
Abstract
Although lung diseases typically result from long-term exposures, even a robust, one-time exposure can result in long-lasting consequences. Endotoxin is a ubiquitous environmental/occupational inflammatory agent often used to model airway inflammation. Using a murine model, the return to lung homeostasis following high dose inhalant lipopolysaccharide (LPS, 10–100 μg) exposure were delineated over 2 weeks. LPS-induced rapid weight loss, release of proinflammatory mediators, and inflammatory cell influx with prolonged persistence of activated macrophages CD11c+CD11b+ and recruited/transitioning CD11cintCD11b+ monocyte-macrophages out to 2 weeks. Next, lung-delivered recombinant (r) interleukin (IL)-10 was intratracheally administered for 3 doses initiated 5 h following LPS (10 μg) exposure for 2 days. IL-10 therapy reduced LPS-induced weight loss and increased blood glucose levels. Whereas there was no difference in LPS-induced bronchoalveolar lavage airway fluid cellular influx, total lung cell infiltrates were reduced (37%) with rIL-10 treatment. Post-LPS exposure treatment with rIL-10 strikingly reduced lavage fluid and lung homogenate levels of tumor necrosis factor-α (88% and 93% reduction, respectively), IL-6 (98% and 94% reduction), CXCL1 (66% and 75% reduction), and CXCL2 (47% and 67% reduction). LPS-induced recruited monocyte-macrophages (CD11cintCD11b+) were reduced (68%) with rIL-10. Correspondingly, LPS-induced lung tissue CCR2+ inflammatory monocyte-macrophage were reduced with rIL-10. There were also reductions in LPS-induced lung neutrophils, lymphocyte subpopulations, collagen content, and vimentin expression. These findings support the importance of studying resolution processes for the development of treatment after unintended environmental/occupational biohazard exposures. Short-term, lung-delivered rIL-10 favorably hastened inflammatory recovery processes following acute, high dose inhalant LPS exposure.
Similar content being viewed by others

Introduction
Whereas occupational-associated lung diseases are typically caused by repeated, long-term exposure to various hazardous agents, even a robust, one-time exposure can damage the lungs resulting in long-term disease consequences. A striking example of the latter would be the lung disease that developed in first responders exposed to the dust cloud following the disastrous World-Trade Center collapse of September 11, 20011,2,3. Whereas efforts to reduce exposures, promote respiratory protective equipment, and continue surveillance are needed, there are currently no therapies for the management of post-occupational exposure induced lung injury/inflammation, underscoring an unmet need to identify alternative approaches. Additional factors that have hampered progress consist of the complexity of the exposome including various potential occupational derivates or microorganisms4 as well as limited information understanding the post-exposure normative recovery and resolution of consequences in the lung. Investigations to understand the resolution processes are needed for the development of treatment options following unintended environmental and/or occupational biohazard exposures. The objective of this study was to investigate the cellular mediators of lung recovery/resolution following a standard inflammatory agent insult and investigate a potential therapeutic lung-targeted approach to reduce disease burden using an in vivo model that could be shared among investigators.
Endotoxins are characterized as lipopolysaccharides (LPS) in gram-negative bacteria with very high pro-inflammatory properties4,5. Human inhalant exposure to endotoxins occurs constantly, as LPS can easily bind to dust6. High LPS exposure induces systemic, and airway inflammatory diseases marked by influx of neutrophils, dyspnea, and chest tightness7. Endotoxins have been considered a main factor contributing to occupational lung diseases including textile workers, dairy workers, animal feed and grain workers, sewage treatment plant workers, and organic dust toxic syndrome8,9,10,11,12. Additional advantages of utilizing LPS as a representative inflammatory agent in experimental designs is that it is highly reproducible, commercially available, elicits a pro-inflammatory lung response in humans and rodents, and can be compared to existing published data for occupation-induced lung disease.
A potential strategy to hasten post-exposure lung recovery/resolution processes is through targeting the clinically relevant anti-inflammatory and pro-resolving interleukin (IL)-10 cytokine pathway. In a cross-sectional study of 625 veterans with farming experience, baseline IL-10 concentrations from a whole blood assay were inversely associated with ΔTNF-⍺ (− r = 0.63) and ΔIL-6 (− r = 0.37) levels with results remaining highly significant (p < 0.0001) after adjusting for potential confounders13. In a murine model of repetitive agriculture dust extract (LPS-enriched)-induced lung inflammation, neutrophil influx and lung pathology was elevated in IL-10 deficient mice and recombinant (r)IL-10 treatment reversed these effects14. Correspondingly, rIL-10 treatment reduced dust extract-induced TNF-⍺ release in cultured murine alveolar macrophages and peritoneal macrophages14. Prior studies also demonstrated that airway injury outcomes are increased following chronic, high concentration-endotoxin in IL-10-deficient mice15,16, and that airway injury was reduced when human IL-10 expression was delivered by adenoviral vector treatment to the murine lung prior to LPS exposure16. Therapeutic application of IL-10 (via systemic delivery) has been tested in chronic diseases including Crohn’s disease, rheumatoid arthritis, psoriasis, hepatitis C, and HIV, with partial efficacy and potential untoward reactions preventing application to clinical practice related to its long-term use17.
Here, we first aimed to delineate the time continuum of cell mediator events of normal recovery following acute LPS exposure, and then we hypothesized that a lung-delivered, short-term approach of IL-10 treatment could have beneficial responses following acute LPS-induced airway inflammation to hasten lung recovery.
Methods
Mice and exposure model
C57BL/6 mice (at 8-weeks of age) were purchased from The Jackson Laboratory (Bar Harbor, ME), randomized upon arrival, and were allowed to acclimate for one week prior to initiation of experiments. AJN, RG, and facility staff were aware of the randomization. All other authors were blinded. Animals were housed in a dedicated room away from all other animals in the facility. Male mice were utilized for all studies because we have previously demonstrated that female mice were less susceptible to inhalant endotoxin-induced airway and systemic inflammatory effects18. Airway inflammation was induced using intranasal or intratracheal instillation of lipopolysaccharide (LPS) of Escherichia coli (O55:B5) from Sigma (St. Louis, MO) whereby mice were lightly sedated under isoflurane and received treatment with either 50 µL of sterile saline or LPS (10 μg or 100 μg). Recombinant (r)IL-10 was purchased from R&D Systems (Minneapolis, MN). After determining safety dose-responses in mice, rIL-10 was intratracheally administered at 1 μg in 50 μl of sterile saline per mouse in post-LPS exposure studies. Vehicle control of 50 μl of sterile saline was also administered in side-by-side studies. Weights were recorded throughout the treatment period. At completion of experiments, mice were sedated and euthanized by isoflurane followed by exsanguination (right axillary blood collection). The study was conducted and reported in accordance with ARRIVE guidelines (https://arriveguidelines.org). Animal procedures were also approved by the University of Nebraska Medical Center (UNMC) Institutional Animal Care and Use Committee and were in accordance with the NIH guidelines for the use of rodents.
