
Abstract
Fetal aberrant right subclavian artery (ARSA) is a relatively common sonographic finding. Congenital heart disease (CHD) is the most common structural abnormality in patients with ARSA. We aimed to assess the prevalence of genetic abnormalities, particularly sequence variants, in fetuses with CHD and ARSA. By clinical phenotyping and genomic sequencing, we retrospectively reviewed all fetuses with a prenatal diagnosis of CHD combined with ARSA at a single center. As a result, we identified 30 fetuses with ARSA combined with CHD, with conotruncal anomalies being the most common (n = 12, 40%), followed by left ventricular outflow tract obstruction (n = 6, 20%) and atrioventricular septal defects (n = 6, 20%). Overall, 18 (60%) cases had a genetic diagnosis. Copy number variation sequencing analysis identified six (20%) fetuses with aneuploidy and seven (23%) with pathogenic copy-number variants. Whole-exome sequencing (WES) analysis of the remaining 17 cases revealed diagnostic genetic variants in five (29%) cases, indicating that the diagnostic yield of WES for the entire cohort was 17% (5/30). Our findings reveal the high burden of genetic abnormalities in fetal CHD with ARSA. Single-gene disorders contribute substantially to the genetic etiology of fetal CHD with ARSA.
Similar content being viewed by others
Introduction
Aberrant right subclavian artery (ARSA) is the most common aortic branching variant. In the left aortic arch with ARSA, the right subclavian artery arises independently from the descending aorta instead of its typical origin from the brachiocephalic trunk1. The incidence of ARSA in the general population is estimated to be 1.2% postnatally and 0.6–1.5% prenatally1,2.
Prenatal demonstration of ARSA raises a concern for genetic aberrations. The association between ARSA and genetic abnormalities in the general population has been widely studied and reported. Most studies found that isolated ARSA probably had no clinical significance and did not serve as a marker for an invasive prenatal chromosomal test3,4,5,6. However, the genetic testing used in these studies, including karyotyping and chromosomal microarray analysis, can only identify chromosomal abnormalities and cannot detect single-gene disorders. The contribution of single-gene disorders to ARSA is unknown but is potentially substantial.
On the other hand, although ARSA is associated with congenital heart disease (CHD), with a significantly higher incidence in the CHD population than in the general population1,7, literature on the comprehensive assessment of genetic abnormalities in fetal CHD with ARSA is lacking. In particular, no data exist regarding the application of exome sequencing to the prenatal phenotype of coexistence of CHD with ARSA.
CHD is the most common congenital defect worldwide, occurs in 1% of live births, and is associated with significantly high perinatal morbidity and mortality8,9. Identifying a genetic diagnosis of fetal CHD can inform prenatal management, optimise post-natal outcomes, and aid in the counselling of parents in both index and subsequent pregnancies10,11.
This study aims to estimate the prevalence of genetic abnormalities in fetuses with ARSA and CHDs, particularly the contribution of single-gene disorders, by assessing results from genomic sequencing, including copy number variation sequencing (CNV-seq) and whole-exome sequencing (WES). This study will also evaluate the potential diagnostic yield of WES for fetal CHD with ARSA.
Methods
Participant recruitment
We retrospectively reviewed our experience with fetuses diagnosed with ARSA coexisting with other congenital heart defects at Beijing Anzhen Hospital, Capital Medical University, from July 2015 and July 2020. Routine fetal ultrasound anatomy scans were performed for pregnant women. If CHD was suspected, echocardiography was subsequently performed in Beijing Anzhen Hospital, a regional and national referral center. Parents who opted for terminating their pregnancies and genetic testing were offered participation in this study. Samples of father-mother-fetus trios were collected for sequencing. Prenatal medical records regarding gestational age at diagnosis, gender, family history, cardiac and extracardiac abnormalities, and the results of genetic testing were collected.
Fetal ultrasound and echocardiography examination
All ultrasound examinations were performed by experienced operators using the General Electric Voluson E8 ultrasound system with transabdominal 2- to 4-MHz curvilinear transducers (GE Healthcare Ultrasound, Milwaukee, WI, USA) or the Aloka SSD ultrasound system (Aloka, Tokyo, Japan) using transabdominal 3- to 6-MHz curvilinear transducers. A complete fetal echocardiographic examination, including twodimensional (2D), M-mode, color, and pulse Doppler echocardiography, was performed according to the American Society of Echocardiography guidelines and standards for performance of the fetal echocardiogram to ascertain the presence of CHD12.
