
Abstract
Purpose
Congenital heart defects (CHD) are associated with genetic syndromes. Rapid aneuploidy testing and chromosome microarray analysis (CMA) are standard care in fetal CHD. Many genetic syndromes remain undetected with these tests. This cohort study aims to estimate the frequency of causal genetic variants, in particular structural chromosome abnormalities and sequence variants, in fetuses with severe CHD at mid-gestation, to aid prenatal counselling.
Methods
Fetuses with severe CHD were extracted from the PRECOR registry (2012–2016). We evaluated pre- and postnatal genetic testing results retrospectively to estimate the frequency of genetic diagnoses in general, as well as for specific CHDs.
Results
919 fetuses with severe CHD were identified. After exclusion of 211 cases with aneuploidy, a genetic diagnosis was found in 15.7% (111/708). These comprised copy number variants in 9.9% (70/708). In 4.5% (41/708) sequence variants were found that would have remained undetected with CMA. Interrupted aortic arch, pulmonary atresia with ventricular septal defect and atrioventricular septal defect were most commonly associated with a genetic diagnosis.
Conclusion
In case of normal CMA results, parents should be offered exome sequencing sequentially, if time allows for it, especially if the CHD is accompanied by other structural malformations due to the large variety in genetic syndromes.
Similar content being viewed by others

Introduction
Congenital heart defects (CHDs) are the most common congenital birth defects with a prevalence of 5–8 per 1.000 liveborns.1 Approximately a third of these cases involve severe CHDs.2 Although survival rates have increased significantly over the past decades, the extent of (disease-related) morbidity is considerable and life-expectance remains reduced.3
It is well known that aneuploidies (numerical chromosome abnormalities), such as trisomy 13, 18, 21 and Turner syndrome, are associated with CHDs.4,5 Submicroscopic deletions or duplications, commonly referred to as copy number variants (CNVs), have been reported in approximately 10–15% of children with CHD.6,7 As these CNVs can have a major impact on development, quality of life and life expectancy of children with CHDs,7,8 chromosome microarray analysis (CMA) is the standard of care in the fetal setting in most developed countries.9 Nevertheless, it is not uncommon in cases with CHD, that a genetic syndrome is diagnosed after birth or during childhood, as sequence variants remain undetected with CMA.
Although CHDs have been described as part of many genetic syndromes, there are only a few cohort studies available that report on genetic diagnoses (chromosome abnormalities or sequence variants) in fetuses diagnosed with a CHD. The studies that have been performed in fetal cohorts are outdated, as they mainly focus on aneuploidy.10,11,12,13 This restricts prenatal counseling, because currently available data are mostly based on postnatal studies. The prevalence of genetic diagnoses is expected to be lower in the postnatal cohorts, as cases with termination of pregnancy (TOP), intra uterine fetal demise or early neonatal death are often not included in these cohorts.5 More importantly postnatal sequencing is mostly requested due to the evolving clinical phenotype, which makes it difficult to estimate a true prenatal prevalence of genetic syndromes at mid-gestation.
Antenatal knowledge on the prevalence of genetic variants, which may either be considered the genetic cause for the CHD or a secondary finding, can improve prenatal counseling in cases with severe CHDs. Accurate figures on the occurrence of these genetic diagnoses will aid professionals to make decisions regarding the use of currently available additional tests, such as exome sequencing (ES) or genetic testing for specific genetic syndromes.
This study aims to provide a conservative estimate of the prevalence of chromosome abnormalities and sequence variants in fetuses with severe CHDs, in particular after an aneuploidy is excluded, by assessing results from both pre- and postnatal genetic testing. The potential diagnostic yield of ES for fetal CHDs, and factors that potentially increase the chance of an underlying genetic diagnosis, will be evaluated as well.
Materials and Methods
In the northwestern region of the Netherlands, the care for children with CHDs is centralized in three tertiary referral centers in Amsterdam and Leiden. All fetuses and infants diagnosed with a severe CHD within this region have been registered in the PRECOR registry since 2002. Severe CHD was defined as the need for surgery or therapeutic intervention in the first year of life. Data collection for this registry has been previously described.14 The Leiden University Medical Center has a general privacy statement informing patients that their data can be used for (retrospective) scientific research.