Inflammatory marker analysis
Bronchoalveolar lavage fluid (BALF) was collected using 3 × 1 mL PBS. Total cell numbers from the combined recovered lavage were enumerated and differential cell counts determined from cytospin-prepared slides (cytopro cytocentrifuge, ELITech Group, Logan, UT) stained with DiffQuick (Siemens, Newark, DE). Lung tissue homogenates were prepared by homogenizing lung samples in 500 μl of sterile phosphate buffered saline (PBS). From cell-free supernatant of the first lavage fraction and lung tissue homogenates, tumor necrosis factor-alpha (TNF-α), IL-6, IL-10, murine neutrophil chemokines (CXCL1 and CXCL2), and the epidermal growth factor amphiregulin (AREG) were quantitated by ELISA (R&D Systems, Minneapolis, MN; minimal detectable dose (MDD) of 1.88, 1.6, 31.3, 2.0, 1.5, and 15.6 pg/ml, respectively.
Lung cell staining and flow cytometry
Following vascular perfusion and BALF removal, harvested lungs (right lungs) were subjected to an automated dissociation procedure using a gentleMACS Dissociator instrument (Miltenyi Biotech, Auburn, CA)19. Lung cells from each animal were incubated with a LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen, Carlsbad, CA) and CD16/32 (Fc Block, Biolegend, San Diego, CA) to minimize non-specific antibody staining. Cells were stained with monoclonal antibody against rat anti-mouse CD45 (clone: 30-F11; BD Biosciences, Franklin Lake, NJ), CD11b (clone: M1/70; BD Biosciences), Ly6G (clone 1A8; BD Biosciences), CD11c (clone: N418; Invitrogen), CD4 (clone RM4-5, BD Biosciences), CD8 (clone 53-6.7, BD Biosciences), CD19 (clone 1D3, Invitrogen), hamster anti-mouse CD3e (clone 145-2C11, BD Biosciences), and mouse anti-mouse NK1.1 (clone PK136, BD Biosciences). Gating strategies for Ly6G+ neutrophils, CD3+CD4+ T cells, CD3+CD8+ T cells, CD19+ B cells, and NK cells (depicted in Supplemental Fig. 1) and monocyte/macrophage populations were as previously reported18,20,21,22,23,24. The percentage of all respective cell populations was determined from live CD45+ lung leukocytes after excluding debris and doublets. This percentage was multiplied by the respective total lung cell numbers to determine specific cell population numbers for each animal.

Normal homeostasis recovery trends in airway inflammatory endpoints over time following a one-time inhalation exposure to high-dose LPS. C57BL/6 mice treated once with 100 ug of LPS and allowed to recover without additional treatment up to one week. Line graphs depict mean with SEM bars over time of LPS-induced weight changes (A), airway cellular influx (B), and levels of pro-inflammatory TNF-⍺, IL-6, and CXCL1 (neutrophil chemoattractant), anti-inflammatory (IL-10), and amphiregulin (AREG) from bronchoalveolar lavage fluid (BALF). N = 24 mice (4 mice/time point). Statistical significance denoted versus saline/baseline as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Lung histopathology
Following lavage, left lungs were excised and inflated to 15 cm H2O pressure with 10% formalin (Fisher Scientific, Fair Lawn, NJ) for 24 h to preserve pulmonary architecture19. Sectioned (4–5 μm) lungs were H&E stained, and slides were reviewed at all scanning magnifications and semi-quantitatively assessed for the degree and distribution of lung inflammation by experimental pathologist (T.A.W.) blinded to the treatment conditions, utilizing a previously published scoring system14. This scoring system evaluates the spectrum of inflammatory changes for alveolar compartment inflammation and bronchiolar compartment inflammation. Each parameter was independently assigned a value from 0 to 5 with a higher score indicating greater inflammatory changes in the lung.
To determine CCR2+ inflammatory monocyte-derived macrophages, neutrophils, vimentin expression, and macrophages, lungs sections were stained with anti-CCR2 (1:100, NBP267700, Lot HM0537, Novus, Littleton, CO), anti-myeloperoxidase (MPO, 1:100, Cat#ab9535, Lot#GR331736-4; Abcam), anti-vimentin (Abcam, ab92547, Lot #GR3258719-9, 1:200), and anti-CD68 (pan-murine macrophage and scavenger receptor marker, Abcam ab31630, Lot #GR3305929-3, 1:100). Cross absorbed with donkey anti-rabbit (AlexaFluor488, A21206, Lot #2156521) or goat-anti rat AF 555 (Cat#A21434, Lot#2184321) from Thermo Fisher, Waltham, MA or goat anti-mouse IgG Alexa Fluor Plus 555 (Invitrogen A32727, Lot #UL287768) all used at 1:100 as secondary antibodies and processed as previously described25,26. Slices were mounted with VECTASHIELD® Antifade Mounting Medium with DAPI (4′6-diamindino-2-phenylindole; to identify nuclei)(Cat#H-1200, Lot#ZG1014, Curlingame, CA) and visualized under Zeiss fluorescent microscope. Photographs (10 per lung images per mouse) of lung parenchyma were taken from the entire section under a Zeiss fluorescent microscope (Zeiss Observer.Z1 [Zeiss, White Plains, NY]). CCR2+ monocyte-derived macrophages, MPO+ neutrophils, vimentin expression, and CD68 + macrophages were quantified by Image J FIJI plugin (version: 2.9.0/1.53t).
Lung sections were also stained with modified Masson’s Trichrome and scanned with Aperio scanner (Leica Biosystems, Deer Park, IL) by the institution’s Tissue Sciences Core Facility. VENTANA image viewer (Roche Diagnostics, Indianapolis, IN, version 3.1.4) was used to take 10 images per section at 20 × from the scanned sections. The tiff images were plugged into FIJI plugin of Image J. Collagen content in the trichrome images were quantified as described by Chen and colleagues27. Briefly, trichrome images were deconvoluted using the Masson’s Trichrome setting. The green component identified as the collagen fibers was used for thresholding at 170/215 to avoid cell nucleus and focus on the collagen fibers. Integrated density was measured and plotted as a measure of collagen content.
