
Abstract
Atypical antipsychotics, despite their rapid dissociation from dopamine receptors and reduced tendency to induce oxidative stress, have been associated with difficult-to-manage movement disorders, including tardive dyskinesia (TD). The study set out to investigate the effects of cannabidiol (CBD), a potent antioxidant, on risperidone-induced behavioural and motor disturbances; namely vacuous chewing movements (VCM), and oxidative stress markers (e.g. superoxide dismutase (SOD), catalase (CAT), reduced glutathione (GSH), malondialdehyde (MDA), Nitric oxide (NO), and DPPH (2,2-diphenyl-1-picrylhydrazyl)). Oral risperidone (10 mg/kg) or oral CBD (5 mg/kg) were administered to six experimental groups. While risperidone alone was administered for 28 days, CBD concomitantly or in sequential order with risperidone, was administered for 28 days; and CBD alone was administered for 21 days. Behavioural, motor, and specific biochemical parameters, which included VCM, muscle tone, fasting blood sugar (FBS), and oxidative stress markers were assessed at different time points after the last dose of medication. Oral CBD (5 mg/kg) significantly reduced risperidone-induced elevated FBS when given after the administration of risperidone. Oral CBD also had effects on VCM when administered before risperidone and similarly, attenuated risperidone-induced increased muscle tone. It was also established that concomitant or sequential administration of CBD and risperidone did not have any adverse effects on cognition or locomotion. Both CBD and risperidone increased the activity of antioxidant enzymes and decreased the activity of pro-oxidant enzymes. This study suggests CBD could mitigate metabolic dysregulation and extrapyramidal side effects associated with risperidone without producing cognitive impairments.
Similar content being viewed by others


Introduction
The reintroduction of clozapine to the market in 1989 after the first attempt in 19701,2 heralded a new generation of antipsychotics referred to as atypical antipsychotics (AAPs) or second-generation antipsychotics, which include clozapine, olanzapine, risperidone, quetiapine, lurasidone, and ziprasidone among others. These antipsychotics are called atypical because of their serotonin 5-HT2A receptor antagonism in combination with D2 receptor antagonism properties3,4. Initially, they were believed to carry a lower burden of extrapyramidal side effects (EPS) such as parkinsonism, acute dystonia, akathisia, and akinesia compared to the typical or first-generation antipsychotics; but more recent studies have disagreed with this assertion. Parkinsonism, for example, has been observed to increase in frequency with high doses of atypical antipsychotics5,6,7. In addition to EPS, antipsychotics have significant metabolic side effects like weight gain, diabetes mellitus, and hyperlipidaemia which can increase the risk of cardiovascular mortality8,9,10.
Apart from the acute motor disorders, other movement disorders associated with atypical antipsychotics include neuroleptic malignant syndrome, tardive dystonia, and tardive dyskinesia (TD)11,12. Tardive dyskinesia is a late-onset movement disorder that is potentially irreversible. Though the pathophysiology is not yet clearly elucidated, dopamine receptor upregulation and supersensitivity, dysfunction of other neurotransmitters such as glutamate, γ-amino butyric acid (GABA), serotonin, neurotoxicity, oxidative stress, changes in synaptic plasticity, defective neuroadaptive signalling, and defective antipsychotic metabolising enzymes have been implicated in the aetiology13,14,15. Risk factors for TD include duration of antipsychotic use, age, genetics, and ethnicity16,17,18,19.
It was initially thought that the incidence of TD would decline with increasing use of AAPs, because of the rapid dissociation of most AAP molecules from receptor sites, reduced tendency to induce oxidative stress, and the ability of most AAPs to bind 5HT2 receptors thereby reducing the tonic effects of dopamine in the basal ganglia20,21,22. Despite a 30% reduction in the prevalence of TD with AAPs compared with typical antipsychotics23,24, the rate of 20% of TD with AAP is still high.
While AAP-induced movement disorders have been observed, there appears to be little or no association with oxidative stress indicators25,26,27. Some authors have also suggested that AAPs, following chronic administration, can reduce pro-oxidants and increase GSH28. Another study observed an increase in antioxidant defence enzymes and lipid peroxidation in rat brains after chronic haloperidol administration but there was no change after chronic risperidone, clozapine, and olanzapine administration25. The mechanisms by which AAPs induce movement disorders, such as TD, remain a puzzle considering that these disorders have been associated with an imbalance between pro-oxidants and antioxidants in various studies17,29,30,31. It is therefore important to investigate the association between VCM, AAPs, antioxidants, and pro-oxidants in an animal model of TD using risperidone as the prototype APP.
Though the mechanism by which AAPs influenced oxidative stress is poorly understood, AAPs have been observed to alleviate microglia-induced oxidative stress by reducing free radical production in the microglia32. Risperidone has been postulated to reduce reactive oxygen species (ROS) production through the regulation of antioxidant enzymes in the brain33. In contrast, risperidone has been shown to induce oxidative stress in the liver through increased reactive oxygen species (ROS) production, thus leading to mitochondrial collapse, lysosomal membrane disruption, GSH depletion and lipid peroxidation34. Pancreatic damage, including pancreatitis, is also associated with risperidone35,36,37, and oxidative stress may play a role here as pancreatitis is associated with oxidative stress38,39,40. The damage to pancreatic β cells following AAP-induced glucose intolerance may be related to increased free radical production41,42. In addition, amelioration of oxidative stress through the administration of a potent antioxidant may also reduce the risk of metabolic syndrome and improve medication adherence and overall quality of life. Identifying an ideal potent antioxidant that can alleviate the side effects of the AAP is an avenue of research that is worth pursuing.
There is much interest in Cannabidiol (CBD), a non-psychoactive and non-reinforcing cannabis sativa constituent43,44, which several studies have confirmed to have anti-oxidative, anti-inflammatory, and neuroprotective properties45,46. It has also shown promise in the prevention and treatment of movement disorders, including tardive dyskinesia, in animal models47,48. Furthermore, it is devoid of any major adverse reactions in both animal and human studies49,50,51, thus making CBD the ideal antioxidant for this study. CBD may, however, be similar to AAPs in its pharmacological profile because in animal models it has antipsychotic-like effects without inducing extrapyramidal-like effects52.
Meanwhile, clinical studies on the glycaemic effects of CBD have yielded conflicting results as one study observed an improvement in insulin secretion while another did not observe an improvement in glycaemic control53. As a best practice, it is prudent and essential to study the effects of CBD in combination with AAPs using risperidone as a prototype to affirm if synergistic actions of the two compounds (CBD and risperidone) can mitigate adverse effects, especially cognitive and metabolic side effects. Our aim was to determine if CBD can ameliorate risperidone induced VCM and reduce the metabolic side effects of risperidone. Our secondary objective was to explore the association between risperidone-induced VCM and oxidative stress indices.
