
Abstract
Here, we report the synthesis, carbonic anhydrase-II (CA-II) inhibition and structure–activity relationship studies of cinnamaldehyde-clubbed thiosemicarbazones derivatives. The derivatives showed potent activities in the range of 10.3 ± 0.62–46.6 ± 0.62 µM. Among all the synthesized derivatives, compound 3n (IC50 = 10.3 ± 0.62 µM), 3g (IC50 = 12.1 ± 1.01 µM), and 3h (IC50 = 13.4 ± 0.52 µM) showed higher inhibitory activity as compared to the standard inhibitor, acetazolamide. Furthermore, molecular docking of all the active compounds was carried out to predict their behavior of molecular binding. The docking results indicate that the most active hit (3n) specifically mediate ionic interaction with the Zn ion in the active site of CA-II. Furthermore, the The199 and Thr200 support the binding of thiosemicarbazide moiety of 3n, while Gln 92 supports the interactions of all the compounds by hydrogen bonding. In addition to Gln92, few other residues including Asn62, Asn67, The199, and Thr200 play important role in the stabilization of these molecules in the active site by specifically providing H-bonds to the thiosemicarbazide moiety of compounds. The docking score of active hits are found in range of − 6.75 to − 4.42 kcal/mol, which indicates that the computational prediction correlates well with the in vitro results.
Similar content being viewed by others

Introduction
Carbonic anhydrases are broadly distributed metalloenzymes in both eukaryotes and prokaryotes. They efficiently catalyze the reversible hydration of CO2 to HCO3− and H+ ions and perform a significant function in the regulation of many pathological and physiological processes1,2,3,4,5. As a result, these enzymes are arousing targets for the treatment of pathological diseases6,7,8. CA-II is greatly associated with maintaining the concentration of bicarbonate in the eyes9,10,11,12. These inhibitors have been examined as a supplement in cancer chemotherapy12. Besides, CA-II is also reported in renal, pancreatic, and gastritis carcinomas and malignant brain tumors13,14,15. Carbonic anhydrase activity from bovine source has a central role in corneal endothelial function by buffering corneal endothelial fluid transport. Carbonic anhydrases support one-third of active ion transport in bovine corneal endothelium but do not in human16,17.
Thiosemicarbazones are synthesized by the use of simple and economical methods through the condensation reaction of thiosemicarbazides with various aldehydes and ketones18. They are a type of N, S-donor ligands, possessing an important position in therapeutic chemistry research because of their structural assortment, modifiable synthesis, and valuable donating ability19. Thiosemicarbazones and their derivatives have a significant awareness in the organic field, pharmacology, and biology; appreciations to their various applications20,21,22,23,24,25,26,27,28,29. Several pieces of research have testified the antiviral, antioxidant30, antitumor, antimalarial31,32, antibacterial33,34, antifungal35,36, anticancer37,38,39,40,41, antiviral42,43 and anti-diabetic44,45,46 actions of thiosemicarbazones47. Moreover, the reputation of thiosemicarbazones has been much improved by the entry of α-N-heterocyclic TSCs derivates (Tripane, COTI-2, DpC) in the medical hearings for cancer and HIV-148.
Biological interaction has been upholding one of the greatest substantial goals in the growth of human strength i.e., providing organically energetic unrelated molecules that can be found over simple synthetic ways with the non-poisonous catalysts and can be reformed and improved for better pharmacological offers, in the midst are thiosemicarbazones (TSCs). The pharmacological properties of thiosemicarbazones assets are due to the presence of thiourea based functional nucleus, having NH and C=S functional groups for metallic coordination49. Research has revealed that therapeutic characteristics of thiosemicarbazones can be enhanced by utilizing vigorous biological moieties as the building blocks of molecules50.
The main chemical constituent obtained from Cinnamomum cassia is cinnamaldehyde51. In the earlier research, cinnamaldehyde has displayed noticeable characteristics in persuading in vivo or in vitro apoptosis of numerous tumor cells52. Our preceding research has exposed that cinnamaldehyde can persuade non-small cell lung cancer (NSCLC) apoptosis and control of Wnt/b-catenin path in A549, YTMLC-90, and H1299 cells beneath the conditions of cinnamaldehyde. Despite all these signs, additional studies are required to offer awareness into the possible anti-tumor mechanism complications in the cinnamaldehyde regulation. It is expected that the cinnamaldehyde derivatives might display extra energy anti-FtsZ result with a difficult antibacterial worth and a larger antibacterial spectrum53,54.