Copy-number variation sequencing
Both CNV-seq and WES were performed as described previously13,14. We performed CNV-seq as follows. RNA-free genomic DNA isolated from the umbilical cord was used for library construction. The DNA library was sequenced on Illumina Hiseq 4000 or Illumina Novaseq. Burrows-Wheeler Aligner15 (BWA) was used to compare and analyze the sequence reading information with the human reference genome (hg19/GRCh37) to obtain the bioinformatics results and determine the existence of chromosomal aneuploidy variation and CNVs. According to the American College of Medical Genetics (ACMG) standards and guidelines for interpretation of CNVs, CNVs were classified into five categories: pathogenic, likely pathogenic, uncertain significance, likely benign, or benign16.
Whole-exome sequencing
WES was performed on DNA extracted from the fetal umbilical cord or parental blood13,14. The captured library was sequenced on Illumina Hiseq 4000 or Illumina Novaseq. The sequencing reads were aligned to the NCBI human reference genome (hg19/GRCh37) using BWA. Variant calling and quality filtering of variants were performed with Genome-Analysis-Toolkit17. ANNOVAR was used for annotation of genes and functional variant effects18. Pathogenicity of variants was determined according to current ACMG guidelines19. Variants classified as pathogenic or likely pathogenic were considered positive genetic results. Sanger sequencing was used to validate the presence of positive genetic results.
Statistical analysis
Categorical variables are presented as frequencies (percentage) and were compared using the Pearson χ2 test or Fisher’s exact test. Statistical analysis was performed using SPSS version 23 (SPSS, Chicago, IL, USA). Two-sided P values < 0.01 were considered significant.
Ethical approval
This retrospective study was approved by the institutional review board of the Medical Ethics Committee of Beijing AnZhen Hospital. All parents agreed to participate in this study and provided signed informed consent.
Results
Cohort characteristics
The demographics, cardiac and extra-cardiac characteristics, and sequencing information of the entire cohort are summarized in Table 1. This cohort was composed of 14 males and 16 females. The median maternal age was 29 (range 20–38) years, and the fetuses were assessed at a median gestational age of 24 (range 16–29) weeks. Twelve (40%) cases had ultrasound-detected extra-cardiac malformations and 18 (60%) were isolated CHD. Most of our CHD patients had a complex disease consisting of more than one distinct anomaly. Patients were categorized to the single-most relevant anomaly to limit overlap between groups. The commonest CHD in this study was conotruncal anomalies, followed by left ventricular outflow tract obstruction (LVOTO) then atrioventricular septal defects (AVSD) (Table 1).
Genetic results
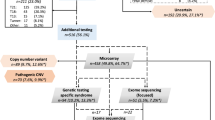
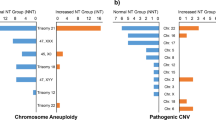
The flowchart of genetic analysis progression is shown in Fig. 1. Overall, a genetic cause was identified in 18(60%) cases, comprising six (20%) cases with aneuploidies, seven (23%) cases with pathogenic CNVs, and five (17%) cases with sequence variants, respectively. The genotype and phenotype information of the 18 cases are shown in Tables 2, 3 and 4. Four of the six aneuploidy cases were trisomy 21 (T21), of which three had conotruncal anomalies (Table 2). Among the seven cases with pathogenic CNVs, four were 22q11.2 deletion syndrome (22q11.2DS), and the corresponding cardiac phenotype was all conotruncal anomaly (Table 3). In addition, there were two cases with unbalanced translocation whose parents were balanced translocation carriers. We further found the diagnostic sequence variants in five cases through WES analysis (Table 4), accounting for 29% of those without chromosome abnormalities and 17% of the entire cohort. Among the five cases of sequence variation, four cases occurred in the KMT2D gene.