Materials and methods
Animals
Male Wistar adult rats used in this study were obtained from the colony of the Nigerian Institute of Medical Research (NIMR), Yaba, Lagos, Nigeria. The animals were kept in clean polypropylene cages in well-ventilated, and hygienic compartments maintained under standard environmental conditions. The rats were also fed with standard rodent pellets (produced by Ladokun Feed Plc, Ibadan, Nigeria), and with water ad libitum. The animals were acclimatized for a period of 2 weeks before experimental procedures were undertaken in accordance with the United States National Institutes of Health Guidelines for Care and Use of Laboratory Animals in Biomedical Research54. This study was also approved by the Institutional Review Board (IRB) of the Nigerian Institute of Medical Research, Yaba, Lagos, Nigeria (IRB/16/329) and Stellenbosch University’s Health Research Ethics Committee: Animal Care and Use (SU-ACUD16-00137).
Drugs
Cannabidiol [(–)-Cannabidiol, GMP (Cannabidiolum)] (CBD) (VAKOS X, a.s., Permova 28a, 186 00 Praha 8, in Czech Republic. Company number: 04801938). The drug was supplied in fine granule form with the amount administered weekly calculated and dissolved in 70% ethanol as recommended by the manufacturers. It was also diluted with distilled water. CBD was administered orally. Risperidone tablets (manufactured by Janseen Pharmaceuticals Beerse, Belgium) were dissolved in distilled water and administered orally.
Experimental design
The experimental groups (n = 51) consisted of six groups: A (n = 10), Group B (n = 9), Group C (n = 8), Group D (n = 8), Group E (n = 8) and Group F (n = 8).
Pharmacological administration was as follows: Group A (Control (Distilled water) 2 ml p.o.), Group B (Oral CBD 5 mg/kg/oral), Group C (Risperidone 10 mg/kg/oral), Group D (CBD 5 mg/kg/oral + Risperidone 10 mg/kg/oral), Group E (Risperidone 10 mg/kg/oral then CBD 5 mg/kg/oral), Group F (CBD 5 mg/kg/oral then Risperidone 10 mg/kg/oral). Some researchers used 10 mg/kg of risperidone to induce VCM55. According to some investigators, effective doses of CBD in rats range between 2.5 and 10 mg/kg56.
In Group D, CBD was administered once daily for 28 days, while risperidone was administered concomitantly but in divided doses for 28 days. In Group C, Risperidone 10 mg/kg/oral only was administered daily in divided doses for 28 days, while administration for Groups A (Control (Distilled water only) 2 ml p.o.) and B (Oral CBD 5 mg/kg/oral only) was once daily for 21 days. In Group E, Risperidone 10 mg/kg/oral was administered for 28 days before the administration of CBD 5 mg/kg/oral for 28 days and in Group F, CBD (5 mg/kg/oral) was administered for 28 days before the administration of risperidone (10 mg/kg/oral) for 28 days. VCM was assessed at 8 h, 24 h, and on Day 8 after the last dose of the medication. Day 8 VCM was used in the analysis because persistent VCM after withdrawal of medications is the appropriate model of TD, though VCM observed after Day 8 have been shown to persist for a long time55.
The rats were fasted overnight after the last meal at 7 pm until 7 am when blood was taken for FBS measurement. A fine sterile disposable hypodermic needle was used to puncture the rats at the end of their tails, and enough blood was dropped on a portable glucometer device, Accu-Chek Performa® (Roche Diagnostics, Mannheim, Germany), to obtain a FBS reading.
For all groups, weight and FBS were measured the day before administration of medication and a day after administration of the last dose of medication.
Notably, risperidone-induced FBS changes in rats have been shown to be stable over time57.
Behavioural assays
Elevated plus maze, open field, rota-rod, and object recognition tests were used to assess behavioural responses. For all groups, behavioural assessments were carried out 24 h after the last dose of medication or distilled water (control group).
VCM were assessed by placing each animal in individual transparent glass plexiform cages. Each animal was allowed to acclimatize for 5 min before counting started. The number of VCM (mouth openings in the vertical plane not directed toward physical material) was counted for 10 min58. Day 8 VCM was used in the analysis because persistent VCM after withdrawal of medications is the appropriate model of TD; though VCM observed after Day 8 have been shown to persist for a long time55. Methodologies used in behavioural assays were described in detail in our previously published study48.
Elevated plus-maze test
Anxiety was monitored using this test59. The elevated plus-maze consisted of two open arms (30 × 5 cm), and two closed arms (30 × 5 × 15 cm) that extended from the central platform (5 × 5 cm). The entire maze was elevated to 40 cm above the floor. During the first 5 min of free exploration, the number of entries into and the time spent in the open and closed arms were recorded. An entry was defined as the point at which the animal placed all four paws onto the arm.
Open-field test
This test was used to assess locomotion in the study60,61. The number of line crossings is used as a measure of locomotor activity. The number of line crosses and the frequency of rearing are usually used as a measure of locomotor activity, exploration, and anxiety. A high frequency of these behaviours indicates increased locomotion and exploration and/or a lower level of anxiety60. The open-field box is a rectangular area composed of a hard floor measuring 36 cm × 36 cm × 26 cm and made of a white painted wood. The floor was divided by permanent red markings into 16 equal squares at the bottom. Each rat was introduced singly into one corner of the field and after each session, the area was cleaned with 70% alcohol to eliminate olfactory bias, and the area was thereafter allowed to dry before introducing a new animal. The locomotor activity was measured for 10 min. A high frequency of these behaviours indicates increased locomotion60.
Object recognition test
This test was used to study drug-induced cognitive effects62. Each animal was placed in a plexiform glass transparent cage and was allowed to acclimatise for 5 min before two similar spherical objects (Balls) were introduced. The animal was allowed to spend 10 more minutes in the cage with the objects and the time the animal spent exploring the objects was measured with a stopwatch by an observer before the animal was removed. The animal was re-introduced into the cage after an hour, and a different object (square shaped) was added into the cage before the animal was re-introduced, while the previous objects (Balls) were removed. Time spent exploring both new and old objects was measured for 10 min by two different observers using stopwatches. The time spent by the animal using its nose to touch the object was measured while the time spent using other parts of the body were ignored.
Rota-rod test
The method was used for the assessment of motor coordination, balance, and grip strength in rodents63. A rota-rod treadmill device (Ugo Basile No. 7600 Varese, Italy) was used for this purpose. The rats were trained to remain on slowly moving (16 revolutions min−1) rods of 5 cm in diameter for 150 s by walking. The animals were then placed on the treadmill after training and the time spent on the treadmill before falling was measured for each animal.
Antioxidant indices assays
Animals were sacrificed on Day 8 after the administration of the last dose of pharmacological agent by first anesthetizing them with phenobarbitone before cervical dislocation. Methodologies used in determining antioxidant indices were also described in detail in our previously-published study48. The following antioxidant indices were determined spectrometrically.
Malondialdehyde (MDA)
MDA is an index of lipid peroxidation, and it was determined using the method of Buege64.
Reduced glutathione (GSH)
The GSH content of the brain as non-protein sulfhydryl was estimated according to the method described by Sedlak and Lindsay65.
Catalase (CAT)
CAT activity was determined according to the method of Sinha66.
Superoxide dismutase (SOD)
SOD activity was determined as described by Sun and Zigma67.
Nitric oxide (NO) scavenging activity
This was measured by the Griess reagent assay.
DPPH scavenging assay
This was determined using the methods described by Kedare and Singh68.