Recent studies have also shown out the pathogenetic characters of oxidative strain in vitiligo and bad skin, both of which are conditions complex to ecological roughness. Furthermore, cinnamaldehyde exhibited strong antioxidant activity consequently since its activation of the NRF2/HO1 pathway. These representatives are gifted candidates for the action of oxidative stress-related sicknesses55,56. There have been some bits of intelligence on cinnamon oil impeding the development of shapes, cinnamaldehyde hydroxyl sulfonic sodium, which was non-volatile, stood also formed but displayed much lower anti-mold activity as compared to cinnamaldehyde. An assessable structure–activity association (QSAR) demonstrating the anti-mold activity of cinnamaldehyde correspondents against Aspergillus niger and Paecilomyces variotii stood offered57,58. There are numbers of sulfonamides derivatives are reported as CA-II inhibitors29,59,60,61,62,63,64,65,66,67. Some examples of known CA-II inhibitors are given in Fig. 1.

Recently, the inhibitory potential of thiosemicarbazones also reported for bovine and human CA-II68. We hereby report the syntheses of a novel 4-(dimethylamino)-cinnamaldehyde-based thiosemi-carbazones derivatives as potent CA-II inhibitors. Moreover, molecular docking studies were also carried to know the interaction of these derivatives at atomic level. We concluded that most of the compounds exhibited a stronger inhibitory effect as compared to acetazolamide which was used as a reference.
Experimental
Chemistry
All the chemicals used in this research were bought in extra purified from Sigma-Aldrich. FT-IR spectra (KBr disks) were recorded on a Bruker FT-IR IFS48 spectrophotometer. Melting points were taken on the melting point apparatus (Büchi 434). The CHN analysis was done on a Carlo Erba Strumentazione-Mod-1106. NMR spectra were taken on Bruker Avance 400 spectrometers in DMSO-d6. The chromatographs were seen under UV light irradiation.
General method for the synthesis of thiosemicarbazones (3a–q)
A series of seventeen TSCs (3a–q) was prepared by adding suitable thiosemicarbazide (2a–q) (5 mmol) and 4-(dimethylamino)-cinnamaldehyde (1) (5 mmol) in 10 mL of methanol and glacial acetic acid in catalytic quantity. After refluxing for 3–6 h at 80 °C, the reaction progress was observed by the TLC analysis. The reaction mixture was left to cool at room temperature and the solid products obtained in all cases were filtered, washed with hot methanol, and dried at room temperature. The targeted TSCs were further recrystallized from the methanol-chloroform mixture (1:1) in very good to excellent yields.
The targeted compounds are characterized as follows:
(E)-2-((E)-3-(4-(Dimethylamino)phenyl)allylidene)-N-(p-tolyl)hydrazine-1-carbothioamide (3a)
Yield: 97%, color: brownish yellow; M.P: 232 °C; 1H NMR (DMSO-d6, δ, ppm): 2.28 (s, 3H, Ar–CH3), 2.93 (s, 6H, CH3), 6.63–6.72 (m, 3H, C=CH, Ar–H), 6.9 (d, J = 16.2 Hz, 1H, Ar–CH), 7.11 (d, J = 7.8 Hz, 2H, Ar–H), 7.38 (d, J = 9.0 Hz, 2H, Ar–H), 7.45 (d, J = 8.4 Hz, 2H, Ar–H), 7.91 (d, J = 9.6 Hz, 1H, N=CH), 9.7 (s, 1H, NH–CS), 11.59 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 20.6, 39.5, 112.1, 119.9, 123.6, 124.7, 128.4, 128.6, 134.0, 136.5, 140.3, 146.1, 150.8, 174.9, FT-IR (υmax, cm−1): 1184 (C=S), 1580 (C=N), 3290 (N–H); ESI MS (m/z) = M+H = 339.16.