Blood and serum studies
Whole blood collected from the axillary artery at the time of euthanasia and serum collected as previously described19. Blood glucose levels were determined by glucometer (True Metrix Air, Nipro Diagnostics, Fort Lauderdale, FL). Serum levels of pentraxin-2 (murine acute phase reactant protein) and IL-6 were assessed by Quantikine ELISA according to the manufacturer’s instructions (R&D, MDD of 0.159 ng/ml and 1.6 pg/ml).
Statistical analysis
Data are presented as the mean ± standard error of mean (SEM). To detect significant changes among 3 or more groups, a one-way analysis of variance (ANOVA) was utilized and a post-hoc test (Tukey/LSD) was performed to account for multiple comparisons if the p value was < 0.05. The nonparametric Mann–Whitney test was used to detect significant changes among 2 groups. All statistical analyses were performed using GraphPad Prism (version: 9.0.0) software and statistical significance accepted at a two-sided p < 0.05.
Ethics approval and consent to participate
Neither human participants, human data nor human tissue were used in these studies. Animal procedures were approved by the Institutional Animal Care and Use Committee and were in accordance with the NIH guidelines for the use of rodents.
Results
Normal homeostasis recovery trends in airway inflammatory endpoints over time following a one-time inhalation exposure to high-dose LPS
In these first set of studies, mice were intranasally treated once with 100 μg of LPS and allowed to recover without additional treatment up to 1 week. High concentration LPS induced significant weight loss at 24, 48, and 72 h (− 12% to − 17% loss, p < 0.0001) as compared to baseline with recovery of weight at one-week post-exposure (Fig. 1A). Total cellular airway influx driven by neutrophils was increased at 5 h post-exposure with peak neutrophil counts demonstrated at 24 h post-exposure, persisted to 72 h, and diminished by 1-week post-exposure (Fig. 1B). Correspondingly, airway influx of macrophages increased at 48 h and persisted at increased counts at 1-week post-exposure vs. baseline. Lymphocyte influx increased at 24 h, and lymphocyte counts remained elevated above baseline measurements at 1-week post-exposure. LPS-induced levels of pro-inflammatory TNF-⍺, IL-6, CXCL1, and amphiregulin peaked in the BALF at 5 h post-LPS exposure with a relatively rapid recovery to baseline by 24–48 h for TNF-⍺, CXCL1, and amphiregulin (Fig. 1C). IL-6 levels remained elevated until 72 h with return to baseline at 1-week post-exposure. In contrast, IL-10 levels were reduced at 48 h and increased at 1 week post exposure as compared to baseline (Fig. 1C).
Normal homeostasis recovery trends in lung cell infiltrates over time following a one-time inhalation exposure to high-dose LPS
Lung tissues were also processed for lung cell populations by flow cytometry. Total lung cell numbers increased and peaked at 48 h post-LPS exposure with return to normative numbers at 1 week (Fig. 2A). High dose LPS exposure modulated lung macrophage-monocyte populations and numbers over time (Fig. 2B,C). Namely, there were decreased numbers of alveolar macrophages (CD11c+CD11blo) at 5, 24, 48 and 72 h with return to normative levels at 1 week. Activated/exudative macrophages (CD11c+C11bhi) increased significantly (p = 0.03) above baseline at 24 h with further rise at 48 to 72 h and remained elevated at 1-week post-exposure. At 24 h post-LPS exposure, there was an increase in the transitioning monocyte/macrophage population (CD11cintCD11bhi) that peaked at 24 h and remained increased at 72 h and 1 week. Lung Ly6G+ neutrophils increased at 5 h post-LPS exposure, remained elevated at 48 h, and returned to baseline values at 1 week (Fig. 2D). Numbers of lung lymphocytes including CD4 + T cells, CD8 + T cells, CD19 + B cells and NK cells demonstrated increase at 48 h-72 h with reduction to baseline values at 1 week (Fig. 2E).

Normal homeostasis recovery trends in lung cell infiltrates over time following a one-time inhalation exposure to high-dose LPS. Lung cell populations determined by flow cytometry after removal of debris, doublets, and gating on live CD45 + cells (% population multiplied by total lung cells). Line graphs depict mean with SEM bars over time of total lung cells (A) and lung monocyte/macrophage subpopulations (B). (C) Gating strategy of lung monocyte/macrophage subpopulations : 1: Alveolar macrophages CD11c+CD11blo, 2: Activated macrophages CD11c+CD11bhi, 3: Transitioning monocyte/macrophages CD11cintCD11bhi, 4: Monocytes CD11bhi CD11c- after exclusion of neutrophils. (D) Ly6G+ neutrophil, and (E) lymphocyte populations shown. N = 24 mice (4 mice/time point). Statistical significance denoted versus saline/baseline as *p < 0.05, **p < 0.01, ***p < 0.001.
Collectively, these studies describe the post-exposure time course of cellular and mediator populations in the lung up to one week following a one-time exposure to high concentration LPS and demonstrated that approximately 48 h is optimal time point for investigating lung cellular and mediator responses with a return to normal homeostasis for many endpoints except monocyte/macrophage subpopulations. Because high concentration of LPS (100 μg) induced an excessive lung inflammatory response with weight loss approaching 20% of body mass, lower concentrations of LPS (i.e., 100 ng and 10 μg) were instilled once with endpoints measured at 2 weeks post-exposure. These modified dosing studies allowed us to capture whether LPS-induced lung monocyte-macrophage populations would return to baseline/normative levels. However, even at 2 weeks post-exposure with the modified high dose LPS (10 μg) exposure, activated lung macrophages (CD11c+CD11bhi), transitioning lung monocyte/macrophages (CD11cintCD11bhi) and lung monocytes (CD11c-CD11b+) remained significantly elevated as compared to saline control (Table 1). Blood glucose levels were also significantly elevated at 2 weeks post exposure with LPS 10 μg, but not 100 ng, as compared to saline control (Table 1). There were no significant differences (p > 0.05) between LPS 10 μg and saline treatment groups in numbers of BALF cells, lung neutrophils, or lung lymphocytes at 2 weeks post-treatment (Table 1 and data not shown).
