antibiotics, age and time-point as fixed effects, and individual subjects as random effect to control for repeated measures. None of the genera were statistically significantly altered upon antibiotic use upon correction for multiple comparisons (FDR > 0.05) (see Supplementary—Table S1). This result suggested that the abundance of taxa was not disturbed, including those of bacteria susceptible to cephalosporins, even immediately after exposure to antibiotics.
Lastly, the linear mixed effects models analysis showed that the number of ARGs in faecal sample collected before antibiotic use was not associated with the subsequent shifts in observed richness and Shannon diversity upon antibiotic exposure (all p-value > 0.05, Supplementary, Tables S2 and Table S3). Also, ordination analysis did not show any correlation between the overall microbial community structure and the number of ARGs in the faecal samples (Fig. S8).
Discussion
This study examined the effect of commonly used antibiotics on gut microbiota among residents of a rural community with frequent antibiotics consumption and high levels of antibiotic resistance24,30,49. We observed modest reduction of gut microbial diversity within less than 31 days of antibiotic consumption, contrasting with a more pronounced reduction in cohorts treated by cephalosporins50 or other antibiotics12,51. Detailed analysis of microbial diversity showed that exposure to common antibiotics only resulted in the reduction of microbial richness but did not affect the biodiversity of microbiota in general, in contrast to observations in other cohorts17,44. These results highlight less diversity in microbiota at baseline as compared to Dutch individuals, from a setting with less antibiotic use.
Previously a strong link between the baseline microbiota and alterations in the resistome upon antibiotic treatment52. On the other hand, it is conceivable that the baseline resistome would also impact the shifts in microbiota composition upon antibiotic exposure since antibiotics do not eliminate antibiotic resistant bacteria in the microbiota6. In our previous study, we showed the extreme abundance of ARGs conferring resistance to beta lactam antibiotics in this community. Likely due to this high resistance background, we did not observe an alteration of Klebsiella.spp and other Enterobacterales after a course of antibiotics, in contrast with a previous report conducted in a healthy Caucasian population using one course of broad-spectrum antibiotics12.
Perhaps, repeated exposure to cephalosporin antibiotics over a short period has resulted in the microbiota reaching a state of tolerance to the antibiotic treatments. Consequently, the further antibiotic perturbations, particularly on the microbiota of our study cohort with a high level of resistance genes, would be transient, and rapidly return to their prior state.
However in contrast to this hypothesis, the microbiota of individuals with no or low number of ARGs against beta-lactam antibiotics and colistin was not more strongly perturbated by antibiotic exposure than the microbiota of individuals containing a high number of ARGs. It should however be noted that there are many more beta-lactamases, including penicillinases and cephalosporinases than the extended-spectrum beta-lactamases and carbapenemases quantified in our study. It is therefore possible that the classification of low and high ARGs does not adequately reflect the true diversity of beta-lactamases in the respective fecal samples. Future studies using shotgun metagenomics should be conducted to disentangle this bi-directional impact of the microbiota and its resistome under antibiotic exposure in more detail.
Low vaccination coverage, coupled with poor water, sanitation, and hygiene (WASH) infrastructure, residents vulnerable to infection and dependent on antibiotics for treatment17, have been recognized as major factors driving antibiotic consumption. Indeed, a research conducted in Vietnam and other LMICs, has shown proportion of households reporting use of antibiotics in the previous month in the household survey was 45% (416/925)49. Possibly, individuals in the control group have used antibiotics without being aware or recording, leading to an alternative microbiota composition with a similar richness and diversity to their prior composition. Consequently, it is difficult to observe the difference in richness and biodiversity between them and antibiotics users. In addition, people of rural communities of LMICs like Vietnam, normally have a diet with higher content than western diets, while studies having shown fiber rich diets were associated with higher diversity of microbiota53,54. When comparing the microbiota of participants of the Ha Nam cohort to that the microbiota of Dutch individuals, we observed differences in the abundance of specific microbial genera in line with previous studies comparing traditional and industrialized populations48. However, in contrast, our results showed a significantly lower diversity in the microbiota of the participants of the Ha Nam cohort as compared to those from a Dutch cohort with less antibiotic use, supporting our hypothesis that Ha Nam cohort participants have chronically disrupted microbiota, with a lower diversity, due to frequent antibiotic exposure.