(E)-2-((E)-3-(4-(Dimethylamino)phenyl)allylidene)-N-(2,6-dimethylphenyl)hydrazine-1-carbothioamide (3b)
Yield: 77%, color: yellow; M.P: 239 °C; 1H NMR (DMSO-d6, δ, ppm): 2.1 (s, 6H, Ar–CH3), 2.93 (s, 6H, CH3), 6.64–6.67 (m, 1H, C=CH, Ar–H), 6.7 (d, J = 8.4 Hz, 2H, Ar–H), 6.89 (d, J = 16.2 Hz, 1H, Ar–CH), 7.05–7.07 (m, 3H, Ar–H), 7.37 (d, J = 8.4 Hz, 2H, Ar–H), 7.91 (d, J = 9.6 Hz, 1H, N=CH), 9.44 (s, 1H, NH–CS), 11.51 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 18.1, 39.5, 112.1, 120.9, 123.7, 126.8, 127.6, 128.3, 136.4, 137.2, 139.8, 145.8, 150.7, 176.1, FT-IR (υmax, cm−1): 1186 (C=S), 1581 (C=N), 3292 (N–H); ESI MS (m/z) = M+H = 353.17.
(E)-2-((E)-3-(4-(Dimethylamino)phenyl)allylidene)-N-(4-ethylphenyl)hydrazine-1-carbothioamide (3c)
Yield: 75%, color: orange yellow; M.P: 180 °C; 1H NMR (DMSO-d6, δ, ppm): 2.87 (t, 2H, Ar–CH2), 2.93 (s, 6H, CH3), 3.71–3.74 (m, 2H, N–CH2), 6.58–6.62 (m, 1H, C=CH), 6.68 (d, J = 8.4 Hz, 2H, Ar–H), 6.84 (d, J = 15.6 Hz, 1H, Ar–CH), 7.20 (t, J = 7.2 Hz, 1H, Ar–H), 7.24 (d, J = 9.6 Hz, 2H, Ar–H), 7.30 (t, J = 7.2 Hz, 2H, Ar–H), 7.36 (d, J = 8.4 Hz, 2H, Ar–H), 7.84 (d, J = 9.0 Hz, 1H, N=CH), 8.17 (s, 1H, NH–CS), 11.28 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 34.9, 39.5, 44.9, 112.1, 119.8, 123.6, 126.2, 128.3, 128.4, 128.6, 139.3, 139.8, 145.6, 150.7, 176.3, FT-IR (υmax, cm−1): 1183 (C=S), 1579 (C=N), 3289 (N–H); ESI MS (m/z) = M+H = 353.18.
(E)-N-(2,3-Dichlorophenyl)-2-((E)-3-(4-(dimethylamino)phenyl)allylidene)hydrazine-1-carbothioamide (3d)
Yield: 95%, Color: Brownish Yellow; M.P: 192 °C; 1H NMR (DMSO-d6, δ, ppm): 2.98 (s, 6H, CH3), 6.62–6.67 (m, 3H, C=CH, Ar–H), 6.84 (d, J = 15.6 Hz, 1H, Ar–CH), 7.21–7.24 (m, 1H, Ar–H), 7.27 (d, J = 7.8 Hz, 1H, Ar–H), 7.34 (d, J = 9.0 Hz, 2H, Ar–H), 7.69 (d, J = 9.6 Hz, 1H, N=CH), 8.49 (d, J = 7.8 Hz, 1H, Ar–H), 9.57 (s, 1H, NH–CS), 10.03 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 40.2, 77.1, 111.1, 119, 123.5, 125.6, 126.7, 128.8, 129.7, 132.8, 136.9, 142.4, 146.9, 151.2, 174.4, FT-IR (υmax, cm−1): 1180 (C=S), 1575 (C=N), 3284 (N–H); ESI MS (m/z) = M+H = 393.07.