Statistical analysis
Descriptive statistics of relevant study variables (weight, FBS, VCM, and behavioural and biochemical assays) are presented using measures of centrality and dispersion (mean and standard deviation). The normality of distribution of the data was evaluated using histograms and supported by the results of the Kolmogorov–Smirnov test. Comparison of means was done using one way-ANOVA. When group differences were statistically significant (p-value < 0.05), pairwise comparisons or post-hoc tests were done. Omnibus tests were also used with post-hoc comparisons. Bonferroni corrections were used to adjust for multiple comparisons when necessary.
Statement on reporting experimental procedures
Reporting in this study follows the recommendations in the ARRIVE guidelines.
Ethical statement
The study was approved by the Institutional Review Board (IRB) of NIMR, Yaba, Lagos, Nigeria (IRB/16/329) and Stellenbosch University’s Health Research Ethics Committee: Animal Care and Use (SU-ACUD16-00137).
Results
Effects of interventions on fasting blood sugar (FBS) and weight gain
Before the administration of medications, there was a statistically significant difference in FBS between groups (F = 14.337, p < 0.001, df = 5). On post-hoc analysis, there was a significant between-group difference in FBS pre-medication in Groups D and E (p < 0.001), Groups C and E (p < 0.001), Groups E and F (p < 0.001), Groups E and A (p < 0.001), Groups E and B (p = 0.001), and Groups F and B (p = 0.016).
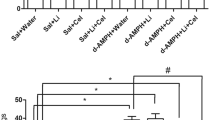
Similarly, after the last dose of medication administration, there was a statistically significant difference in the mean FBS between groups (F = 34.079, p < 0.001, df = 5). On post-hoc analysis, there was a significant difference in FBS between Groups D and E (p = 0.009), D and A (p < 0.001), D and B (p < 0.001), C and E (p = 0.004), C and A (p < 0.001), C and B (p = 0.006), E and F (p < 0.001), E and A (p < 0.001), F and A (p < 0.001), F and B (p = 0.006) (Fig. 1).

Fasting blood sugar across groups after the last dose of medications. Observations of the post-pharmacological interventions FBS values in each group indicated CBD influenced the FBS values. *Statistically significant difference.
There was no statistical difference in mean weight before (F = 0.469, p = 0.797, df = 5) and after (F = 2.837, p = 0.062, df = 5) the last dose of administration of pharmacological agents. After the last dose of medications, subjects in Group D had the highest mean weight gain (45.5 ± 40.0), while those in Group F had the lowest mean weight gain of the six groups (3.2 ± 38.4). However, there was no statistically significant difference in the weight between the groups (F = 1.831, p = 0.127, df = 5) (Table 1).
Behavioural assays after the last dose of pharmacological interventions
VCM
After the last dose of pharmacological intervention, there was a statistically significant difference in VCM between the groups (F = 9.131, p < 0.001, df = 5). On post-hoc analysis, there were a significant difference between Groups D and B (p = 0.043), C and F (p = 0.003), C and A (0.000), C and B (p = 0.047), and E and B (p = 0.038) after the last dose of the medication. See Fig. 2.

Vacuous chewing movements (VCM) across groups after the last dose of medications. CBD had effects on risperidone induced VCM. *Statistically significant difference.
Calculated open arm
There was no significant difference in the mean difference of the duration spent in the open arm of the elevated plus maze across the groups (F = 0.334, p = 0.890, df = 5).
Discrimination index
There was no significant difference in the mean difference of the discrimination index across the groups (F = 1.611, p = 0.178, df = 5).
Line crossing
There was no significant difference in the mean difference of locomotor activity across the groups (F = 2.262, p = 0.092, df = 5).
Rota rod
There was a statistically significant difference between the Rota Rod values between groups (F = 2.738, p = 0.031, df = 5). This difference was noted between Groups A and B (p = 0.019) and groups B and F (p = 0.027) (Fig. 3).

Inter-group comparison of the observed time spent on the treadmill in each group during the Rota-rod experiment. There was increase in time spent on the treadmill in Group B compared to the rest of the groups. *Statistically significant difference.
Brain oxidative stress parameters
Brain GSH
There was a statistically significant difference in brain GSH between groups (F = 6.156, p < 0.001, df = 5). On post-hoc analysis, the differences were significant between Groups C and A (p = 0.001), E and A (p = 0.020), F and A (p = 0.000), and A and B (p = 0.003).
Brain SOD
There was a statistically significant difference in brain SOD between groups (F = 11.534, p < 0.001, df = 5). On post-hoc analysis, there were significant differences between Groups D and F (p = 0.006), D and A (p = 0.016), D and B (p = 0.012), C and F (p = 0.005), C and A (p = 0.012), C and B (p = 0.011), and F and A (p = 0.010).
Brain CAT
Significant differences were observed in the brain CAT values between groups (F = 13.870, p < 0.001, df = 5). On post-hoc analysis, these differences were observed between D and F (p = 0.002), D and A (p = 0.004), D and B (p = 0.002), C and F (p = 0.005), C and A (p = 0.008), C and B (p = 0.004), E and F (p = 0.047), E and B (p = 0.041), and A and B (p = 0.001).
Brain DPPH
Similarly, a significant difference was observed in the brain DPPH values between groups (F = 4.421, p = 0.004, df = 5). On post-hoc analysis, these differences were observed between Groups D and E (p = 0.033), D and A (p = 0.013), D and B (p = 0.000).
Brain NO
Values significantly differed between groups (F = 4.620, p = 0.006, df = 5). On post-hoc analysis, the differences were observed between Groups D and F (p = 0.041), D and B (p = 0.006), C and B (p = 0.002), A and B (p = 0.037).
Brain MDA
Brain MDA values did not significantly differ between groups (F = 0.617, p = 0.688, df = 5) (Fig. 4).

Inter-group comparison of assay of antioxidant indices assay after the last dose of medications. There were intergroup differences in the Brain’s GSH, SOD, CAT, SOD and DPPH results, but there were no intergroup differences in the brain’s MDA. Group 1: Control Group; Group 2: Cannabidiol only; Group 3: Risperidone only; Group 4: Cannabidiol + Risperidone; Group 5: Risperidone then Cannabidiol; Group 6: Cannabidiol then Risperidone. P1 = P-value for brain CAT differences between groups; P2 = P-value for brain DPPH differences between groups; P3 = P-value for brain GSH differences between groups; P4 = P-value for brain NO differences between groups; P5 = P-value for brain SOD differences between groups; P6 = P-value for brain MDA differences between groups.
Discussion
The present study is a continuation of our studies on the effectiveness of CBD in the treatment of symptoms of TD in an animal model. In this set of analyses, we focused on the synergistic effects of oral CBD and oral risperidone on weight, FBS, behavioural and oxidative stress parameters in the brain. We also assessed the effect of oral CBD on oral risperidone-induced VCM. We did not observe a significant increase in weight after the last dose of medication when risperidone alone and risperidone co-administered with CBD were compared. Concomitant administration of risperidone and CBD did not ameliorate VCM, unlike what we observed with haloperidol-induced VCM in our previous study48; however, there were distinct changes in antioxidant and pro-oxidant compounds, thus suggesting that both risperidone and CBD reduced oxidative stress in our experiments.