Flowchart showing the genetic testing and findings of next-generation sequencing analysis in the study of fetal congenital heart disease with aberrant right subclavian artery. In this cohort, ‘syndromic’ and ‘non-syndromic’ refer to presence and absence, respectively, of fetal structural abnormalities detected on ultrasound. CNV copy-number variant, CNV-seq CNV sequencing, WES whole-exome sequencing.
When considering the associated heart defects with ARSA, the prevalence of a genetic diagnosis was highest in AVSD (5/6, 83%), followed by conotruncal defects (8/12, 67%) then LVOTO (2/6, 33%). Genetic causes were identified more frequently in fetuses with extra-cardiac malformations (8/12; 66.7%) than in those without extra-cardiac malformations (10/18; 55.6%). However, the difference was not significant (Fisher's exact test; p = 0.7).
Besides the findings presented above, we also detected two uncertain significance variants and two novel candidate genes for CHD (Supplementary Table 1). All these variants met the following conditions: (1) were absent in the gnomAD database, (2) were LoF variants occurring in the LoF-intolerant genes (pLI score > 0.99)20 or non-synonymous variants predicted to be damaging by all silico predictions (SIFT, PolyPhen-2, MutationTaster). Further research is required to evaluate the pathogenicity of these variants.
Discussion
This study represents the first cohort to assess the sequence variants in fetuses with CHD and ARSA. The diagnostic yield of 60% in our study is substantially higher than that of 10–30% in unselected CHD studies21,22,23,24, reflecting the significant burden of genetic abnormalities underlying CHD complicated with ARSA. Our findings show that single-gene disorders, especially KMT2D gene abnormality, also play an essential role in CHD combined with ARSA; AVSD and conotruncal anomalies are definite signs for genetic testing among the cardiac defects coexistence with ARSA. Its clinical implication is that prenatal WES should be performed to identify genetic diagnoses to facilitate perinatal decision-making and management when conventional tests (karyotype and microarray) are not diagnostic.
Our study's significant strengths are combining CNV-seq and WES to identify genetic diagnoses, including chromosomal abnormalities and sequence variants. In contrast, all previous studies used karyotype analysis or chromosomal microarray analysis, focusing only on chromosomal abnormalities3,25. This genetic testing strategy allowed us to reveal a more comprehensive genetic profile of ARSA. More importantly, we found for the first time that nearly 30% of patients without chromosomal abnormalities carried diagnostic sequence variants. Another strength of this study is that it focused on a specific phenotype, namely fetal CHD with ARSA, rather than unselected CHD or ARSA. While narrow in its focus, our approach is more effective and accurate in assessing the predictive value of ARSA for genetic abnormalities in fetuses with CHD. The yield of 60% in our series is substantially higher than the 10–30% yield in studies of unselected CHD21,22,23,24, suggesting that demonstration of ARSA in pregnancies with CHD may serve as a solid red flag for genetic abnormalities. Pregnancies with CHD and ARSA should be referred for invasive prenatal testing.
Although several recent studies have researched aneuploidy or 22q11.2DS in ARSA cases3,4,5,6,25,26,27,28, evidence on the prevalence of chromosome abnormalities and sequence variants in ARSA with specific heart defects is limited. In our study, the commonest cardiac phenotype associated with ARSA was conotruncal anomalies, followed by LVOTO and AVSD. This association is in line with the previous literature1.
The prevalence of genetic causes varied significantly between CHD subgroups. The AVSD group was most associated with a genetic diagnosis, and the total positive rate was 83% (5/6), among which T21 was the most common. The conotruncal defects group was also frequently accompanied by a genetic diagnosis, of which 22q11.2DS is the most common. On the other hand, genetic diagnoses were rarely encountered among ARSA fetuses with a diagnosis of LVOTO. On the other hand, in terms of genotype, the most common chromosomal abnormalities in this study were T21 and 22q11.2DS, respectively. The association between chromosomal abnormalities, especially T21 and 22q11.2DS, and CHD is well documented in the literature25. This study also identified two fetuses with unbalanced chromosomal rearrangement and confirmed their parents to be carriers of a balanced translocation. Notably, the offspring of balanced translocation carriers are at high risk of unbalanced rearrangement, and they should consider assisted reproduction.