From this registry, all cases with a diagnosis of a severe CHD, born between 2012 and 2016, were extracted. We chose this period, as CMA became a routine diagnostic test from 2012 onwards, if parents opted for invasive testing in pregnancy. In order to ensure a reasonable follow-up period after birth, we included cases born before 2017 and followed them until September 2019. From a clinical perspective, we chose not to exclude cases in which genetic testing was not performed pre- or postnatally to avoid a substantial selection bias, and consequently an overestimation of prevalence of genetic diagnosis. All numbers reported in this cohort are therefore conservative estimates of the prevalence of chromosome abnormalities and sequence variants. Cases with an aneuploidy were not included in subsequent analyses, as the prevalence of aneuploidy in fetal CHD cases has already been well-documented in the literature.12 To estimate the minimum prevalence of chromosome abnormalities and sequence variants in fetuses with CHDs, results from pre- and postnatal genetic testing, postnatal clinical evaluation and postmortem reports were assessed.
Antenatal characteristics
The obstetric databases were evaluated to collect information on maternal and fetal characteristics, including maternal age, obstetric history, obesity, smoking, alcohol use, positive family history of CHDs, self-reported consanguinity, multiple pregnancy, the presence of additional fetal extra-cardiac malformations (ECM) and prenatal genetic testing. Routine prenatal genetic testing, which involves Quantitative Fluorescence-Polymerase Chain Reaction (QF-PCR) analysis and chromosome microarray analysis (CMA), was offered in all cases with a prenatal diagnosis of a CHD. Genetic testing for specific genetic diagnoses, not detectable with routine prenatal genetic testing, was only performed in fetuses with features that raised suspicion of a specific syndrome. The CHD was classified as either isolated or non-isolated, as it can be expected that the presence of additional structural malformations in the fetus would increase the probability of an underlying genetic cause. Isolated was defined as the absence of significant structural ECMs or intra-uterine growth restriction (according to consensus-based definition15) prenatally, whereas non-isolated comprised cases diagnosed with one or multiple structural ECMs. Minor antenatal extra-cardiac abnormalities, also referred to as ‘soft markers’, such as an echogenic bowel or single umbilical artery, were considered non-significant ECMs.
Postnatal course
Data on pregnancy and neonatal outcome, such as gestational age at birth, birth weight, presence of dysmorphic features postnatally, mortality and need for medication were retrieved from electronic patient records. Until the end of the study period we assessed whether signs of developmental delay, such as neurocognitive or speech and language disorders, were reported in the patient records. Clinical records were evaluated for results of CMA, genetic testing for specific syndromes (e.g. if CHARGE, Noonan, Kabuki or Alagille syndrome was suspected) and focused or exome-wide analysis of ES, if parents chose to delay (additional) genetic testing until after birth. If additional signs for a genetic syndrome, such as dysmorphic features or developmental delay, were apparent but less specific and CMA results were normal, ES was considered. ES was only performed in postnatal cases, as this was not yet available in a prenatal setting during the study period. The specific ES based CHD gene panel used in our facilities for focused analysis of sequence data, consisted of a panel of 129 genes that are known to be associated with CHD.16 A clinical geneticist was consulted in both pre- and postnatal cases before genetic testing for a specific syndrome or exome sequencing was performed.
Classification of all genetic variants
Structural chromosome abnormalities and sequence variants were classified using the guidelines for interpretation of CNV and sequence variants, developed by The American College of Medical Genetics and Genomics (ACMG).17,18 This classification allocates abnormal results into the following five categories, based on their expected clinical relevance: pathogenic, likely pathogenic, uncertain significance, likely benign and benign. Likely pathogenic was only used when there was a certainty of at least 90% that a variant was disease-causing.17 Pathogenic and likely pathogenic variants, later referred to as ‘genetic diagnoses’, will therefore be reported together in the subsequent analysis.