Lung-delivered rIL-10 treatment hastens recovery of systemic and airway inflammatory events following a one-time LPS exposure
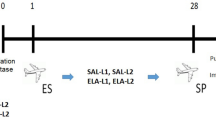
In these set of studies, we sought to determine whether treatment with lung-delivered rIL-10 (1 μg) would hasten recovery from LPS-induced inflammation. Using the modified high dose LPS (10 μg) and the optimal time point of 2 days following exposure, three doses of rIL-10 or vehicle were intratracheally instilled at 5, 24, and 48 h following the one-time LPS exposure (schematic of experimental design, Fig. 3A). Treatment with rIL-10 significantly reduced the LPS-induced weight loss at day 2 (69% difference, p = 0.0003, Fig. 3B,C). Blood glucose levels were correspondingly increased in the rIL-10 treatment group as compared to vehicle control (Fig. 3D). Serum levels of the murine acute phase reactant protein, pentraxin-2, and IL-6 were both strikingly reduced with rIL-10 treatment (Fig. 3E,F, respectively). In contrast, there was no difference between groups in the numbers of total cells, neutrophils, macrophages, lymphocytes in BALF compartment (Fig. 3G and data not shown). However, airway inflammatory makers including TNF-α, IL-6, CXCL1, and CXCL2 were reduced with rIL-10 treatment, and this finding was particularly profound for TNF-α and IL-6 (Fig. 3H). These same inflammatory markers were determined in lung tissue homogenates with similar trends demonstrated (Fig. 3I). Namely, treatment with rIL-10 nearly abrogated detection of lung TNF-α and IL-6 as well as reducing CXCL1 and CXCL2 levels. Consistent with administration of IL-10, IL-10 levels were elevated in both BALF and lung homogenates (Fig. 3J).

Instilled rIL10 treatment hastens recovery of systemic and airway inflammatory events following a one-time LPS exposure. (A) Schematic of experimental design (Created with BioRender.com). (B) Line graph depicts mean with SEM bars of weight changes over time. Scatter plot graphs depict mean with SEM bars between treatment groups of percent change in weight at day 3 versus baseline (C), blood glucose levels (D), serum pentraxin-2 levels (E), serum IL-6 levels (F). Total cellular influx depicted in bronchoalveolar lavage fluid (BALF) (G). Levels of airway inflammatory markers determined by ELISA from BALF (H) and lung homogenates (I) and levels of BALF and lung homogenate IL-10 (J). N = 7 (vehicle) and N = 8 (rIL-10) mice/group from 2 experimental runs. Statistical significance denoted by asterisks (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
rIL10 treatment following a one-time high dose LPS exposure modulates specific lung monocyte/macrophage populations
In these same mice, lung tissues were also processed for specific lung cell infiltrate determination by flow cytometry. First, total lung cell numbers were reduced following LPS exposure by rIL-10 treatment as compared to vehicle control (Fig. 4A). Of 4 monocyte/macrophage subpopulations examined, there was a marked reduction in the recruited, transitioning CD11cintCD11bhi monocyte/macrophage subpopulation in mice receiving rIL-10 treatment as compared to vehicle control following LPS exposure (Fig. 4B–D). This was evident by a decrease in both the percentage (Fig. 4C) and numbers (Fig. 4D) of these CD11cintCD11bhi cells. There was an associated increase in the percentages, but not numbers, of activated macrophages (CD11c+CD11bhi) and monocytes (CD11c-CD11bhi). There was no difference in alveolar macrophages (CD11c+CD11blo) between groups. Correspondingly, there was also no significant difference in the expression of the murine macrophage CD68 marker (also recognized as a scavenger receptor) by immunohistochemical staining of lung sections between treatment groups (mean ± SEM, integrated density; LPS + vehicle: 5.86 × 104 ± 1.81 × 104, N = 7 mice/group; and LPS + rIL-10: 11.96 × 104 ± 3.10 × 104, N = 8 mice/group; p = 0.19).

Instilled rIL10 treatment following a one-time LPS exposure modulates specific lung monocyte/macrophage populations. (A) Scatter plot depicts mean with SE bars of total lung cells of vehicle vs. rIL-10 treatment groups post-LPS (10 μg) exposure. (B) Representative image of gates for lung macrophage/monocyte subpopulations between treatment groups: 1: Alveolar macrophages CD11c+CD11blo, 2: Activated macrophages CD11c+CD11bhi, 3: Transitioning monocyte/macrophages CD11cintCD11bhi, 4: Monocytes CD11bhi CD11c- after exclusion debris, doublets, dead cells, CD45- cells, and neutrophils (Supplemental Fig. 1). Percentage of monocyte/macrophage subpopulations from live CD45+ lung cells depicted by scatter plot graphs (C). This percentage then multiplied by total lung cells enumerated to denote number of these subpopulations (D). N = 7 (vehicle) and N = 8 (rIL-10) mice/group from 2 experimental runs. Statistical significance denoted by asterisks (**p < 0.01, ***p < 0.001).
Effect of rIL-10 treatment following a one-time high dose LPS exposure in modulating neutrophils and lymphocyte lung populations
Lung-delivered rIL-10 also reduced the percentage (Fig. 5A) and number (Fig. 5B) of Ly6G+ lung neutrophils as compared to vehicle control following LPS exposure. For lymphocyte subpopulations, rIL-10 treatment resulted in an increase in percentage, but not number, of CD4 + T cells, decrease in number, but not percentage, of CD8 + T cells, and no difference in NK cell percentage or number (Fig. 5A,B). However, there was an increase in both percentage and number of CD19+ B cells following rIL-10 treatment (Fig. 5A,B).

Lung-delivered rIL10 treatment following a one-time LPS exposure modulates neutrophils and lymphocyte lung populations. A, Scatter plot depicts mean with SE bars of percentage of neutrophil and lymphocyte populations of live CD45+ lung cells. Next, neutrophil and lymphocyte lung cell % populations multiplied by total lung cells (B). Gating strategy depicted in Supplemental Fig. 1. N = 7 (vehicle) and N = 8 (rIL-10) mice/group from 2 experimental runs. Statistical significance denoted by asterisks (*p < 0.05, **p < 0.01, ***p < 0.001).