The study has several limitations. The objective, to evaluate the effect of antibiotic use on microbiota in a community, may require a longer follow-up study, and the sample size was constrained by monthly sample collection. Currently, the definition of a healthy microbiome has not been established. Therefore, we could not conclude with certainty that the gut microbiota of this cohort is in an unhealthy steady state. Moreover, recorded antibiotic use in our community was predominantly with earlier generation cephalosporins (access group), which are less potent compared to later generation (watch group) or other restricted antibiotic (reverse group) classes55. This contrasts with another recent study, where the use of both access- (59.0%) and watch (39.3%) group antibiotics was commonly observed in rural Vietnam56.This suggest that it needs an investigation the response of the human gut microbiota to the new patterns of antibiotic use. Nonetheless, the results presented here support the hypothesis that frequent or chronic antibiotic exposure may push the microbiota to a different steady state that is less diverse but more resilient to disruption by antibiotic use. Further research including comparisons between populations with different exposures is needed to provide more evidence for this hypothesis. Furthermore, the potential health consequences of this altered microbiota composition should be considered.
Conclusions
This study has added a line to the long-term impacts of antibiotic use that, with frequent exposure to antibiotic, the gut microbiota adapts to a state that is little influenced by antibiotics of longitudinal study targeting long term influence of antibiotics on microbiota in similar settings.
Data availability
The data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB53339 (https://www.ebi.ac.uk/ena/browser/view/ PRJEB53339). Accession Numbers have been listed in Supplementary (Table S5: Accession Number).
Abbreviations
- LMICs:
-
Lower and middle income countries
- PCA:
-
Principal component analyses
- ARGs:
-
Antibiotic resistance genes
- AMR:
-
Antimicrobial resistance
- DNA:
-
Acid deoxyribonucleic
References
Adelman, M. W. et al. The gut microbiome’s role in the development, maintenance, and outcomes of sepsis. Critical care 24, 278–278 (2020).
Cryan, J. F., O’Riordan, K. J., Sandhu, K., Peterson, V. & Dinan, T. G. The gut microbiome in neurological disorders. Lancet Neurol. 19, 179–194 (2020).
Penders, J. et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521 (2006).
Faith, J. J. et al. The long-term stability of the human gut microbiota. Science 341, 1237439–1237439 (2013).
Zimmermann, P. & Curtis, N. The effect of antibiotics on the composition of the intestinal microbiota—a systematic review. J. Infect. 79, 471–489 (2019).
Buffie, C. G. & Pamer, E. G. Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol. 13, 790–801 (2013).
Ng, K. M. et al. Recovery of the gut microbiota after antibiotics depends on host diet, community context, and environmental reservoirs. Cell Host Microbe 26, 650-665.e4 (2019).
Dethlefsen, L. & Relman, D. A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl 1), 4554–4561 (2011).
Fassarella, M. et al. Gut microbiome stability and resilience: Elucidating the response to perturbations in order to modulate gut health. Gut 70, 595–605 (2021).
Francino, M. P. Antibiotics and the human gut microbiome: Dysbioses and accumulation of resistances. Front. Microbiol. 6, 1543–1543 (2016).
Liu, L. et al. Amoxicillin increased functional pathway genes and beta-lactam resistance genes by pathogens bloomed in intestinal microbiota using a simulator of the human intestinal microbial ecosystem. Front. Microbiol. 11, 1213–1213 (2020).
Palleja, A. et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 3, 1255–1265 (2018).
Knecht, H. et al. Effects of β-lactam antibiotics and fluoroquinolones on human gut microbiota in relation to Clostridium difficile associated diarrhea. Plos One 9, e89417–e89417 (2014).