The Affymetrix Cytoscan HD array or Agilent CGH 180 K oligo array (Amadid 023363) platforms were used for CMA, as described earlier by Jansen et al.19 ES was performed using the Agilent SureSelectXT Human all Exon v5 or Clinical Research Exome v2 capture library kit (Agilent, Santa Clara, USA) accompanied by paired end Sequencing on an Illumina sequencing platform (Illumina, San Diego, USA), generating reads with at least 80x median coverage. The sequence analysis pipeline and tool for annotation of variants has extensively been reported on before.20 Trio samples of the fetus and both parents were assessed for sequencing to optimize variant filtering, when available. Reported variants were submitted to the DECIPHER database.
Statistical analysis
A conservative estimate of the prevalence of chromosomal abnormalities and sequence variants in mid-gestation fetuses diagnosed with a CHD was determined by complementing genetic diagnoses made in pregnancy with those detected after birth, based on postnatal clinical evaluation and follow-up assessment. We assessed the proportion of sequence variants, not detectable with CMA, as this is currently recommended for all fetuses with CHDs.9 Information on the presence of clinical features for a genetic syndrome is limited on prenatal ultrasound. This reduces the applicability of genetic testing for a specific syndrome in utero. We therefore evaluated the potential diagnostic yield of ES, either analyzed using our specific CHD gene panel16 or exome-wide, for the detection of (likely) pathogenic sequence variants in fetal CHD cases. The probability of structural chromosome abnormalities and sequence variants, after aneuploidy is excluded, was determined separately for each heart defect that encompassed at least 1% of this cohort. We also calculated whether the probability of genetic diagnoses in fetuses with CHDs is affected by maternal age, obstetric history, family history of CHDs, consanguinity, multiple pregnancy and additional structural fetal malformations. The clinical impact of structural chromosome abnormalities and sequence variants was assessed by comparing neonatal outcome of fetuses with and without abnormal genetic testing results.
Numeric variables were studied for significant differences using an independent t-test, whereas a χ2-test was used to test associations between categorical variables. A Fisher’s exact test was used, if the expected number was < 5. A p-value <0.05 was considered statistically significant. All statistical analyses were performed using IBM SPSS Statistics 25.0 (IBM, Armonk, NY, USA). This study was approved by the Leiden University Medical Ethics Committee.
Results
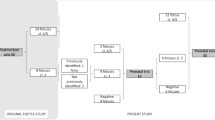
A total of 919 cases, diagnosed with a severe CHD between January 2012 and December 2016, were extracted from the PRECOR registry. Parents chose invasive prenatal testing in 542/919 cases (59.0%), whereas in 185/919 cases (20.1%) genetic testing was performed after birth. Aneuploidy testing was performed in all these cases. Genetic testing was not performed pre- or postnatally in 192/919 cases (20.9%). In these cases parents either declined genetic testing or it was not indicated, because dysmorphic features were absent and the CHD was expected not to be associated with genetic syndromes. After clinical assessment by a geneticist, signs of a genetic syndrome were absent after birth in the majority of these cases (173/192, 90.1%), whereas 9.9% (19/192) did have additional ECMs (Fig. 1).

*Adjusted prevalence after exclusion of aneuploidy cases. CHD congenital heart defect, CNV copy number variant, T21 trisomy 21, T18 trisomy 18, T13 trisomy 13.
An aneuploidy was found in 211/919 (23.0%) cases. As we were mainly interested in the prevalence of structural chromosome abnormalities and sequence variants, rather than aneuploidy, only the remaining 708 euploid cases were included in further analyses. Genetic testing in these cases involved CMA in 64.7% (458/708), genetic testing for specific syndromes in 13.3% (94/708) and focused (7.2%, 51/708) or exome-wide analysis of exome sequencing data (8.1%, 57/708). Baseline characteristics of these cases are enclosed as Supplementary Material (Table S1).