Lung-delivered rIL-10 treatment reduces lung histopathology, CCR2 + inflammatory monocytes/macrophages, and MPO + neutrophils
By microscopic review, there was also a reduction in acute LPS-induced bronchiolar and alveolar inflammation in mice that were treated with rIL-10 as compared to vehicle control (Fig. 6A). This histopathologic observation was confirmed by semi-quantitative assessment by pathologist blinded to treatment conditions (Fig. 6B) corresponding to the decrease in overall total lung cells with rIL-10 treatment (Fig. 4). Based upon studies demonstrating a reduction in specific LPS-induced recruited monocyte-macrophage (CD11cintCD11bhi) subpopulations with rIL-10 treatment as evident by flow cytometry studies (Fig. 4), lung sections were stained for CCR2 expression (Fig. 6C). CCR2 is a chemokine receptor primarily found on monocyte-derived macrophages and is important for monocyte-derived macrophage tracking from peripheral blood to injured tissues. By confocal microscopy, CCR2+ inflammatory monocyte-macrophages were reduced in rIL-10 treated mice as compared to vehicle control treated mice (Fig. 6C,D). Lung tissue MPO+ neutrophils were also reduced in rIL-10 treated mice as compared to vehicle control treated mice (Fig. 6E,F), consistent with the flow cytometry studies (Fig. 5).

Treatment with rIL-10 reduces lung inflammation, CCR2 + inflammatory monocytes, and MPO + neutrophils following a one-time high dose LPS exposure. Representative H&E-stained lung section images from both treatment groups and sham (saline control) (A) with semi-quantitative lung inflammatory score (B) shown. Confocal images of lung tissues stained for CCR2+ inflammatory monocytes/macrophages (green) (C) and myeloperoxidase (MPO)+ neutrophils (red) (E) with nuclei staining by DAPI (blue) and integrated density of CCR2 (D) and MPO (F) quantified per each mouse. Individual values are averaged from 8 to 10 images per section per mouse. Statistical significance vs. sham (####p < 0.0001). Asterisks (*p < 0.05, **p < 0.01, ***p < 0.001). Line scale is 100 μm.
Lung-delivered rIL-10 treatment reduces LPS-induced lung collagen in trichrome staining and lung vimentin expression.
Collagen content as measured by trichrome staining was increased in LPS + vehicle treated mice as compared to the sham group, and this response was significantly reduced with lung-delivered rIL-10 therapy at 48-h post-LPS exposure (Fig. 7A,B). Furthermore, vimentin expression, an extracellular matrix protein with expression attributable to mesenchymal cells (i.e. fibroblasts, smooth muscle cells, and endothelial cells) and macrophages, was also strikingly increased post-LPS exposure versus sham mice, and this LPS-induced vimentin expression was also reduced with IL-10 therapy (Fig. 7C,D). These data support that lung-delivered IL-10 therapy post-LPS exposure reduces pro-fibrosis processes to hasten recovery.

Treatment with rIL-10 reduces lung collagen and vimentin expression following a one-time high dose LPS exposure. Representative Masson’s trichrome lung section images from both treatment groups and sham (saline control) (A) with integrated densities from deconvoluted trichrome images as measure of collagen content (B) shown. Confocal images of lung tissues stained for vimentin (green) (C) with nuclei staining by DAPI (blue) and integrated density of vimentin (D) quantified per each mouse. Individual values are averaged from 8 to 10 images per section per mouse. Statistical significance vs. sham (####p < 0.0001). Asterisks (****p < 0.001). Line scale is 100 μm.
Lung-delivered rIL-10 treatment hastens recovery of inflammatory events without compensatory increases in pro-fibrogenic or inflammatory processes at one week following a one-time LPS exposure
In these set of studies, mice were similarly treated once with high dose LPS (10 μg) followed by three doses of rIL-10 or vehicle at 5, 24, and 48 h and then euthanized at 1-week post-LPS exposure to capture potential compensatory adverse versus resolution events with IL-10 therapy. There was no potential compensatory increase in experimental inflammatory indices, cellular recruitment, or pro-fibrogenic processes measured, and instead, several outcomes remained reduced in the IL-10 treatment group, with overall trends toward normative states in both treatment groups (Table 2). To highlight findings, there were significant reductions in the number of BALF lymphocytes and lung total cells and subpopulations of lung activated macrophages, monocyte-macrophages, monocytes, and NK cells with IL-10 therapy. LPS-induced collagen and vimentin expression (Table 2 and Supplemental Fig. 1), weight loss, and serum pentraxin levels were also reduced with IL-10 treatment. There were also no differences in lung inflammatory scores (Table 2 and Supplemental Fig. 1) or IL-10 levels from BALF and lung homogenates between groups. There was no detection of TNF-⍺, IL-6, CXCL1, CXCL2 in BALF or lung homogenates and no detection of IL-13, IL-4, or IL-12p70 in lung homogenates in LPS + vehicle or LPS + rIL-10 treatment groups (data not shown).
Discussion
Occupational exposure-induced lung diseases are the primary cause of occupation-associated illness in the United States28. The rates of acute inhalation incidents are not well known but have been largely attributable to lack of adequate respiratory or ventilation equipment, equipment failure, leaks/spills, accidents, fires, disasters, among others with treatment consisting of supportive care approaches including oxygen, bronchodilators, and sometimes corticosteroids29,30. Using high concentration acute LPS inhalation animal modeling, these current time continuum studies demonstrated a rapid rise/recovery in weight loss, proinflammatory mediators, neutrophils, and lymphocytes, but also a prolonged persistence of activated macrophage and recruited/transitioning monocyte-macrophage subpopulations. Short-term, lung-delivered post-LPS exposure treatment with rIL-10 demonstrated several beneficial responses including blunting weight loss, reduction in pro-inflammatory cytokines/chemokines, reduction in lung neutrophils, reduction in lung collagen and vimentin expression, and striking reduction in recruited/transitioning monocytes-macrophages with corresponding decrease in CCR2+ inflammatory monocytes-macrophages. These findings support that lung-targeted IL-10 therapy could represent a potential therapeutic benefit.
Animal models are increasingly utilized to understand lung recovery/resolution processes as they can be more easily manipulated and tested. In the setting of acute LPS exposure (100 μg), others reported that lung mechanics deteriorate with the first 4 days, fully recover by day 10, and that neutrophils and classic pro-inflammatory mediators resolve quickly31. Those authors had also hypothesized that lymphocytes were the key cellular players in the recovery response, but their investigations failed to show a role for T cells leading them to speculate on the potential role for other cells such as macrophages31. Consistently, our findings demonstrated rapid recovery of TNF-⍺ (by 48 h), IL-6 (by 1 week), and murine neutrophil chemoattractant CXCL1 (by 48 h) with corresponding recovery of neutrophils, T and B lymphocytes, and NK cells by 1 week. However, the monocyte-macrophage subpopulations as defined by CD11c/CD11b expression were profoundly modulated within 24 h post-LPS exposure and these changes persisted for up to 2 weeks, the duration of the current investigations.