Bhalodi, A. A., van Engelen, T. S. R., Virk, H. S. & Wiersinga, W. J. Impact of antimicrobial therapy on the gut microbiome. J. Antimicrob. Chemother. 74, i6–i15 (2019).
de Lalla, F. et al. Third generation cephalosporins as a risk factor for Clostridium difficile-associated disease: A four-year survey in a general hospital. J. Antimicrob. Chemother 23, 623–631 (1989).
Lee, H. Y. et al. Risk factors and outcomes of Clostridium difficile infection in hospitalized patients. Biomed. J. 42, 99–106 (2019).
Reijnders, D. et al. Effects of gut microbiota manipulation by antibiotics on host metabolism in obese humans: A randomized double-blind placebo-controlled trial. Cell Metab. 24, 63–74 (2016).
Kabbani, T. A. et al. Prospective randomized controlled study on the effects of Saccharomyces boulardii CNCM I-745 and amoxicillin-clavulanate or the combination on the gut microbiota of healthy volunteers. Gut Microbes 8, 17–32 (2017).
Rashid, M. U. et al. Ecological effect of ceftazidime/avibactam on the normal human intestinal microbiota. Int. J. Antimicrob. Agents 46, 60–65 (2015).
Pletz, M. W. R. et al. Ertapenem pharmacokinetics and impact on intestinal microflora, in comparison to those of ceftriaxone, after multiple dosing in male and female volunteers. Antimicrob. Agents Chemother. 48, 3765–3772 (2004).
Minh, N. N. Q. et al. Antibiotic use and prescription and its effects on enterobacteriaceae in the gut in children with mild respiratory infections in Ho Chi Minh city Vietnam. a prospective observational outpatient study. Plos One 15, e0241760 (2020).
Edlund, C., Stark, C. & Nord, C. E. The relationship between an increase in beta-lactamase activity after oral administration of three new cephalosporins and protection against intestinal ecological disturbances. J. Antimicrob. Chemother. 34, 127–138 (1994).
Lu, S., Huang, Q., Wei, B. & Chen, Y. Effects of β-lactam antibiotics on gut microbiota colonization and metabolites in late preterm infants. Curr. Microbiol. 77, 3888–3896 (2020).
Sriram A, Kapoor G, Craig J, Balasubramanian R, Brar S, Criscuolo N, Hamilton A, Klein E, Tseng K, Boeckel TPV, Laxminarayan R. The State of the World's Antibiotics 2021 - A Global Analysis of Antimicrobial Resistance and Its Drivers. The Center for Disease Dynamics, Economics & Policy, INC. (2021).
De Filippo, C. et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U. S. A. 107, 14691–14696 (2010).
Thang, N. M. & Popkin, B. M. Patterns of food consumption in Vietnam: Effects on socioeconomic groups during an era of economic growth. Eur. J. Clin. Nutr. 58, 145–153 (2004).
le Dien, N., Thang, N. M. & Bentley, M. E. Food consumption patterns in the economic transition in Vietnam. Asia Pac. J. Clin. Nutr. 13, 40–47 (2004).
Naja, F. et al. A Western dietary pattern is associated with overweight and obesity in a national sample of Lebanese adolescents (13–19 years): A cross-sectional study. Br. J. Nutr. 114, 1909–1919 (2015).
Smits, S. A. et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 357, 802–806 (2017).
Bich, V. T. N. et al. An exploration of the gut and environmental resistome in a community in northern Vietnam in relation to antibiotic use. Antimicrob. Resist. Infect. Control 8, 194 (2019).
Arcilla, M. S. et al. The carriage of multiresistant bacteria after travel (COMBAT) prospective cohort study: Methodology and design. BMC Public Health 14, 410 (2014).
Costea, P. I. et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 35, 1069–1076 (2017).
Galazzo, G. et al. How to count our microbes? the effect of different quantitative microbiome profiling approaches. Front. Cell Infect. Microbiol. 10, 403 (2020).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624 (2012).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. Plos One 5, e9490 (2010).
Pruesse, E. et al. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196 (2007).
Wickham, H. et al. Welcome to the Tidyverse. J. Open Source Softw. 4, 1686 (2019).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Patil, I. Visualizations with statistical details: The “ggstatsplot” approach. J. Open Source Softw. 6, 3167 (2021).