Prevalence genetic variants
An estimate of the prevalence of chromosome abnormalities and sequence variants amongst fetuses with a CHD in this cohort was determined by complementing all genetic variants reported (including ‘uncertain significance’) in pregnancy with those detected after birth. An overview of all genetic variants encountered in this cohort, is shown in Table 1.
Genetic diagnoses were encountered in 111/708 (15.6%) euploid cases with severe CHD. These comprised copy number variants (CNVs) diagnosed with CMA in 70/708 cases (9.9%), of which 63/70 were classified as pathogenic and 7/70 likely pathogenic. In 2.7% of cases (19/708) a CNV of uncertain significance was found. The 22q11.2 deletion syndrome was the most common microdeletion syndrome encountered, with a prevalence of 4.2% (30/708), encompassing 42.9% (30/70) of all (likely) pathogenic CNVs detected.
In 41/708 (5.8%) fetuses with a CHD, a potentially causal sequence variant was detected with additional testing. These variants were classified pathogenic in 33/41 and likely pathogenic in 8/41. A sequence variant of uncertain significance was found in 2.8% of cases (20/708). Frequently encountered genetic syndromes included CHARGE (5/41, 12.2%), Kabuki (3/41, 7.3%) and Noonan syndrome (3/41, 7.3%).
Diagnostic yield additional testing
In 65.9% (27/41) of (likely) pathogenic sequence variants, genetic testing for a specific syndrome (e.g. Kabuki or Noonan syndrome) had been performed based on clinical suspicion. These variants were diagnosed postnatally in the majority of cases (16/27, 59.3%). The remaining 34.1% (14/41) of (likely) pathogenic sequence variants were detected with either focused or exome-wide analysis of exome sequencing data.
To compare the strengths and limitations of a gene panel approach to an exome-wide analysis of ES data, we analyzed the diagnostic yield in relation to all reported sequence variants (including ‘uncertain’ significance). In 24.6% (15/61) of all sequence variants, a (likely) pathogenic variant was encountered that did not involve genes known to be related with CHDs. Significant variants, such as a RASA1 sequence variant causing capillary malformation-arteriovenous malformation syndrome or a variant in the TSC1 gene leading to tuberous sclerosis (not suspected prenatally), will thus remain undetected if ES data are only analyzed using a targeted CHD gene panel. The disadvantage of exome-wide analysis of ES data is the increased identification of variants of ‘uncertain significance’, as identified in 19.7% (12/61) of all sequence variants.
Genetic cause for CHD
Eighty-five percent (94/111) of the encountered structural chromosome abnormalities and sequence variants were considered a definite explanation for the development of the heart defect. This means that in 13% (94/708) of euploid CHD cases, a genetic cause for the heart defect was found, which was 18% (94/516) for those who underwent genetic testing. Secondary findings involved genetic causes for severe hemophilia, Charcot-Marie-Tooth Disease and psychomotor retardation.
Structural chromosome abnormalities and sequence variants that were classified pathogenic or likely pathogenic, were both considered a pathogenic diagnosis in subsequent analyses, and therefore reported together. The remaining cases without genetic testing results were regarded as having a normal test result, whereas those with a variant of uncertain significance were reported separately.
Specific heart defects
Heart defects that were most frequently accompanied by a genetic diagnosis included an interrupted aortic arch (IAoA), pulmonary atresia with a ventricular septal defect (PA-VSD), (un)balanced atrioventricular septal defect (AVSD), truncus arteriosus and Tetralogy of Fallot. Genetic diagnoses were, on the other hand, barely encountered amongst fetuses with a diagnosis of transposition of the great arteries with intact ventricular septum (simple TGA), tricuspid atresia, double inlet left ventricle or total anomalous pulmonary venous connection. The genetic diagnoses encountered in two cases with simple TGA were both secondary findings that did not explain the heart defect. In one case hemophilia type A was diagnosed, and the mother turned out to be a carrier, whereas the other comprised a de novo distal 16q11.2 deletion which was very unlikely to be associated with the heart defect. For each common CHD, and isolated cases separately, the probability for genetic diagnoses is presented in Table 2.