Lung monocytes/macrophages are considered guardians of the lung and can alter plasticity and function in response to environmental irritants to regulate lung homeostasis32. Although over-simplified, the classic M1-M2 macrophage dynamic encompasses opposing roles of macrophages in the lung with classically activated M1 macrophages promoting the development of acute lung injury/inflammation while activated M2 macrophages limit inflammation or can promote fibrosis33. The recruited and/or inflammatory lung monocyte-macrophage has emerged as driving the transition from acute inflammation to lung fibrosis34. Our studies strongly support the appearance, progression, and persistence of a recruited/transitional monocyte-macrophage population defined by CD11cintCD11bhi cells first appearing at 24 h post-LPS exposure. Moreover, this subpopulation was profoundly reduced with post-exposure rIL-10 treatment. Recruited inflammatory monocyte-macrophage subpopulations have also been defined by expression of CCR2 as CCR2 is critical for mobilization of monocytes from the bone marrow to various tissues during systemic inflammation35. Repeated exposure to an LPS-enriched (hog barn) organic dust extract has also been previously shown to increase lung CCR2+ monocytes-macrophages36. Here, we demonstrated that rIL-10 treatment reduced tissue CCR2+ monocytes/macrophages following acute LPS exposure. As others have suggested more research is necessary to target pathways to minimize recruitment of these lung monocytes/macrophages (as they are long-lasting) to prevent lung fibrosis34, our data support that exploiting the IL-10 pathway may accomplish this goal. Furthermore, future studies to inform potential therapeutic approaches are also warranted to fully understand the phenotype (e.g. expression of various markers including F4/80, Ly6C, CD64, MHC Class II, CD80, CD86, CD14, CD206, among others), function, and potential transcriptome profile of these macrophage-monocyte subpopulations. Other proof-of principle strategies to inhibit this LPS-induced cell subpopulation include antibody blocking studies (e.g., anti-CCR2, -CD11c, -CD11b), monocyte depletion (e.g., systemic clodronate liposome reduction techniques), or potentially conditional animal knockout approaches to further understand their role.
IL-10 is a well-recognized anti-inflammatory cytokine that is naturally produced following activation of multiple pattern recognition receptor signaling pathways that exerts its functions to restrict excessive inflammatory responses and promote tissue repair37. In general, IL-10 exerts major suppressive effects on myeloid cells by inhibition of pro-inflammatory cytokines, directly inhibits effects on T cells, and has immune stimulatory effects on B cells37. Consistent with prior in vitro macrophage studies demonstrating reduction in organic dust extract-stimulated TNF-⍺ release with rIL-1014, lung levels of LPS-induced TNF-⍺ were essentially abrogated with rIL-10 treatment. The mechanisms of this bi-directional regulation of TNF-⍺ and IL-10 in macrophages following organic dust extract exposures was shown to be attributable to scavenger receptor A signaling pathway induction of protein kinase C zeta resulting in IL-10 release that fed-back to inhibit TNF-⍺ converting enzyme (ADAM-17) activity14. Others have also demonstrated that pretreatment, as opposed to post-exposure treatment, of mice with IL-10 over-expressing mesenchymal stromal/stem cells protected mice against acute lung injury to LPS and this corresponded to increased lung infiltration of IL-10 producing T cells and B cells, less weight loss, and lower TNF production38. Our studies also strongly support that IL-10 suppressed other pro-inflammatory mediators including IL-6 and neutrophil chemoattractants. Corresponding to the decrease in the neutrophil chemoattractants, lung neutrophils were also reduced with rIL-10 treatment. Moreover, rIL-10 therapy reduced numbers of CD8 T cell infiltrates, but increased B cell numbers, consistent with previously described IL-10 actions37.
Although IL-10 is well-recognized as an anti-inflammatory cytokine with ability to facilitate wound healing, it has also been suggested that the IL-10 cytokine family (e.g., IL-10, -19, -20, -22, -24, -26, -28A, -28B, and -29) also govern fibrogenesis39. Consistent with studies demonstrating that IL-10 inhibits the proliferation and collagen synthesis of myofibroblasts39, these current studies also support that lung-delivered IL-10 therapy reduced LPS-induced collagen content, and moreover, LPS-induced lung vimentin expression was also reduced. With extending studies to 1-week post-LPS exposure with and without short-term IL-10 therapy, studies did not suggest compensatory adverse or pro-fibrotic processes but supported hastening of recovery/resolution. The current studies only employed lung-directed IL-10 therapy for 2 days, and therefore, caution is necessary in future studies that might explore longer duration of treatment or use in other disease scenarios due to its potential fibrogenic properties.
Endotoxin exposure (particularly systemic) induces cachexia and weight loss and can contribute to features of the metabolic syndrome40. Acute LPS inhalation induced weight loss of 10–20% within the first few days with return to normal by one week. IL-10 treatment blunted LPS-induced weight loss, and this was correspondingly associated with increased blood glucose levels. Stress-related hyperglycemia is associated with increased morbidity and mortality in critical illness41,42, and IL-10 treatment has been shown to attenuate and prevent insulin resistance, improve insulin signaling, and beneficially affect peripheral glucose metabolism43,44. An explanation for the increase in blood glucose levels with rIL-10 treatment at day 2 is that it does not reflect a negative outcome, but mostly likely reflects weight differences between the 2 groups.