Moreover, our investigation did not reveal a significant change in weight when pre- and post-medication weights of all groups were compared, though association of risperidone with weight increase has been documented in animal and human studies69,70,71,72. Risperidone-induced weight gain may be associated with histamine H1 receptor antagonism, leptin resistance, and consequent disinhibition of food intake73. CBD has also been shown to decrease weight in animal studies74, but other studies contradict these observations75,76.
Comparison of FBS values across groups strongly suggests that oral CBD on its own can stabilize blood glucose. Most studies on the hypoglycaemic effects of CBD have reported similar findings, though experimental designs and doses varied77,78,79. Mechanisms of action may involve stimulation of insulin production by the beta-pancreatic cells and protective and regenerative effects on the beta cells80,81. When given concurrently with risperidone, CBD may not be effective as a hypoglycaemic agent, but CBD given after risperidone as in Group E (Risperidone before CBD) obviously reduced FBS and appeared to have similar effects as CBD alone, indicating that CBD may be effective in lowering blood sugar if given after the administration of risperidone. Results of CBD given before risperidone (Group F) were similar to those of risperidone alone, suggesting that CBD may not be effective in lowering blood sugar if given before the administration of risperidone. These findings require confirmation with a pharmacokinetic/pharmacodynamic (PK/PD) study. Meanwhile, risperidone has been shown to increase total and active ghrelin levels as well as leptin, and prolactin levels in some studies; and these are associated with increased appetite and metabolic dysfunction72, while CBD was shown to reduce appetite and insulin resistance by lowering resistin levels in some studies81.
In this study, risperidone alone (Group C) induced more VCM than all the groups, but this was only significant with CBD before risperidone, CBD alone, and the control group. We also observed that CBD administered concomitantly and after risperidone resulted in less VCM count than risperidone alone, though this was not significant. Our observations suggested that though CBD ameliorated the VCM induced by risperidone, and the effect was not quite pronounced, it may have a protective effect if administered before risperidone, as in Group F. This is contrary to what was observed with haloperidol-induced VCM in an earlier study, where CBD (5 mg/kg) administered concomitantly with haloperidol significantly reduced VCM48. We therefore propose that the aetiology and pathophysiology of risperidone-induced VCM may be different from that of haloperidol. Further investigations are needed on the aetiology and pathophysiology of risperidone-induced VCM.
In our model of motor coordination, balance, and grip strength, we observed that combinations of risperidone with CBD prevented the motor incoordination and increase in muscle tone associated with risperidone alone. The rats in the risperidone-alone group spent the least time on the treadmill, though this was only significant when compared with the group that received risperidone and CBD concomitantly (Group D), and the control (Group A). Risperidone is known to have a high affinity for D2 receptors and M4 receptors. D2 receptors are involved in EPS, while M4 receptors play a significant role in catalepsy, a condition where rodents have difficulty in changing externally-imposed posture82,83.
CBD is known to interact with 5HT1A and 5HT2A receptors subtypes in the basal ganglia to ameliorate the EPS induced by D2 receptor occupation84,85. CBD also influences phosphoinositide metabolism stimulated by activation of muscarinic receptors86. These factors may explain the effects of CBD in our experiment. To our knowledge, there are no published data on the effects of CBD on risperidone-induced movement disorders.
We could not confirm an anxiolytic effect for risperidone, either alone or in combination with CBD in our study using the elevated plus maze. This contrasts with a study which showed that risperidone at low dose induced an anxiolytic effect87. CBD at a higher dose than what we used in this study has reported anxiolytic effects47,86. We may not have observed an anxiolytic effect of different combinations of risperidone and CBD in our work because of the relatively low dose of CBD and high dose of risperidone. We expected the high dose of risperidone to affect the explorative activity and locomotion of the animals87 in the open field test because the receptor occupancy of risperidone at a high dose is above 60% and at this level, 5-HT2 blockade may not be protective against EPS with the development of parkinsonism and motor retardation88. At this dose however, we did not observe any differences when risperidone alone was compared with CBD only and the control groups, thus indicating that risperidone alone may not produce motor retardation or decrease locomotion at a high dose in rats. This suggests that 5-HT2 blockade may offer some protection.
In this study, there was no evidence of deterioration of cognitive function despite the high dose of risperidone. There was also no evidence for enhancement of cognitive function when risperidone was combined with CBD. Some clinical studies have observed an improvement in cognitive function in patients with schizophrenia on risperidone89. Results from animal studies indicate improved cognition with CBD alone47, but these results are contrasted by clinical studies that have found no clear clinical evidence of the positive benefits of CBD on cognition75. There are no published studies on the cognitive effects of combining risperidone and CBD.
Effects of interventions on oxidative stress indices in the brain
Glutathione peroxidase (GPx) is the enzyme that catalyses the reaction between reduced GSH and hydrogen peroxide (H2O2) to form GSSG (glutathione disulfide), its oxidized form and water90, and elevated GSH therefore indicated less activity of GPx. The control group (Group A) had a significantly higher GSH level in the brain compared to other groups, with either CBD or risperidone alone, or in combination at different points, thus indicating that GPx is not active in the control. Lower GSH levels were observed in CBD and risperidone groups compared with the control, suggesting that these groups stimulated glutathione peroxidase production, leading to increased use of reduced GSH, which is converted to oxidised GSH (GSSG), and therefore lower GSH levels, compared to the control group.
SOD activities increased in the risperidone alone group (Group C), risperidone plus CBD (Group D), and risperidone before CBD (Group E) when compared with CBD before risperidone (Group F), CBD alone, and the control. These observations suggest increased antioxidant activities in the risperidone alone group (Group C), risperidone plus CBD (Group D), and risperidone before CBD (Group E) compared to others. CAT activities in the brain reflected a similar pattern except that risperidone administered before CBD produced higher activity than CBD before risperidone and CBD alone.
Though the effect of atypical antipsychotics on oxidative stress enzymes is still being explored, one study suggests atypical antipsychotics increase the expression of SOD1 gene leading to an increase in SOD91. We know that SOD acts as a first line of defence against oxidative stress by converting superoxide radicals to hydrogen peroxide, which is in turn converted to water and oxygen by catalase and glutathione peroxidase. An increase in SOD activity may indicate antioxidant potential or a compensatory mechanism92,93, depending on the level of pro-oxidants in the organ.
The lower level of GSH, coupled with the observed increased activity of SOD and CAT enzymes in risperidone alone, risperidone concomitantly administered with CBD, and risperidone before CBD groups suggests that both risperidone and CBD may have antioxidant effects. Meanwhile, CBD before risperidone antioxidant status (reduced SOD, CAT, and high GSH concentrations) compared to risperidone concomitantly administered with CBD and risperidone before CBD, suggests that CBD did not have a positive effect on oxidative stress when given before risperidone. We are not able to clearly explain our observation, which may be related to the ability of CBD to inhibit cytochrome P450 group of enzymes49,94 involved in the metabolism of antipsychotics and an increased concentration of risperidone to a level where it behaves like a typical antipsychotic95.