(E)-2-((E)-3-(4-(Dimethylamino)phenyl)allylidene)-N-(2-fluorophenyl)hydrazine-1-carbothioamide (3e)
Yield: 75%, color: yellow; M.P: 189 °C; 1H NMR (DMSO-d6, δ, ppm): 2.98 (s, 6H, CH3), 6.62–6.67 (m, 3H, C=CH, Ar–H), 6.83 (d, J = 15.2 Hz, 1H, Ar–CH), 7.11–7.17 (m, 3H, Ar–H), 7.34 (d, J = 8.4 Hz, 2H, Ar–H), 7.64 (d, J = 8.4 Hz, 1H, N=CH), 8.41–8.44 (m, 1H, Ar–H), 9.21 (s, 1H, NH–CS), 9.66 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 40.2, 77.0, 111.9, 115.3, 119.1, 123.5, 125.4, 126.2, 126.3, 128.7, 142.0, 146.4, 151.1, 154.1, 174.9, FT-IR (υmax, cm−1): 1181 (C=S), 1577 (C=N), 3286 (N–H); ESI MS (m/z) = M+H = 343.13.
(E)-2-((E)-3-(4-(Dimethylamino)phenyl)allylidene)-N-(3-methoxyphenyl)hydrazine-1-carbothioamide (3f)
Yield: 85%, color: brownish yellow; M.P: 198 °C; 1H NMR (DMSO-d6, δ, ppm): 2.98 (s, 6H, CH3), 3.8 (s, 3H, O-CH3), 6.62–6.6 (m, 3H, C=CH, Ar–H), 6.73 (d, J = 7.8 Hz, 1H, Ar–H) 6.82 (d, J = 15.6 Hz, 1H, Ar–CH), 7.14 (d, J = 8.4 Hz, 1H, Ar–H), 7.23–7.26 (t, J = 7.8 Hz, 1H), 7.34 (d, J = 8.4 Hz, 2H, Ar–H), 7.44 (s, 1H, Ar–H), 7.67 (d, J = 9.6 Hz, 1H, N=CH), 9.12 (s, 1H, NH–CS), 9.95 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 40.2, 65.4, 77.1, 109.1, 110.1, 116.1, 119.1, 123.7, 128.7, 129.3, 139.2, 141.9, 146.2, 151.1, 159.8, 174.6, FT-IR (υmax, cm−1): 1187 (C=S), 1587 (C=N), 3297 (N–H); ESI MS (m/z) = M+H = 355.16.
(E)-N-(4-Bromophenyl)-2-((E)-3-(4-(dimethylamino)phenyl)allylidene)hydrazine-1-carbothioamide (3g)
Yield: 80%, color: brownish yellow; M.P: 240 °C; 1H NMR (DMSO-d6, δ, ppm): 2.93 (s, 6H, CH3), 6.60–6.64 (m, 3H, C=CH, Ar–H), 6.78 (d, J = 16.2 Hz, 1H, Ar–CH) 7.27 (d, J = 8.4 Hz, 2H, Ar–H), 7.36 (d, J = 7.8 Hz, 2H, Ar–H), 7.58 (d, J = 8.4 Hz, 2H, Ar–H), 7.86 (d, J = 9.6 Hz, 1H, N=CH), 9.49 (s, 1H, NH–CS), 11.59 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 39.7, 78.3, 111.7, 117.1, 119.6, 123.5, 128.1, 130.7, 137.9, 140.4, 146.4, 150.6, 174.4, FT-IR (υmax, cm−1): 1185 (C=S), 1582 (C=N), 3291 (N–H); ESI MS (m/z) = M+H = 403.6.
(E)-N-(2,4-Difluorophenyl)-2-((E)-3-(4-(dimethylamino)phenyl)allylidene)hydrazine-1-carbothioamide (3h)
Yield: 95%, color: brownish yellow; M.P: 202 °C; 1H NMR (DMSO-d6, δ, ppm): 2.98 (s, 6H, CH3), 6.62–6.65 (m, 3H, C=CH, Ar–H), 6.83–6.90 (m, 3H, Ar–CH, Ar–H), 7.33 (d, J = 8.4 Hz, 2H, Ar–H), 7.67 (d, J = 9.0 Hz, 1H, N=CH), 8.17–8.21 (m, 1H, Ar–H), 9.0 (s, 1H, NH–CS), 9.94 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 40.2, 77.0, 103.9, 110.0, 112.0, 119.0, 122.7, 123.7, 127.5, 128.7, 129.7,142.2, 146.7, 151.2, 175.4, FT-IR (υmax, cm−1): 1182 (C=S), 1576 (C=N), 3286 (N–H); ESI MS (m/z) = M+H = 361.12.