The prevalence of sequence variants (29.4%) was higher in our fetal CHD cohort than in the previous fetal CHD cohort. Several studies have reported diagnostic rates of sequence variants in fetal CHD ranging from 5.2 to 13.5%24,29,30,31. This difference reflects the population selection bias, as we only included fetal CHD with ARSA, while other studies included all cases with and without ARSA. Therefore, the estimated diagnostic yield in our cohort may be more accurate and more suitable for prenatal counseling for fetal CHD associated with ARSA. The coexistence of ARSA in fetal CHD can be a crucial anatomic marker for genetic diagnosis. Another interesting finding was that four of the five sequence variants occurred in KMT2D, the causative gene of Kabuki syndrome-1 (OMIM:147920). This high proportion of KMT2D mutations may reflect the high incidence of KMT2D mutations in CHD because an increasing number of exome sequencing studies have shown that KMT2D is the most common mutated gene in CHD14,32. More importantly, this finding suggests that ARSA may be an indicator of Kabuki syndrome. However, more evidence is needed to support this hypothesis.
Limitations
Major limitations of the study include the small sample size, its retrospective observational nature, and the absence of a control group with fetal CHD without ARSA. We acknowledge that there may be a selection bias because this is a small series study from a tertiary hospital. Our center is one of the major referral hospitals for fetal CHD. Therefore the most severe cardiac malformation, such as conotruncal anomalies and AVSD, may be found more frequently.
Conclusions
In conclusion, we used CNV-SEq and WES to comprehensively investigate genetic abnormalities, including aneuploidy, CNV, and single-gene disorder, in a cohort of fetal CHD with ARSA. Our results indicate a high burden of genetic abnormalities in fetal CHD with ARSA and suggest that ARSA in pregnancies with CHD may serve as a solid red flag for genetic abnormalities. A single-gene disorder diagnosis would not have been found in about 30% of cases without chromosome abnormalities if WES had not been performed. In genotype-phenotypic association, KMT2D is the most frequently mutated gene, while AVSD and conotruncal anomalies are most commonly associated with genetic abnormalities. However, future research, which offers WES in prospective extensive cohort studies on fetal CHD with ARSA, is needed to obtain more reliable estimates.
Data availability
All datasets generated for this study are included in the article/Supplementary Material. This study is compliant with the “Guidance of the Ministry of Science and Technology (MOST) for the Review and Approval of Human Genetic Resources”, which requires formal approval for the export of human genetic material or data from China. The phenotypic data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Zapata, H., Edwards, J. E. & Titus, J. L. Aberrant right subclavian artery with left aortic arch: Associated cardiac anomalies. Pediatr. Cardiol. 14, 159–161 (1993).
De León-Luis, J. et al. Second-trimester fetal aberrant right subclavian artery: Original study, systematic review and meta-analysis of performance in detection of down syndrome. Ultrasound Obst. Gyn. 44, 147–153 (2014).
Sagi-Dain, L. et al. Microarray analysis has no additional value in fetal aberrant right subclavian artery: Description of 268 pregnancies and systematic literature review. Ultrasound Obst. Gyn. 53, 810–815 (2019).
Willruth, A. et al. Fetal aberrant right subclavian artery (ARSA)—a potential new soft marker in the genetic scan?. Ultraschall Med. Eur. J. Ultrasound 33, E114–E118 (2012).
Scala, C. et al. Aberrant right subclavian artery in fetuses with down syndrome: A systematic review and meta-analysis. Ultrasound Obst. Gyn. 46, 266–276 (2015).
Fehmi Yazıcıoğlu, H. et al. Aberrant right subclavian artery in down syndrome fetuses. Prenatal Diag. 33, 209–213 (2013).
Tawfik, A. M., Sobh, D. M., Ashamallah, G. A. & Batouty, N. M. Prevalence and types of aortic arch variants and anomalies in congenital heart diseases. Acad. Radiol. 26, 930–936 (2019).
Zaidi, S. & Brueckner, M. Genetics and genomics of congenital heart disease. Circ. Res. 120, 923–940 (2017).