Although our study suggests a potential novel, lung-targeted, short-term approach to using rIL-10 therapy following airway injury/inflammation (Fig. 8), there are a few limitations that should be addressed. While we utilized LPS as a representative inflammatory agent, there are a wide number of other agents in our exposome that could be explored including industrial chemical toxins (e.g., chlorine, phosgene, hydrogen sulfide, and ammonia), heavy metals (e.g., cadmium, mercury), microbial agents (e.g., gram positive bacteria, mold spores), other real-world/complex exposures (e.g., burn pit exposures, wild-fire smoke, organic dust), and even potentially overwhelming lung infectious process as seen in viral (e.g., SARS-CoV-2) infections or bacterial infections. Whereas systemic delivery of IL-10 as treatment in various chronic diseases has shown partial efficacy, long-term use has been associated with anemia and thrombocytopenia17. We did not evaluate extrapulmonary organs (e.g., spleen bone marrow, liver, kidney, etc.), which should be investigated in future studies to determine potential effects. However, these potential adverse effects may be negated by short-term use applications as in settings of acute exposures (e.g., first responders, military, disasters). Next, pulmonary drug delivery options are currently limited to metered dose inhalers, nebulizers, and dry powder inhalers with recent efforts investigating novel nanoparticle formulations to maximize lung deposition and minimize pulmonary clearances of drugs45. Thus, future studies investigating lung-targeted IL-10 therapy should also focus on optimal lung-delivery strategies.

Schematic of lung-delivered rIL-10 treatment effects upon acute LPS-induced airway inflammatory consequences. Post-LPS exposure induced lung pro-inflammatory cytokine/chemokine release with corresponding recruitment of lung monocyte-macrophage subpopulation (CD11cintCD11b+) and/or CCR2 + monocytes/macrophages and neutrophils is reduced with rIL-10 therapy (Created with BioRender.com).
Conclusion
In conclusion, the present findings strongly support the potential use of lung-delivered therapy of IL-10 in human applications, particularly in a short-term approach following untoward and extreme occupational and/or environmental inhalation exposures.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files]. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- LPS:
-
Lipopolysaccharides
- IL:
-
Interleukin
- R:
-
Recombinant
- BALF:
-
Bronchoalveolar lavage fluid
- PBS:
-
Phosphate buffered saline
- TNF-α:
-
Tumor necrosis factor-alpha
- AREG:
-
Amphiregulin
- MDD:
-
Minimal detectable dose
- SEM:
-
Standard error of mean
- ANOVA:
-
A one-way analysis of variance
References
Glaser, M. S. et al. Estimating the time interval between exposure to the World Trade Center disaster and incident diagnoses of obstructive airway disease. Am. J. Epidemiol. 180, 272–279 (2014).
Niles, J. K. et al. The respiratory pyramid: From symptoms to disease in World Trade Center exposed firefighters. Am. J. Ind. Med. 56, 870–880 (2013).
Guidotti, T. L., Prezant, D., de la Hoz, R. E. & Miller, A. The evolving spectrum of pulmonary disease in responders to the World Trade Center tragedy. Am. J. Ind. Med. 54, 649–660 (2011).
Kim, K. H., Kabir, E. & Jahan, S. A. Airborne bioaerosols and their impact on human health. J. Environ. Sci. (China) 67, 23–35 (2018).
Armstrong, M. T., Rickles, F. R. & Armstrong, P. B. Capture of lipopolysaccharide (endotoxin) by the blood clot: A comparative study. PLoS ONE 8, e80192 (2013).
Wallace, R. J. et al. Risks associated with endotoxins in feed additives produced by fermentation. Environ. Health 15, 5 (2016).
Thilsing, T. et al. Dust, endotoxin, fungi, and bacteria exposure as determined by work task, season, and type of plant in a flower greenhouse. Ann. Occup. Hyg. 59, 142–157 (2015).
Park, S. M., Kwak, Y. S. & Ji, J. G. The effects of combined exercise on health-related fitness, endotoxin, and immune function of postmenopausal women with abdominal obesity. J. Immunol. Res. 2015, 830567 (2015).
Paudyal, P., Semple, S., Niven, R., Tavernier, G. & Ayres, J. G. Exposure to dust and endotoxin in textile processing workers. Ann. Occup. Hyg. 55, 403–409 (2011).
Mitchell, D. C. et al. Particulate matter, endotoxin, and worker respiratory health on large Californian dairies. J. Occup. Environ. Med. 57, 79–87 (2015).
Cyprowski, M., Sobala, W., Buczynska, A. & Szadkowska-Stanczyk, I. Endotoxin exposure and changes in short-term pulmonary function among sewage workers. Int. J. Occup. Med. Environ. Health 28, 803–811 (2015).
Davidson, M. E. et al. Personal exposure of dairy workers to dust, endotoxin, muramic acid, ergosterol and ammonia on large-scale dairies in the high plains western United States. J. Occup. Environ. Hyg. 15, 182–193 (2017).
LeVan, T. D. et al. Relationship of systemic IL-10 levels with proinflammatory cytokine responsiveness and lung function in agriculture workers. Respir. Res. 19, 1–7 (2018).
Wyatt, T. A. et al. Organic dust-induced lung injury and repair: Bi-directional regulation by TNFalpha and IL-10. J. Immunotoxicol. 17, 153–162 (2020).
Quinn, T. J., Taylor, S., Wohlford-Lenane, C. L. & Schwartz, D. A. IL-10 reduces grain dust-induced airway inflammation and airway hyperreactivity. J. Appl. Physiol. 88, 173–179 (2000).
Garantziotis, S. et al. Leukocyte-derived IL-10 reduces subepithelial fibrosis associated with chronically inhaled endotoxin. Am. J. Respir. Cell Mol. Biol. 35, 662–667 (2006).
Fioranelli, M. & Roccia, M. G. Twenty-five years of studies and trials for the therapeutic application of IL-10 immunomodulating properties. From high doses administration to low dose medicine new paradigm. J. Integr. Cardiol. 1, 2–6 (2014).
Nelson, A. J. et al. Sex differences impact the lung-bone inflammatory response to repetitive inhalant lipopolysaccharide exposures in mice. J. Immunotoxicol. 15, 73–81 (2018).
Poole, J. A. et al. Combined collagen-induced arthritis and organic dust-induced airway inflammation to model inflammatory lung disease in rheumatoid arthritis. J. Bone Miner. Res. 34, 1733–1743 (2019).
Poole, J. A. et al. A role for B cells in organic dust induced lung inflammation. Respir. Res. 18, 214 (2017).
Poole, J. A. et al. alphabeta T cells and a mixed Th1/Th17 response are important in organic dust-induced airway disease. Ann. Allergy Asthma Immunol. 109, 266-273.e262 (2012).
Mikuls, T. R. et al. The impact of airborne endotoxin exposure on rheumatoid arthritis-related joint damage, autoantigen expression, autoimmunity, and lung disease. Int. Immunopharmacol. 100, 108069 (2021).
Robbe, P. et al. Distinct macrophage phenotypes in allergic and nonallergic lung inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, L358-367 (2015).