(E)-2-((E)-3-(4-(Dimethylamino)phenyl)allylidene)-N-(4-isopropylphenyl)hydrazine-1-carbothioamide (3i)
Yield: 70%, color: dark brown; M.P: 220 °C; 1H NMR (DMSO-d6, δ, ppm): 1.22 (d, J = 7.2 Hz, 6H, CH3), 2.8 (m, 1H, CH3-CH), 2.98 (s, 6H, CH3), 6.64 (m, 3H, C=CH, Ar–H), 6.82 (d, J = 15.6 Hz, 1H, Ar–CH), 7.21 (d, J = 7.8 Hz, 2H, Ar–H), 7.33 (d, J = 8.4 Hz, 2H, Ar–H), 7.54 (d, J = 8.4 Hz, 2H, Ar–H), 7.66 (d, J = 9.6 Hz, 1H, N=CH,), 9.04 (s, 1H, NH–CS), 9.81 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 24.02, 33.7, 40.2, 77.0, 112.0, 119.1, 123.8, 124.3, 126.7, 128.6, 135.7, 141.7, 146.5, 151.1, 174.9, FT-IR (υmax (cm−1): 1195 (C=S), 1592 (C=N), 3298 (N–H); ESI MS (m/z) = M+H = 367.1.
(E)-2-((E)-3-(4-(Dimethylamino)phenyl)allylidene)-N-(3-fluorophenyl)hydrazine-1-carbothioamide (3j)
Yield: 85%, Color: Yellow; M.P: 218 °C; 1H NMR (DMSO-d6, δ, ppm): 2.99 (s, 6H, CH3), 6.63- 6.67 (m, 3H, C=CH, Ar–H), 6.84–6.89 (m, 2H, Ar–H, Ar–CH), 7.28 (dd, J = 7.2 Hz, 7.8 Hz, 1H, Ar–H), 7.34–7.37 (m, 3H, Ar–H), 7.63 (d, J = 9.0 Hz, 1H Ar–H), 7.67 (d, J = 10.2 Hz, 1H, N=CH), 9.16 (s, 1H, NH–CS), 9.58 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 40.2, 77.2, 110.8, 119.2, 112.2, 118.8, 119.0, 123.6, 128.7, 129.7, 139.7, 142.1, 146.3, 151.2, 174.6, FT-IR υmax (cm−1): 1182 (C=S), 1578 (C=N), 3289 (N–H); ESI MS (m/z) = M+H = 343.10.
(E)-N-(4-(Chloromethyl)phenyl)-2-((E)-3-(4-(dimethylamino)phenyl)allylidene)hydrazine-1-carbothioamide (3k)
Yield: 80%, color: orange yellow; M.P: 196 °C; 1H NMR (DMSO-d6, δ, ppm): 2.97 (s, 6H, CH3), 4.86 (d, J = 5.4 Hz, 2H, Ar–CH2), 6.53–6.57 (m, 1H, C=CH), 6.62 (d, J = 8.4 Hz, 2H, Ar–H), 6.77 (d, J = 15.6 Hz, 1H, Ar–CH), 7.29–7.30 (m, 6H, Ar–H), 7.59 (m, 2H, N=CH, NH–CS), 9.63 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 40.2, 47.4, 77.0, 112.0, 119.1, 123.7, 128.6, 128.8, 129.2, 136.3, 141.4, 146.2, 151.0, 177.0, FT-IR (υmax, cm−1): 1183 (C=S), 1580 (C=N), 3290 (N–H); ESI MS (m/z) = M+H = 373.12.