Risk factors for a genetic diagnosis
The probability of a genetic diagnosis was significantly lower for prenatally isolated (11.6%, 63/541) compared to non-isolated cases (28.7%, 48/167) (p < 0.001). Self-reported consanguinity (35.5% vs 2.9%, p = 0.002) and a positive parental history of a CHD (32.3% vs 3.8%, p = 0.03) were also significantly associated with genetic diagnoses. Other variables of interest, such as maternal age, gravidity, obesity or multiple pregnancy were not significantly correlated with genetic diagnoses (Table 3).
Impact on outcome
The effect of genetic diagnoses on the pregnancy outcome of all included cases is depicted in Table 3. The postnatal outcome was compared between cases with and without a genetic diagnosis. First of all, parents opted for TOP significantly more often when the heart defect was accompanied by a genetic diagnosis compared to those without a genetic diagnosis (36.9% vs 24.4%; p = 0.01). The detection of variants of ‘uncertain significance’ did not lead to an increase in TOPs, as the proportion of parents that terminated pregnancy was even lower in these cases compared to those with normal genetic testing results (10.3% vs 24.4%; p = 0.04). This indicates that the specific diagnosis rather influences the parental decision for TOP than the detection of variants of ‘uncertain significance’. The proportion of cases with a birth weight < 3rd centile was higher amongst CHD cases with a genetic diagnosis (20.6% vs 6.5%; p = 0.01). Postnatal mortality was also increased in cases with a genetic diagnosis (32.8% vs 9.0%, p < 0.001). Signs of developmental delay were significantly more often present in children with (75.6%) compared to those without a genetic diagnosis (9.7%) (p < 0.001). Cases with genetic diagnoses required medication for other reasons than the cardiac defect itself, more often (30.0% vs 5.4%, p = 0.001), as well as (medical) support, such as speech therapy, physical therapy or special education (79.5% vs 9.5%, p < 0.001).
Prevalence including aneuploidy
The minimum prevalence of (likely) pathogenic CNVs in all fetuses with a CHD appears 7.6% (70/919). For sequence variants, not detectable with standard micro-array testing, the prevalence of (likely) pathogenic variants is estimated at 4.5% (41/919) of all fetuses with a CHD (Fig. 1). A genetic cause for the CHD was encountered in 33% (305/919) of all CHD cases, which was 42% (305/708) for those who underwent genetic testing (Supplementary Material, Figure S1).
Discussion
This is the largest cohort study on the total prevalence of genetic diagnoses in fetuses with a severe CHD that included results from genetic testing for specific syndromes and ES as well. In the setting of prenatal counseling concerning a fetal heart defect, a 15.7% probability should be counseled for clinically significant genetic diagnoses, other than aneuploidy. These involved CNVs in 9.9%, whereas 5.8% had a sequence variant not detectable with QF-PCR and CMA. These numbers, however, comprise conservative estimates, as not all patients underwent genetic testing and exome sequencing was not performed in the majority of cases. More importantly, a genetic diagnosis worsened the prognosis significantly, both on surgical outcome and, not unexpectedly, (neuro-)development.21
Our results show that a genetic diagnosis has a substantial impact on neonatal outcome in fetuses with a severe CHD. Not only does it significantly increase the risk of mortality, but also morbidity, as these cases showed more often developmental delay (75.6% vs 9.7%) and required more (medical) support and medication, other than for the heart defect itself. The estimated prevalence of pathogenic CNVs in 7.6% of our entire cohort corresponds to three previously described cohorts of fetuses with CHDs, as these report pathogenic CNVs in 8–11%.22,23,24 The proportion of CNVs appeared slightly lower in postnatal cohorts that reported CNVs in 5–8% of neonates with CHDs.25,26 One recent fetal cohort reported a prevalence of 16% pathogenic CNVs.27 This proportion, however, reflects a selected population, as they only included those referred for invasive genetic testing.