There were no significant group differences in MDA, a known marker of lipid peroxidation concentration in the brain, when all six groups were compared in this study. Another study also did not observe a change in lipid peroxidation in the brains of rats treated with risperidone after 45 and 90 days25. A group of investigators, however, observed that risperidone decreased MDA levels in rat brains suggesting that it could have antioxidant effects28. Relatedly, a clinical study claimed an increase in MDA levels in patients with schizophrenia, which was modified by atypical antipsychotics92. Some researchers have also observed a decrease in glutathione peroxidase (GPX) activity with an increase in MDA levels, suggesting an increase oxidative stress in schizophrenia patients on risperidone96. Our study concurs with other studies showing that risperidone has antioxidant properties92. This may explain our observation that MDA activity in risperidone-alone animals was not increased when compared with CBD alone, or any of the CBD combinations with risperidone.
This study also observed reduced NO production in the risperidone-alone (Group C), and risperidone given concomitantly with CBD (Group D) groups compared with the control and CBD-alone groups. Both risperidone and CBD have been shown to reduce inducible NO synthase, and NO production by some researchers97,98,99,100,101,102. Other studies that observed increased NO production in risperidone, however, contradict these reports103,104,105,106. In this study, risperidone appears to be an effective antioxidant, but risperidone and CBD given concomitantly is as effective as risperidone alone. CBD alone (Group B) appeared to be less effective in reducing NO level, compared to risperidone alone (Group C), and risperidone given concomitantly with CBD (Group D). We are not aware of any studies that have examined the antioxidant status of CBD combined with risperidone.
Our observations of the antioxidant effects of risperidone support our proposal that the aetiology and pathophysiology of risperidone-induced VCM may be different from that of haloperidol because risperidone had antioxidant effects in our study and did not induce oxidative stress, in contrast to haloperidol55. Earlier researchers also observed that sub-chronic treatment with risperidone did not increase the population of D2 receptors in the rat striatum, in contrast to the actions of haloperidol107,108. These observations suggest that oxidative stress has a role to play in the manifestation of movement disorders induced by haloperidol but may not be important in movement disorders produced by risperidone. The DPPH radical scavenging activities in the brain of all the groups were within the same range, but the risperidone plus CBD group (Group D) had fewer scavenging activities than the other groups. There is no known published study on DPPH radical scavenging activities in animals administered risperidone and CBD, but an in-vitro study used a DPPH assay to confirm the antioxidant status of risperidone109.
In summary, we found that CBD ameliorated the increase in FBS induced by risperidone if given before or after risperidone, and improved muscle incoordination and tone induced by risperidone, but it did not significantly reduce risperidone-induced VCM. CBD may, however, have a protective effect against VCM if given before risperidone. Concomitant or sequential administration of CBD and risperidone did not have any adverse effects on cognition or locomotion. Both CBD and risperidone therefore exhibited antioxidant properties.
Limitations
First, 21 days of administration of CBD, and distilled water in the control group instead of 28 days is an important limitation of this study. All treatments should have the same duration; however, these data were drawn from a larger study that included haloperidol that was administered for 21 days. The IRB at the institute at which the experiments were conducted considered the same control to be adequate for the study involving haloperidol and risperidone.
Second, the animal model of TD used in this study may not be specific enough for TD, and may closely resemble EPS (e.g. Parkinsonian tremor, dystonia), even though only VCM that persisted after the 8th day was used in this study to reduce the probability that what was observed is EPS. Furthermore, the experimental paradigm used in this study closely resembles that of Bishnoi and Boparai110, and Marchese55.
Another limitation observed in this study is that automated behavioural apparatus would have produced more precise and less biased measurements, but financial constraints prevented us from procuring this apparatus. It was not possible to measure oxidative stress indices in specific regions of the brain due to technical difficulties.
Conclusion
Our results suggest that a combination drug formulation of risperidone and CBD in clinical practice may have less metabolic dysregulation and extrapyramidal side effects than with risperidone alone, without producing cognitive impairments as seen with the olanzapine and samidorphan111 combination. Such a combination may also improve the overall safety and tolerability of risperidone. Clinical studies will be required to prove the efficacy of risperidone and CBD combination. Pharmacokinetic and pharmacodynamic studies in both humans and animals are also necessary to further study risperidone and CBD drug-drug interactions.
References
Crilly, J. The history of clozapine and its emergence in the US market: A review and analysis. History of Psychiatry, Vol. 18 (2007).
Cloud, L. J., Zutshi, D. & Factor, S. A. Tardive dyskinesia: Therapeutic options for an increasingly common disorder. Neurotherapeutics 11(1), 166–176 (2014).
Mauri, M. C. et al. Clinical pharmacology of atypical antipsychotics: An update. EXCLI J. 13, 1163–1191 (2014).
Li, P., Snyder, G. L. & Vanover, K. E. Dopamine targeting drugs for the treatment of schizophrenia: Past, present and future. Curr. Top. Med. Chem. 646, 3385–3403 (2016).
Peluso, M. J., Lewis, S. W., Barnes, T. R. E. & Jones, P. B. Extrapyramidal motor side-effects of first and second-generation antipsychotic drugs. Br. J. Psychiatry 200(5), 101485. https://doi.org/10.1192/bjp.bp.111.101485 (2012).
Divac, N., Prostran, M., Jakovcevski, I. & Cerovac, N. Second-generation antipsychotics and extrapyramidal adverse effects. Biomed. Res. Int. 2014, 656370 (2014).
Shireen, E. Experimental treatment of antipsychotic-induced movement disorders. J. Exp. Pharmacol. 8, 1–10 (2016).
Ozasa, R. et al. The antipsychotic olanzapine induces apoptosis in insulin-secreting pancreatic β cells by blocking perk-mediated translational attenuation. Cell Struct. Funct. 38(2), 183–194 (2013).
Fatani, B. Z., Aldawod, R. A. & Alhawaj, F. A. Schizophrenia: Etiology, pathophysiology and management: A review. Egypt. J. Hosp. Med. 69(6), 2640–2646 (2017).
Stroup, T. S. & Gray, N. Management of common adverse effects of antipsychotic medications. World Psychiatry 17, 341–356 (2018).
Casey, D. E. Pathophysiology of antipsychotic drug-induced movement disorders. J. Clin. Psychiatry 65, 25–28 (2004).
Caroff, S. N., Hurford, I., Lybrand, J. & Campbell, E. C. Movement disorders induced by antipsychotic drugs: Implications of the CATIE schizophrenia trial. Neurol. Clin. 29, 127–148 (2011).
Berger, P. A. & Rexroth, K. Tardive dyskinesia: Clinical, biological, and pharmacological perspectives. Schizophr Bull. 6(1), 102–116 (1980).
Mahmoudi, S., Lévesque, D. & Blanchet, P. J. Upregulation of dopamine D3, not D2, receptors correlates with tardive dyskinesia in a primate model. Mov. Disord. 29(9), 1125–1133 (2014).
Cornett, E. M., Novitch, M., Kaye, A. D., Kata, V. & Kaye, A. M. Medication-induced tardive dyskinesia: A review and update. Ochsner J. 17, 162–174 (2017).