(E)-2-((E)-3-(4-(Dimethylamino)phenyl)allylidene)-N-(2,4-dimethylphenyl)hydrazine-1-carbothioamide (3l)
Yield: 85%, color: yellow; M.P: 232 °C; 1H NMR (DMSO-d6, δ, ppm): 2.27 (s, 3H, Ar-CH3), 2.30 (s, 3H, Ar-CH3), 2.98 (s, 6H, CH3), 6.63- 6.65 (m, 3H, C=CH, Ar–H), 6.81 (d, J = 16.2 Hz, 1H, Ar–CH), 7.02–7.05 (m, 2H, Ar–H), 7.32 (d, J = 9.0 Hz, 2H, Ar–H), 7.40 (d, J = 7.8 Hz, 1H Ar–H), 7.66 (d, J = 9 Hz, 1H, N=CH), 8.74 (s, 1H, NH–CS), 9.81 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 17.9, 21.1, 40.2, 77.2, 112.0, 119.2, 123.8, 127.4, 128.6, 129.5, 131.3, 134.2, 137.0, 141.5, 146.0, 151.1, 176.3, FT-IR (υmax, cm−1): 1197 (C=S), 1595 (C=N), 3297 (N–H); ESI MS (m/z) = M+H = 353.17.
(E)-N-(4-Chlorophenyl)-2-((E)-3-(4-(dimethylamino)phenyl)allylidene)hydrazine-1-carbothioamide (3m)
Yield: 90%, color: brownish yellow; M.P: 238 °C; 1H NMR (DMSO-d6, δ, ppm): 2.90 (s, 6H, CH3), 6.54–6.58 (m, 3H, C=CH, Ar–H), 6.60 (d, J = 15.6 Hz, 1H), 7.19 (d, J = 8.4 Hz, 2H, Ar–H), 7.23 (d, J = 8.4 Hz, 2H, Ar–H), 7.56 (d, J = 8.4 Hz, 2H, Ar–H), 7.80 (d, J = 9.0 Hz, 1H, N=CH), 9.2 (s, 1H, NH–CS), 11.3 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 40.0, 77.7, 112.0, 119.8, 123.8, 125.4, 128.5, 130.0, 137.4, 141.1, 146.9, 150.9, 174.9, FT-IR (υmax, cm−1): 1185 (C=S), 1579 (C=N), 3289 (N–H); ESI MS (m/z) = M+H = 359.11.
(E)-2-((E)-3-(4-(Dimethylamino)phenyl)allylidene)-N-(4-(fluorobenzyl)hydrazine-1-carbothioamide (3n)
Yield: 80%, color: brownish yellow; M.P: 192 °C; 1H NMR (DMSO-d6, δ, ppm): 2.96 (s, 6H, CH3), 4.86 (d, J = 4.8 Hz, 2H, Ar–CH2), 6.55–6.58 (m, 1H, C=CH, Ar–H), 6.62 (d, J = 7.8 Hz, 2H, Ar–H), 6.78 (d, J = 15.6 Hz, 1H, Ar–CH), 6.99–7.02 (m, 2H, Ar–H), 7.28–7.33 (m, 4H, Ar–H), 7.59–7.62 (m, 2H, NH–CS, N=CH), 9.7 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 40.2, 47.5, 111.9, 115.5 (ICF = 85.32 Hz), 119.2, 123.7, 128.6, 129.5(ICF = 26.95 Hz), 133.5(ICF = 10.05 Hz), 146.1, 151.0, 161.5, 163.1, 176.9, FT-IR (υmax, cm−1): 1183 (C=S), 1589 (C=N), 3286 (N–H); ESI MS (m/z) = M+H = 357.15.
(E)-N-(3-Cyanophenyl)-2-((E)-3-(4-(dimethylamino)phenyl)allylidene)hydrazine-1-carbothioamide (3o)
Yield: 70%, color: brownish yellow; M.P: 215 °C; 1H NMR (DMSO-d6, δ, ppm): 2.98 (s, 6H, CH3), 6.60–6.64 (m, 3H, C=CH, Ar–H), 6.86 (d, J = 15.6 Hz, 1H, Ar–CH), 7.33 (d, J = 8.4 Hz, 2H, Ar–H), 7.43 (d, J = 5.4 Hz, 2H, Ar–H,), 7.71 (d, J = 9.0 Hz, 1H, N=CH), 7.92–7.93 (m, 1H, Ar–), 8.09 (s, 1H, Ar–H), 9.2 (s, 1H, NH–CS), 10.21 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 40.2, 77.1, 112.0, 112.6, 118.5, 118.6,123.5, 126.8, 128.0,128.8, 129.5, 139.1, 142.7, 147.1, 151.2, 174.4, FT-IR (υmax, cm−1): 1187 (C=S), 1583 (C=N), 3281 (N–H); ESI MS (m/z) = M+H = 350.14.