As CMA is routinely offered in pregnancy, there remains a residual probability of at least 6.6% (41/616) to identify (likely) pathogenic sequence variants after birth. Due to the large variety of genetic syndromes, which can occur in less than 1:1000 CHD cases, exome sequencing should be considered in fetuses with CHDs. Especially, as it may change the prognosis considerably, which is why parents opt for invasive genetic testing in the first place. Our results suggest that the diagnostic yield of ES will be no less than 6.6% if ES is offered routinely for all fetal CHDs, and even higher for those with additional structural malformations. This association with ECMs is in line with previous literature,14,22,23 and therefore an important factor for prenatal counseling and the decision to perform additional genetic testing in some cases. Another recently published fetal cohort found diagnostic sequence variants in as many as 13.6% of fetuses with any cardiac malformation with normal results after aneuploidy testing and CMA.28 These findings can again be explained by the selection of their cohort; ES was not offered to all women and only those with ES results were eligible for inclusion. The estimated diagnostic yield encountered in our cohort is therefore conservative.
The prevalence of sequence variants appeared also higher in fetuses (6.6%) than neonates, as one postnatal cohort study reported genetic syndromes in 5.1% of neonates with normal chromosomes.26 This risk is probably higher in fetal cohorts compared to cohorts that focus exclusively on postnatal cases, as cases with TOP, intra uterine fetal demise or early neonatal death are often not included in postnatal cohorts. It is therefore important that our data are evaluated from a prenatal perspective to enable prenatal counseling at mid-gestation. This is confirmed by the fact that parents in this cohort opted for TOP more often, if a genetic cause was identified, which is similar to our previous findings in prenatal exome sequencing.20
This study shows that if a focused approach is chosen as the method of advanced genetic testing in the setting of normal CMA, 36.6% (15/41) of (likely) pathogenic variants would remain undetected. This method is less preferable in a prenatal setting, as essential clinical symptoms may be impossible to detect in the fetus, which may lead to analysis of the ‘wrong’ gene panel. Exome-wide analysis of ES data can detect changes in the entire exome, but at the expense of the turnaround time or costs. Sequence variants in genes not associated with CHDs particularly, may also increase the risk of additional morbidity instead of being an explanation for the development of the heart defect itself. This might be important for prenatal counseling, as it can affect prognosis and neonatal management significantly. Exome-wide analysis of sequence data is also imperative to identify novel pathogenic genes and consequentially improve currently available gene panels for CHDs. It did lead to the detection of variants of uncertain significance in 19.7% (12/61) of all sequence variants, which may complicate prenatal counseling and parental decision making. We believe, however, that the advantages of an exome-wide analysis of exome sequencing data for fetal CHDs may outweigh its difficulties,20 particularly in the presence of additional structural malformations.
A limitation of this study is that in 20.9% of our entire cohort, genetic testing was not performed. The majority of these cases, however, comprised isolated cases without dysmorphic features after birth that showed a normal development, as all the children do have follow-up visits in our center due to relatively short travel distances and a very low threshold for genetic testing after birth. We therefore chose not to exclude cases, to avoid substantial selection bias, as this might lead to an overestimation of the prevalence.
Due to the large sample size and completeness of our regional CHD registry, we were able to stratify the probability of genetic diagnoses according to the specific heart defect, using not only results from karyotyping and FISH for 22q11.2, but CMA, genetic testing for specific syndromes and exome sequencing in selected cases as well. Although several recent cohorts have studied aneuploidy or 22q11.2 deletion syndrome in CHD cases,22,23,26,29 evidence on the prevalence of other structural chromosome abnormalities and sequence variants for specific heart defects is limited. IAoA, PA-VSD and AVSD were most associated with the presence of genetic diagnoses. The particularly strong correlation between IAoA and submicroscopic genetic changes (69% probability in IAoA cases) was also demonstrated by a large study that evaluated the results of karyotyping and fluorescence in situ hybridization (FISH) for 22q11.2 in a population of infants with CHDs.30 Although we evaluated results from additional diagnostic modalities as well, genetic diagnoses were encountered in a similar proportion (71%) of IAoA cases. This stresses that IAoA is mainly associated with 22q11.2 deletion syndrome. One study on CMA in fetuses with a VSD reported pathogenic CNVs in 12%.13 This might be an overestimation, as our cohort comprised twice as many VSD cases with pathogenic CNVs in 9.3% (10/107). A cohort of Tetralogy of Fallot infants found genetic diagnoses in 25% of cases, after exclusion of aneuploidy, which comprised a heterogenous set of genetic syndromes.31 This is consistent with the 21.1% probability of pathogenic CNVs or sequence variants in our cohort.