Turrone, P., Remington, G. & Nobrega, J. N. The vacuous chewing movement (VCM) model of tardive dyskinesia revisited: Is there a relationship to dopamine D2 receptor occupancy?. Neurosci. Biobehav. Rev. 26, 361–380 (2002).
Cho, C. H. & Lee, H. J. Oxidative stress and tardive dyskinesia: Pharmacogenetic evidence. Prog. Neuro-Psychopharmacol. Biol. Psychiatry [Internet] 46, 207–213. https://doi.org/10.1016/j.pnpbp.2012.10.018 (2013).
Lister, J., Nobrega, J. N., Fletcher, P. J. & Remington, G. Oxidative stress and the antipsychotic-induced vacuous chewing movement model of tardive dyskinesia: Evidence for antioxidant-based prevention strategies. Psychopharmacology 231, 2237–2249 (2014).
Lerner, P. P., Miodownik, C. & Lerner, V. Tardive dyskinesia (syndrome): Current concept and modern approaches to its management. Psychiatry Clin. Neurosci. 69, 321–334 (2015).
Kapur, S. & Seeman, P. Does fast dissociation from the dopamine D2 receptor explain the action of atypical antipsychotics? A new hypothesis. Am. J. Psychiatry 158, 360–369 (2001).
Nandra, K. S. & Agius, M. The differences between typical and atypical antipsychotics: The effects on neurogenesis. Psychiatr. Danubina 24(Suppl 1), 95–99 (2012).
Miyamoto, S., Miyake, N., Jarskog, L. F., Fleischhacker, W. W. & Lieberman, J. A. Pharmacological treatment of schizophrenia: A critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol. Psychiatry 17, 1206–1227 (2012).
Kane, J. M. Tardive dyskinesia rates with atypical antipsychotics in adults: Prevalence and incidence. J. Clin. Psychiatry 65, 16–20 (2004).
Stegmayer, K., Walther, S. & van Harten, P. Tardive dyskinesia associated with atypical antipsychotics: Prevalence, mechanisms and management strategies. CNS Drugs 32, 135–147 (2018).
Parikh, V., Khan, M. M. & Mahadik, S. P. Differential effects of antipsychotics on expression of antioxidant enzymes and membrane lipid peroxidation in rat brain. J. Psychiatr. Res. 37(1), 43–51 (2003).
Kropp, S. et al. Oxidative stress during treatment with first- and second-generation antipsychotics. J. Neuropsychiatr. 17(2), 227–231 (2005).
Noto, C. et al. Oxidative stress in drug naïve first episode psychosis and antioxidant effects of risperidone. J. Psychiatr. Res. 68, 210–216 (2015).
Stojković, T. et al. Risperidone reverses phencyclidine induced decrease in glutathione levels and alterations of antioxidant defense in rat brain. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 39(1), 192–199 (2012).
Wyatt, R. J. Tardive dyskinesia: Possible involvement of free radicals and treatment with vitamin E. Schizophr. Bull. 31, 731–740 (1999).
Teo, J. T., Edwards, M. J. & Bhatia, K. Tardive dyskinesia is caused by maladaptive synaptic plasticity: A hypothesis. Mov. Disord. 27(10), 1205–1215 (2012).
Zuardi, A. W. Cannabidiol: From an inactive cannabinoid to a drug with wide spectrum of action. Revista Brasileira de Psiquiatria [Internet] 30, 271–280 (2008).
Caruso, G. et al. Antioxidant properties of second-generation antipsychotics: Focus on microglia. Pharmaceuticals 13(12), 1–20 (2020).
Al-amin, M., Choudhury, F. R. & Chowdhury, A. S. Pretreatment with risperidone ameliorates systemic LPS-induced oxidative stress in the cortex and hippocampus. Front. Neurosci. 12, 384 (2018).
Eftekhari, A. et al. In vitro/vivo studies towards mechanisms of risperidone-induced oxidative stress and the protective role of coenzyme Q10 and N-acetylcysteine. Toxicol. Mech. Methods 26(7), 520–528 (2016).
Shah, R., Subhan, F., Sultan, S. M. & Ali, G. Short-term oral administration of risperidone induces pancreatic damage and hyperamylasemia in Sprague–Dawley rats. Braz. J. Pharm. Sci. 54(4), e17841 (2018).
Kawabe, K. & Ueno, S. I. A case of acute pancreatitis associated with risperidone treatment. Clin. Psychopharmacol. Neurosci. 12(1), 67–68 (2014).
Shah, R. et al. Comparative evaluation of pancreatic histopathology of rats treated with olanzapine, risperidone and streptozocin. Braz. J. Pharm. Sci. 54(3), e17579 (2018).
Aja, P. M. et al. Hesperidin protects against cadmium-induced pancreatitis by modulating insulin secretion, redox imbalance and iNOS/NF-ĸB signaling in rats. Life Sci. 259, 118268 (2020).
Choi, S. & Kim, H. The remedial potential of lycopene in pancreatitis through regulation of autophagy. Int. J. Mol. Sci. 21, 5775 (2020).
Bansod, S. et al. Borneol protects against cerulein-induced oxidative stress and inflammation in acute pancreatitis mice model. Environ. Toxicol. 36(4), 530–539 (2021).
Miyazaki, Y. et al. Pancreatic B-cell function is altered by oxidative stress induced by acute hyperglycaemia. Diabet. Med. 24(2), 154–160 (2007).
Sakano, D. et al. VMAT2 safeguards β-cells against dopamine cytotoxicity under high-fat diet-induced stress. Diabetes 69(11), 2377–2391 (2020).
Viudez-Martínez, A. et al. Cannabidiol does not display drug abuse potential in mice behavior. Acta Pharmacol. Sin. 40(3), 358–364. https://doi.org/10.1038/s41401-018-0032-8 (2019).
Navarrete, F., García-Gutiérrez, M. S., Gasparyan, A., Austrich-Olivares, A. & Manzanares, J. Role of cannabidiol in the therapeutic intervention for substance use disorders. Front. Pharmacol. 12, 1–24 (2021).
Valvassori, S. S. et al. Effects of cannabidiol on amphetamine-induced oxidative stress generation in an animal model of mania. J. Psychopharmacol. 25(2), 274–279 (2011).
Burstein, S. Cannabidiol (CBD) and its analogs: A review of their effects on inflammation. Bioorg. Med. Chem. 23, 1377–1385 (2015).
Peres, F. F. et al. Cannabidiol prevents motor and cognitive impairments induced by reserpine in rats. Front. Pharmacol. 7, 1–10 (2016).
Kajero, J. A., Seedat, S., Ohaeri, J., Akindele, A. & Aina, O. Investigation of the effects of cannabidiol on vacuous chewing movements, locomotion, oxidative stress and blood glucose in rats treated with oral haloperidol. World J. Biol. Psychiatry 21(8), 612–626 (2020).
Machado Bergamaschi, M., Helena Costa Queiroz, R., Waldo Zuardi, A., Alexandre, S. & Crippa, J. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr. Drug Saf. 6(4), 237–249 (2011).