Barnett, et al. microViz: An R package for microbiome data visualization and statistics. J. Open Source Softw. 6, 3201 (2021).
Lin, H. & Peddada, S. D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 11, 3514 (2020).
Gamage, H. et al. Third generation cephalosporins and piperacillin/tazobactam have distinct impacts on the microbiota of critically ill patients. Sci. Rep. 11, 7252 (2021).
Panda, S. et al. Short-term effect of antibiotics on human gut microbiota. Plos One 9, e95476 (2014).
Kim, S. H. et al. Dietary factors related to body weight in adult Vietnamese in the rural area of Haiphong, Vietnam: The Korean genome and epidemiology study (KoGES). Nurs. Res. Pract. 4, 235–242 (2010).
National Institute for Public Health and the Environment Ministry of Health Was. The diet of the Dutch—results of the Dutch national food consumption survey 2012–2016. (2020).
Liên, N. Người Việt tiêu thụ thịt gần gấp đôi khuyến nghị dinh dưỡng, p In Vietnamnet. https://vietnamnet.vn/vn/suc-khoe/nguoi-viet-tieu-thu-thit-nhieu-gap-doi-khuyen-nghi-723612.html (2021).
Merrill, B. D. et al. Ultra deep sequencing of Hadza HunterGatherers recovers vanishing gut microbes. BioRxiv 78(458), 789 (2022).
Do, N. T. T. et al. Community-based antibiotic access and use in six low-income and middle-income countries: A mixed-method approach. Lancet Glob. Health 9, e610–e619 (2021).
Venturini, C., Bowring, B., Fajardo-Lubian, A., Devine, C. & Iredell, J. Effects of antibiotic treatment with piperacillin/Tazobactam versus Ceftriaxone on the composition of the murine gut microbiota. Antimicrob. Agents Chemother. 65, e01504–e01520 (2021).
Patterson, R. E. & Sears, D. D. Metabolic effects of intermittent fasting. Annu. Rev. Nutr. 37, 371–393 (2017).
Willmann, M. et al. Distinct impact of antibiotics on the gut microbiome and resistome: A longitudinal multicenter cohort study. BMC Biol. 17, 76 (2019).
De Filippo, C. et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696 (2010).
Zhernakova, A. et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science (New York, NY) 352, 565–569 (2016).
WHO. WHO model list of essential medicines (20th list). https://www.who.int/medicines/news/2017/20th_essential_med-list/en/ (2017).
Nguyen, N. V. et al. Community-level consumption of antibiotics according to the AWaRe (Access, Watch, Reserve) classification in rural Vietnam. JAC-Antimicrob. Resist 2, dlaa048 (2020).
Acknowledgements
The authors thank the Ha Nam community health workers who conducted the interviews and sample collection, Ha Nam CDC, and the Ministry of Health of Vietnam for their continuing support of the research collaboration between the Oxford University Clinical Research Unit and the National Institute for Hygiene and Epidemiology.
Funding
This work was supported by Wellcome (106680), a Radboudumc Revolving Research Fund (R3Fund) grant and the Fleming Fund pilot grant Vietnam (Department of Health and Social Care [UK]).
Author information
Authors and Affiliations
Contributions
V.T.N.B., T.H.H., R.v.D., H.W. and J.P. contributed to the study and experimental design. T.D.T., N.T.H.A. contributed to the sample collection. V.T.N.B., N.L.G., and J.P. contributed to the 16S rRNA sequencing. D.B., J.C., N.v.B. contributed to microbiota data analysis and statistics. V.T.N.B., D.B., N.v.B. and J.P. contributed to present the data, tables and figures. VTNB wrote the draft of the manuscript. J.P., R.v.D. and H.W. contributed to edit the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bich, V.T.N., Le, N.G., Barnett, D. et al. Moderate and transient impact of antibiotic use on the gut microbiota in a rural Vietnamese cohort. Sci Rep 12, 20189 (2022). https://doi.org/10.1038/s41598-022-24488-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-24488-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