In conclusion, this cohort study shows that, after an aneuploidy is excluded, structural chromosome abnormalities and sequence variants are identified in a substantial proportion of cases with severe CHDs. In 5.8% of euploid fetuses with a CHD, the genetic diagnosis would not have been found, if only CMA had been performed. ES should therefore be considered for fetal CHDs, especially if accompanied by other structural malformations, because genetic diagnoses can affect neonatal outcomes significantly. Future research, which offers ES to all fetuses with a CHD, is however needed to obtain more reliable estimates.
References
van der Linde D, Konings EE, Slager MA, et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58:2241–7.
Leirgul E, Fomina T, Brodwall K, et al. Birth prevalence of congenital heart defects in Norway 1994-2009-a nationwide study. Am Heart J. 2014;168:956–64.
Hoang TT, Goldmuntz E, Roberts AE, et al. The Congenital Heart Disease Genetic Network Study: Cohort description. PLoS One. 2018;13:e0191319.
Zaidi S, Brueckner M. Genetics and Genomics of Congenital Heart Disease. Circ Res. 2017;120:923–40.
Song MS, Hu A, Dyamenahalli U, et al. Extracardiac lesions and chromosomal abnormalities associated with major fetal heart defects: comparison of intrauterine, postnatal and postmortem diagnoses. Ultrasound Obstet Gynecol. 2009;33:552–9.
de Wit MC, Srebniak MI, Govaerts LC, Van Opstal D, Galjaard RJ, Go AT. Additional value of prenatal genomic array testing in fetuses with isolated structural ultrasound abnormalities and a normal karyotype: a systematic review of the literature. Ultrasound Obstet Gynecol. 2014;43:139–46.
Simmons MA, Brueckner M. The genetics of congenital heart disease… understanding and improving long-term outcomes in congenital heart disease: a review for the general cardiologist and primary care physician. Curr Opin Pediatr. 2017;29:520–8.
Russell MW, Chung WK, Kaltman JR, Miller TA. Advances in the Understanding of the Genetic Determinants of Congenital Heart Disease and Their Impact on Clinical Outcomes. J Am Heart Assoc. 2018;7:e006906. https://doi.org/10.1161/JAHA.117.006906.
Committee on G, the Society for Maternal-Fetal M. Committee Opinion No.682: Microarrays and Next-Generation Sequencing Technology: The Use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology. Obstet Gynecol. 2016;128:e262–e8.
Copel JA, Cullen M, Green JJ, Mahoney MJ, Hobbins JC, Kleinman CS. The frequency of aneuploidy in prenatally diagnosed congenital heart disease: an indication for fetal karyotyping. Am J Obstet Gynecol. 1988;158:409–13.
Paladini D, Calabro R, Palmieri S, D’Andrea T. Prenatal diagnosis of congenital heart disease and fetal karyotyping. Obstet Gynecol. 1993;81(5 (Pt 1)):679–82.
Wimalasundera RC, Gardiner HM. Congenital heart disease and aneuploidy. Prenat Diagn. 2004;24:1116–22.
Du L, Xie HN, Huang LH, Xie YJ, Wu LH. Prenatal diagnosis of submicroscopic chromosomal aberrations in fetuses with ventricular septal defects by chromosomal microarray-based analysis. Prenat Diagn. 2016;36:1178–84.
van Velzen CL, Clur SA, Rijlaarsdam ME, et al. Prenatal detection of congenital heart disease-results of a national screening programme. BJOG. 2016;123:400–7.