Lafuse, W. P. et al. Identification of an increased alveolar macrophage subpopulation in old mice that displays unique inflammatory characteristics and is permissive to mycobacterium tuberculosis infection. J. Immunol. 203, 2252–2264 (2019).
Gaurav, R. et al. IL-33 depletion in COVID-19 lungs. Chest 160, 1656–1659 (2021).
Poole, J. A. et al. Increased susceptibility to organic dust exposure-induced inflammatory lung disease with enhanced rheumatoid arthritis-associated autoantigen expression in HLA-DR4 transgenic mice. Respir. Res. 23, 160 (2022).
Chen, Y., Yu, Q. & Xu, C.-B. A convenient method for quantifying collagen fibers in atherosclerotic lesions by ImageJ software. Int. J. Clin. Exp. Med. 10, 14904–14910 (2017).
De Matteis, S. et al. Current and new challenges in occupational lung diseases. Eur. Respir. Rev. 26, 170080 (2017).
Mirabelli, M. C. et al. Inhalation incidents and respiratory health: Results from the European Community Respiratory Health Survey. Am. J. Ind. Med. 52, 17–24 (2009).
Gorguner, M. & Akgun, M. Acute inhalation injury. Eur. J. Med. 42, 28–35 (2010).
Verjans, E. et al. Initiation of LPS-induced pulmonary dysfunction and its recovery occur independent of T cells. BMC Pulm. Med. 18, 174 (2018).
Puttur, F., Gregory, L. G. & Lloyd, C. M. Airway macrophages as the guardians of tissue repair in the lung. Immunol. Cell Biol. 97, 246–257 (2019).
Laskin, D. L., Malaviya, R. & Laskin, J. D. Role of macrophages in acute lung injury and chronic fibrosis induced by pulmonary toxicants. Toxicol. Sci. 168, 287–301 (2019).
Misharin, A. V. et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 214, 2387–2404 (2017).
Tsou, C. L. et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Investig. 117, 902–909 (2007).
Gaurav, R. et al. High-throughput analysis of lung immune cells in a combined murine model of agriculture dust-triggered airway inflammation with rheumatoid arthritis. PLoS ONE 16, e0240707 (2021).
Ouyang, W. & O’Garra, A. IL-10 family cytokines IL-10 and IL-22: From basic science to clinical translation. Immunity 50, 871–891 (2019).
Wang, C. et al. Interleukin-10-overexpressing mesenchymal stromal cells induce a series of regulatory effects in the inflammatory system and promote the survival of endotoxin-induced acute lung injury in mice model. DNA Cell. Biol. 37, 53–61 (2018).
Sziksz, E. et al. Fibrosis related inflammatory mediators: Role of the IL-10 cytokine family. Mediat. Inflamm. 2015, 764641 (2015).
Jialal, I. & Rajamani, U. Endotoxemia of metabolic syndrome: A pivotal mediator of meta-inflammation. Metab. Syndr. Relat. Disord. 12, 454–456 (2014).
Eslami, S., Taherzadeh, Z., Schultz, M. J. & Abu-Hanna, A. Glucose variability measures and their effect on mortality: A systematic review. Intensive Care Med. 37, 583–593 (2011).
Edriss, H., Selvan, K., Sigler, M. & Nugent, K. Glucose levels in patients with acute respiratory failure requiring mechanical ventilation. J. Intensive Care Med. 32, 578–584 (2017).
Hong, E. G. et al. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 58, 2525–2535 (2009).
Dagdeviren, S. et al. IL-10 prevents aging-associated inflammation and insulin resistance in skeletal muscle. FASEB J. 31, 701–710 (2017).
Mangal, S., Gao, W., Li, T. & Zhou, Q. T. Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: Challenges and opportunities. Acta Pharmacol. Sin. 38, 782–797 (2017).
Acknowledgements
The authors acknowledge members of the Tissue Sciences Facility at the Department of Pathology and Microbiology (University of Nebraska Medical Center, Omaha, NE, USA) for assistance with tissue processing and staining. We thank Victoria B. Smith, Holly Britton, and Craig Semerad in the Flow Cytometry Research Core Facility at the University of Nebraska Medical Center for aiding with flow cytometry studies. Schematics were created from www.BioRender.com. We also thank Lisa Chudomelka for manuscript submission assistance.
Funding
Department of Defense #PR200793 (JAP). National Institute for Occupational Safety and Health Grant U54OH010162 (JAP, TAW, RG) and R01OH012045 (JAP). Central States Center of Agricultural Safety and Health (CS-CASH, U54OH010162). TAW is the recipient of a Research Career Scientist Award (IK6 BX003781) from the Department of Veterans Affairs. Fred & Pamela Buffett Cancer Center [Flow Cytometry Research Facility and Tissue Sciences Facility] Shared Resource, supported by the National Cancer Institute award P30CA036727.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.A.P., T.A.W., A.J.N., R.G., D.J.R. Methodology: J.A.P., R.G., A.S., A.J.N., A.G., T.A.W., D.J.R. Investigation: J.A.P., R.G., A.S., A.J.N., A.G., T.A.W., D.J.R. Visualization: J.A.P., T.A.W., A.J.N., R.G., A.S., A.N., D.J.R. Funding acquisition: J.A.P., T.A.W., D.J.R. Project administration: J.A.P., T.A.W., D.J.R. Supervision: J.A.P., T.A.W. Writing—original draft: J.A.P., T.A.W., A.J.N., R.G. Writing—review and editing: J.A.P., T.A.W., R.G., A.S., A.J.N., A.G., D.J.R.
Corresponding author
Ethics declarations
Competing interests
JAP has received research support from AstraZeneca (no monies) and is involved in clinical research studies with Takeda and GlaxoSmithKline, all unrelated to this project. TAW, RG, AS, AJN, AG, DJR do not have any competing interests to declare.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Poole, J.A., Gaurav, R., Schwab, A. et al. Post-endotoxin exposure-induced lung inflammation and resolution consequences beneficially impacted by lung-delivered IL-10 therapy. Sci Rep 12, 17338 (2022). https://doi.org/10.1038/s41598-022-22346-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-22346-2
This article is cited by
-
Targeting transitioning lung monocytes/macrophages as treatment strategies in lung disease related to environmental exposures
Respiratory Research (2024)
-
Mechanistic and Therapeutic Approaches to Occupational Exposure-Associated Allergic and Non-Allergic Asthmatic Disease
Current Allergy and Asthma Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