Franco, V. & Perucca, E. Pharmacological and therapeutic properties of cannabidiol for epilepsy. Drugs 79, 1435–1454 (2019).
Huestis, M. A. et al. Cannabidiol adverse effects and toxicity. Curr. Neuropharmacol. 17(10), 974–989 (2019).
Moreira, F. A. & Guimarães, F. S. Cannabidiol inhibits the hyperlocomotion induced by psychotomimetic drugs in mice. Eur. J. Pharmacol. 512(2–3), 199–205 (2005).
Iffland, K. & Grotenhermen, F. An update on safety and side effects of cannabidiol: A review of clinical data and relevant animal studies. Cannabis Cannabinoid Res. 2(1), 139–154 (2017).
National Research Council. Guide for the Care and Use of Laboratory Animals 8th edn. (The National Academies Press, 2011).
Marchese, G. et al. Haloperidol versus risperidone on rat “early onset” vacuous chewing. Behav. Brain Res. 149(1), 9–16 (2004).
Guimarães, F. S., Chiaretti, T. M., Graeff, F. G. & Zuardi, A. W. Antianxiety effect of cannabidiol in the elevated plus-maze. Psychopharmacology 100(4), 558–559. https://doi.org/10.1007/BF02244012 (1990).
Adeneye, A. A., Agbaje, E. O. & Olagunju, J. A. Metformin: An effective attenuator of risperidone-induced insulin resistance hyperglycemia and dyslipidemia in rats. Indian J. Exp. Biol. 49(5), 332–338 (2011).
Crowley, J. J. et al. Antipsychotic-induced vacuous chewing movements and extrapyramidal side effects are highly heritable in mice. Pharmacogenomics J. 12(2), 147–155 (2012).
Pellow, S., Chopin, P., File, S. E. & Briley, M. Validation of open: Closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J. Neurosci. Methods 14(3), 149–167 (1985).
Walsh, R. N. & Cummins, R. A. The open-field test: A critical review. Psychol. Bull. 83(3), 482–504 (1976).
Eweka, A. & Om’Iniabohs, F. The effects of monosodium glutamate on the open field locomotor activities in adult Wistar rats. Internet J. Nutr Wellness 6(2) (2012).
Ennaceur, A. & Delacour, J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 31(1), 47–59 (1988).
Öztürk, Y., Aydin, S., Beis, R., Başer, K. H. C. & Berberoǧlu, H. Effects of Hypericum perforatum L. and Hypericum calycinum L. extracts on the central nervous system in mice. Phytomedicine 3(2), 139–146 (1996).
Buege, J. A. & Aust, S. D. Microsomal lipid peroxidation. Methods Enzymol. 52(C), 302–310 (1978).
Sedlak, J. & Lindsay, R. H. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal. Biochem. 25(1), 192–205 (1968).
Sinha, A. K. Colorimetric assay of catalase. Anal. Biochem. 47(2), 389–394 (1972).
Sun, M. & Zigman, S. An improved spectrophotometric assay for superoxide dismutase based on epinephrine autoxidation. Anal. Biochem. 90(1), 81–89 (1978).
Kedare, S. B. & Singh, R. P. Genesis and development of DPPH method of antioxidant assay. J. Food Sci. Technol. 48(4), 412–422 (2011).
Allison, D. B. et al. Antipsychotic-induced weight gain: A comprehensive research synthesis. Am. J. Psychiatry 156(11), 16686–21696 (1999).
Meyer, J. M. Novel antipsychotics and severe hyperlipidemia. J. Clin. Psychopharmacol. 21(4), 369–374 (2001).
Murashita, M. et al. Glucose and lipid metabolism of long-term risperidone monotherapy in patients with schizophrenia. Psychiatry Clin. Neurosci. 61(1), 54–58 (2007).
Smith, R. C., Rachakonda, S., Dwivedi, S. & Davis, J. M. Olanzapine and risperidone effects on appetite and ghrelin in chronic schizophrenic patients. Psychiatry Res. [Internet] 199(3), 159–163. https://doi.org/10.1016/j.psychres.2012.03.011 (2012).
Reynolds, G. P. & Kirk, S. L. Metabolic side effects of antipsychotic drug treatment—Pharmacological mechanisms. Pharmacol. Ther. [Internet] 125(1), 169–179. https://doi.org/10.1016/j.pharmthera.2009.10.010 (2010).
Ignatowska-Jankowska, B., Jankowski, M. M. & Swiergiel, A. H. Cannabidiol decreases body weight gain in rats: Involvement of CB2 receptors. Neurosci. Lett. [Internet] 490(1), 82–84. https://doi.org/10.1016/j.neulet.2010.12.031 (2011).
Osborne, A. L., Solowij, N. & Weston-Green, K. A systematic review of the effect of cannabidiol on cognitive function: Relevance to schizophrenia. Neurosci. Biobehav. Rev. [Internet] 72, 310–324. https://doi.org/10.1016/j.neubiorev.2016.11.012 (2017).
Carvalho, R. K. et al. Chronic cannabidiol exposure promotes functional impairment in sexual behavior and fertility of male mice. Reprod. Toxicol. 81, 34–40 (2018).
Weiss, L. et al. Cannabidiol lowers incidence of diabetes in non-obese diabetic mice. Autoimmunity 39, 143–151 (2006).
Horvth, B., Mukhopadhyay, P., Hask, G. & Pacher, P. The endocannabinoid system and plant-derived cannabinoids in diabetes and diabetic complications. Am. J. Pathol. [Internet] 180(2), 432–442. https://doi.org/10.1016/j.ajpath.2011.11.003 (2012).
Chaves, Y. C. et al. Two-weeks treatment with cannabidiol improves biophysical and behavioral deficits associated with experimental type-1 diabetes. Neurosci. Lett. [Internet] 729, 135020. https://doi.org/10.1016/j.neulet.2020.135020 (2020).
Bouwens, L. Beta cell regeneration. Curr. Diabetes Rev. 2(1), 3–9 (2006).
Brunt, T. M., Van Genugten, M., Höner-Snoeken, K., Van De Velde, M. J. & Niesink, R. J. M. Therapeutic satisfaction and subjective effects of different strains of pharmaceutical-grade cannabis. J. Clin. Psychopharmacol. 34(3), 344–349 (2014).
Hoffman, D. C. & Donovan, H. Catalepsy as a rodent model for detecting antipsychotic drugs with extrapyramidal side effect liability. Psychopharmacology 120(2), 128–133 (1995).
Fink-Jensena, A. et al. Antipsychotic-induced catalepsy is attenuated in mice lacking the M4 muscarinic acetylcholine receptor. Eur. J. Pharmacol. 23(1), 39–44 (2011).
Russo, E. B., Burnett, A., Hall, B. & Parker, K. K. Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem. Res. 30(8), 1037–1043 (2005).
Nah, S. Y., Saya, D. & Vogel, Z. Cannabinoids inhibit agonist-stimulated formation of inositol phosphates in rat hippocampal cultures. Eur. J. Pharmacol. Mol. Pharmacol. 246(1), 19–24 (1993).