Gordijn SJ, Beune IM, Thilaganathan B, et al. Consensus definition of fetal growth restriction: a Delphi procedure. Ultrasound Obstet Gynecol. 2016;48:333–9.
Exome Gene panel Congenital heart defects version 1, Leiden University Medical Center, 2018. https://www.lumc.nl/sub/4080/att/1768907. Accessed 5 November 2019.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, Working Group of the American College of Medical Genetics Laboratory Quality Assurance C. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13:680–5.
Jansen FA, Hoffer MJ, van Velzen CL, et al. Chromosomal abnormalities and copy number variations in fetal left-sided congenital heart defects. Prenat Diagn. 2016;36:177–85.
de Koning MA, Haak MC, Adama van Scheltema PN, et al. From diagnostic yield to clinical impact: a pilot study on the implementation of prenatal exome sequencing in routine care. Genet Med. 2019;21:2303–10.
Marino BS, Lipkin PH, Newburger JW, et al. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation. 2012;126:1143–72.
Wang Y, Cao L, Liang D, et al. Prenatal chromosomal microarray analysis in fetuses with congenital heart disease: a prospective cohort study. Am J Obstet Gynecol. 2018;218:244 e1–17.
Luo S, Meng D, Li Q, et al. Genetic Testing and Pregnancy Outcome Analysis of 362 Fetuses with Congenital Heart Disease Identified by Prenatal Ultrasound. Arq Bras Cardiol. 2018;111:571–7.
Song T, Wan S, Li Y, et al. Detection of copy number variants using chromosomal microarray analysis for the prenatal diagnosis of congenital heart defects with normal karyotype. J Clin Lab Anal. 2019;33:e22630.
Choi BG, Hwang SK, Kwon JE, Kim YH. Array Comparative Genomic Hybridization as the First-line Investigation for Neonates with Congenital Heart Disease: Experience in a Single Tertiary Center. Korean Circ J. 2018;48:209–16.
Dykes JC, Al-mousily MF, Abuchaibe EC, et al. The incidence of chromosome abnormalities in neonates with structural heart disease. Heart. 2016;102:634–7.
Xia Y, Yang Y, Huang S, Wu Y, Li P, Zhuang J. Clinical application of chromosomal microarray analysis for the prenatal diagnosis of chromosomal abnormalities and copy number variations in fetuses with congenital heart disease. Prenat Diagn. 2018;38:406–13.
Lord J, McMullan DJ, Eberhardt RY, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393:747–57.
Molck MC, Simioni M, Paiva Vieira T, et al. Genomic imbalances in syndromic congenital heart disease. J Pediatr (Rio J). 2017;93:497–507.
Hartman RJ, Rasmussen SA, Botto LD, et al. The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatr Cardiol. 2011;32:1147–57.
Michielon G, Marino B, Formigari R, et al. Genetic syndromes and outcome after surgical correction of tetralogy of Fallot. Ann Thorac Surg. 2006;81:968–75.
Acknowledgements
We would like to thank Maud Zwagers (MD) and Wineke van Seters (MD) (Leiden University Medical Center, Leiden) for their assistance with gathering results from genetic testing in all three affiliations to ensure complete information on genetic status of fetuses with a severe CHD.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
van Nisselrooij, A.E.L., Lugthart, M.A., Clur, SA. et al. The prevalence of genetic diagnoses in fetuses with severe congenital heart defects. Genet Med 22, 1206–1214 (2020). https://doi.org/10.1038/s41436-020-0791-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0791-8
Keywords
This article is cited by
-
Contribution of genetic variants to congenital heart defects in both singleton and twin fetuses: a Chinese cohort study
Molecular Cytogenetics (2024)
-
Genetic abnormalities in fetal congenital heart disease with aberrant right subclavian artery
Scientific Reports (2022)
-
Prenatal and postnatal chromosomal microarray analysis in 885 cases of various congenital heart defects
Archives of Gynecology and Obstetrics (2022)
-
Abnormal Microarray, Clinical Outcomes, and Surgical Risk Scores in Young Children with Cardiac Disease
Pediatric Cardiology (2021)