Liu, Y. et al. Global birth prevalence of congenital heart defects 1970–2017: Updated systematic review and meta-analysis of 260 studies. Int. J. Epidemiol. 48, 455–463 (2019).
Petracchi, F., Sisterna, S., Igarzabal, L. & Wilkins Haug, L. Fetal cardiac abnormalities: Genetic etiologies to be considered. Prenatal Diag. 39, 758–780 (2019).
Best, S. et al. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn. 38, 10–19 (2018).
Rychik, J. et al. American Society of Echocardiography Guidelines and Standards for performance of the fetal Echocardiogram. J. Am. Soc. Echocardiog. 17, 803–810 (2004).
Sun, H. et al. Genetics and clinical features of noncompaction cardiomyopathy in the fetal population. Front. Cardiovasc. Med. 7, 617561 (2021).
Sun, H. et al. Contribution of single-gene defects to congenital cardiac left-sided lesions in the prenatal setting. Ultrasound Obst. Gyn. 56, 225–232 (2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257 (2020).
McKenna, A. et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing DATA. Nucleic Acids Res. 38, e164 (2010).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
De Backer, J. et al. Genetic counselling and testing in adults with congenital heart disease: A consensus document of the ESC Working Group of Grown-Up Congenital Heart Disease, the ESC Working Group On Aorta and Peripheral Vascular Disease and the European Society of Human Genetics. Eur. J. Prev. Cardiol. 27, 1423–1435 (2020).
Mital, S. et al. Enhancing literacy in cardiovascular genetics: A scientific statement from the American Heart Association. Circ. Cardiovasc. Genet. 9, 448–467 (2016).
Cowan, J. R. & Ware, S. M. Genetics and genetic testing in congenital heart disease. Clin. Perinatol. 42, 373–393 (2015).
van Nisselrooij, A. E. L. et al. The prevalence of genetic diagnoses in fetuses with severecongenital heart defects. Genet. Med. 22, 1206–1214 (2020).
Maya, I. et al. Chromosomal microarray analysis in fetuses with aberrant right subclavian artery. Ultrasound Obst. Gyn. 49, 337–341 (2017).
Svirsky, R., Reches, A., Brabbing-Goldstein, D., Bar-Shira, A. & Yaron, Y. Association of aberrant right subclavian artery with abnormal karyotype and microarray results. Prenatal Diag. 37, 808–811 (2017).
Pico, H. et al. Prenatal associated features in fetuses diagnosed with an aberrant right subclavian artery. Fetal Diagn. Ther. 40, 187–194 (2016).
Rembouskos, G. et al. Aberrant right subclavian artery (ARSA) in unselected population at first and second trimester ultrasonography. Prenatal Diag. 32, 968–975 (2012).
Mone, F. et al. COngenital heart disease and the diagnostic yield with exome sequencing (CODE) study: Prospective cohort study and systematic review. Ultrasound Obst. Gyn. 57, 43–51 (2021).
Lord, J. et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): A cohort study. Lancet 393, 747–757 (2019).
Petrovski, S. et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: A prospective cohort study. Lancet 393, 758–767 (2019).
Jin, S. C. et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 49, 1593–1601 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82170301 to Yihua He and No. 82100322 to Tong Yi.), Beijing Advanced Innovation Center for Big Data-based Precision Medicine, Capital Medical University, Beijing, 100069, China (No. PXM2021_014226_000026 to Hongjia Zhang) and Beijing Lab for Cardiovascular Precision Medicine, Beijing, China (No. PXM2020_014226_000017_00377132_FCG to Hongjia Zhang).
Author information
Authors and Affiliations
Contributions
H.Z. and Y.H. designed the study. X.H., T.Y., J.W., Z.C., X.Z., X.G., J.H., Y.Z., L.S., X.L., S.Z. and L.H. collected the study materials or samples. H.S. and T.Y. completed the sequencing experiments. H.S. and X.H. collected and aggregated the data. H.S. analyzed and interpreted the data. H.S. and X.H. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, H., Han, L., Hao, X. et al. Genetic abnormalities in fetal congenital heart disease with aberrant right subclavian artery. Sci Rep 12, 15899 (2022). https://doi.org/10.1038/s41598-022-20037-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-20037-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