Zuardi, A. W., Shirakawa, I., Finkelfarb, E. & Karniol, I. G. Action of cannabidiol on the anxiety and other effects produced by delta 9-THC in normal subjects. Psychopharmacology 76(3), 245–250. https://doi.org/10.1007/BF00432554 (1982).
Rogóz, Z. & Skuza, G. Anxiolytic-like effects of olanzapine, risperidone and fluoxetine in the elevated plus-maze test in rats. Pharmacol. Rep. 63(6), 1547–1552 (2011).
Knable, M. B., Heinz, A., Raedler, T. & Weinberger, D. R. Extrapyramidal side effects with risperidone and haloperidol at comparable D2 receptor occupancy levels. Psychiatry Res. Neuroimaging 75(2), 91–101 (1997).
Désaméricq, G., Schurhoff, F., Meary, A., Szöke, A., Macquin-Mavier, I., Bachoud-Lévi, A. C. et al. Long-term neurocognitive effects of antipsychotics in schizophrenia: A network meta-analysis. Eur. J. Clin. Pharmacol. [Internet] 70, 127–134 (2014). https://www.ncbi.nlm.nih.gov/books/NBK169259/.
Bhabak, K. P. & Mugesh, G. Functional mimics of glutathione peroxidase: Bioinspired synthetic antioxidants. Acc. Chem. Res. 43(11), 1408–1419 (2010).
He, J., Kong, J., Tan, Q. R. & Li, X. M. Neuroprotective effect of atypical antipsychotics in cognitive and non-cognitive behavioral impairment in animal models. Cell Adhes. Migr. https://doi.org/10.4161/cam.3.1.7401 (2009).
Dakhale, G. et al. Oxidative damage and schizophrenia: The potential benefit by atypical antipsychotics. Neuropsychobiology 49(4), 205–209 (2004).
Kunz, M. et al. Elevated serum superoxide dismutase and thiobarbituric acid reactive substances in different phases of bipolar disorder and in schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 32(7), 1677–1681 (2008).
Zendulka, O. et al. Cannabinoids and cytochrome P450 interactions. Curr. Drug Metab. 17(3), 206–226 (2016).
Kudo, S. & Ishizaki, T. Pharmacokinetics of haloperidol. An update. Clin. Pharmacokinet. 37, 435–456 (1999).
Padurariu, M., Ciobica, A., Dobrin, I. & Stefanescu, C. Evaluation of antioxidant enzymes activities and lipid peroxidation in schizophrenic patients treated with typical and atypical antipsychotics. Neurosci. Lett. [Internet] 479(3), 317–320. https://doi.org/10.1016/j.neulet.2010.05.088 (2010).
Esposito, G. et al. Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci. Lett. 399(1–2), 91–95. https://doi.org/10.1016/j.neulet.2006.01.047 (2006).
Esposito, G. et al. Cannabidiol in vivo blunts β-amyloid induced neuroinflammation by suppressing IL-1β and iNOS expression. Br. J. Pharmacol. 151(8), 1272–1279 (2007).
Kato, T., Monji, A., Hashioka, S. & Kanba, S. Risperidone significantly inhibits interferon-γ-induced microglial activation in vitro. Schizophr. Res. 92(1–3), 10815 (2007).
Levkovitz, Y. et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J. Clin. Psychiatry 71(2), 138–149 (2010).
MacDowell, K. S. et al. Risperidone normalizes increased inflammatory parameters and restores anti-inflammatory pathways in a model of neuroinflammation. Int. J. Neuropsychopharmacol. 16(1), 121–135 (2013).
Chen, J. et al. Protective effect of cannabidiol on hydrogen peroxide-induced apoptosis, inflammation and oxidative stress in nucleus pulposus cells. Mol. Med. Rep. 14(3), 2321–2327 (2016).
Tarazi, F. I., Zhang, K. & Baldessarini, R. J. Long-term effects of newer antipsychotic drugs on neuronal nitric oxide synthase in rat brain. Nitric Oxide Biol. Chem. 7(4), 297–300 (2002).
Suzuki, E., Nakaki, T., Shintani, F., Kanba, S. & Miyaoka, H. Antipsychotic, antidepressant, anxiolytic, and anticonvulsant drugs induce type II nitric oxide synthase mRNA in rat brain. Neurosci. Lett. 333(3), 217–219 (2002).
Taneli, F., Pirildar, Ş, Akdeniz, F., Uyanik, B. S. & Ari, Z. Serum nitric oxide metabolite levels and the effect of antipsychotic therapy in schizophrenia. Arch. Med. Res. 35(5), 401–405 (2004).
Da Cruz Jung, I. E. et al. Haloperidol and Risperidone at high concentrations activate an in vitro inflammatory response of RAW 264.7 macrophage cells by induction of apoptosis and modification of cytokine levels. Psychopharmacology 233(9), 1715–1723 (2016).
Liskowsky, D. R. & Potter, L. T. Dopamine D2 receptors in the striatum and frontal cortex following chronic administration of haloperidol. Neuropharmacology 26(5), 481–483 (1987).
Kusumi, I., Takahashi, Y., Suzuki, K., Kameda, K. & Koyama, T. Differential effects of subchronic treatments with atypical antipsychotic drugs on dopamine D2 and serotonin 5-HT2A receptors in the rat brain. J. Neural Transm. (Vienna) 107(3), 295–302. https://doi.org/10.1007/s007020050024 (2000).
Brinholi, F. F. et al. Clozapine and olanzapine are better antioxidants than haloperidol, quetiapine, risperidone and ziprasidone in in vitro models. Biomed. Pharmacother. 81, 411–415 (2016).
Martin, W. F. et al. Mitigation of olanzapine-induced weight gain with samidorphan, an opioid antagonist: A randomized double-blind phase 2 study in patients with schizophrenia. Am. J. Psychiatry 176(6), 457–467 (2019).
Bishnoi, M. & Boparai, R. K. An animal model to study the molecular basis of tardive dyskinesia. Methods Mol. Biol. 829, 193–201 (2012).
Acknowledgements
Dr. Opeyemi Awofeso was involved in data analysis; Mr. Sunday Adenekan and Mr. Abiodun Doherty helped with the biochemical analysis; while Mr. Chiadika Chimeremeze, Mr. Hafeez Shittu, Hasnat Osibote, Damilola Oshunyinka, and Abisola Kolawole were the laboratory assistants who worked with the team in the conduct of the experiments.
Funding
This research was supported by the South African Research Chair in PTSD hosted by the Stellenbosch University and funded by the Department of Science and Technology South Africa (No. 64811) and administered by the National Research Foundation. This work was also supported by Cannabis Science Inc. (No. S005181).
Author information
Authors and Affiliations
Contributions
J.K. and S.S. were responsible for the conceptualization of the study. The experiments were designed by J.K., S.S. and A.A. while J.K., O.A. and A.A. conducted the experiments. Manuscript, figures and tables were prepared by J.K. S.S. and J.O. supervised and validated the study for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kajero, J.A., Seedat, S., Ohaeri, J. et al. Effects of cannabidiol on vacuous chewing movements, plasma glucose and oxidative stress indices in rats administered high dose risperidone. Sci Rep 12, 19718 (2022). https://doi.org/10.1038/s41598-022-24235-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-24235-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