(E)-2-((E)-3-(4-(Dimethylamino)phenyl)allylidene)-N-phenylhydrazine-1-carbothioamide (3p)
Yield: 75%, color: orange yellow; M.P: 218 °C; 1H NMR (DMSO-d6, δ, ppm): 2.98 (s, 6H, CH3), 6.65–6.69 (m, 3H, C=CH, Ar–H), 6.84 (d, J = 15.6 Hz, 1H, Ar–CH), 7.10 (t, J = 7.2 Hz, 1H, Ar–H), 7.35–7.37 (m, 4H, Ar–H), 7.64–7.67 (m, 3H, Ar–H, N=CH), 9.1 (s, 1H, NH–CS), 9.7 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 40.2, 77.3, 112.1, 119.1, 124.0, 125.8, 128.7, 129.6, 138.1, 141.8, 146.1, 151.1, 174.9, FT-IR (υmax, cm−1): 1184 (C=S), 1588 (C=N), 3287 (N–H); ESI MS (m/z) = M+H = 325.14.
(E)-N-Cyclohexyl-2-((E)-3-(4-(dimethylamino)phenyl)allylidene)hydrazine-1-carbothioamide (3q)
Yield: 80%, color: pale yellow; M.P: 252 °C; 1H NMR (DMSO-d6, δ, ppm): 1.17–1.28 (m, 5H, Cyclohexyl), 1.37–1.43 (m, 2H, Cyclohexyl), 1.61–1.63 (m, 1H, Cyclohexyl), 1.71–1.75 (m, 2H, Cyclohexyl), 2.97 (s, 6H, CH3), 4.20–4.27 (m, 1H, Cyclohexyl), 6.58–6.65 (m, 3H, C=CH, Ar–H), 6.76 (d, J = 17.4 Hz, 1H, Ar–CH), 7.20 (t, J = 8.4 Hz, 1H, NH–CS), 7.54 (d, J = 9.0 Hz, 2H, N=CH), 9.25 (s, 1H, N–NH–CS). 13C NMR (δ, ppm): 25.5, 32.8, 40.2, 53.0, 77.0, 111.9, 119.4, 123.9, 128.5, 140.9, 145.4, 151.0, 175.3, FT-IR (υmax, cm−1): 1181 (C=S), 1586 (C=N), 3288 (N–H); ESI MS (m/z) = M+H = 331.19.
Carbonic anhydrase II (CA-II) inhibition protocol
Evaluation of carbonic anhydrase inhibition was done by spectrophotometric technique as narrated by Pocker and Meany with little alteration69,70 as described previously3. The process was carried out in HEPES-Tris buffer (20 mM) having pH 7.4 at 25 °C. Each inhibitory well contained 20 µL of CA-II enzyme solution (0.1 mg/mL HEPES-Tris buffer), 140 µL of HEPES-Tris buffer solution, 20 µL of the compound to be analyzed in HPLC grade DMSO. At 25 °C, the solution mixture was pre-incubated for 15 min. Substrate p-nitrophenyl acetate (p-NPA) (0.7 mM) was prepared in HPLC grade methanol and the reaction was initiated by the addition of 20 µL to a well in a 96-well-plate. The quantity of product formed was measured at 400 nm continually at 1 min gap for 30 min in a 96-well-plate using xMARK microplate spectrophotometer, Bio-Rad (USA). The inhibition activity of the controlled compound was considered as 100%. All the experiments were performed in triplicates of each used concentration, and the conclusions are expressed as a mean of the triplicate.
Molecular docking
The X-ray crystal structure of human carbonic anhydrase II in complex with the standard drug, acetazolamide (PDB code: 3HS4, resolution: 1.10 Å) was selected from RCSB-Protein databank for molecular docking71, which was performed on Molecular Operating Environment (MOE version 2020.0901)72. Previously, we have carried out re-docking to test the docking performance of MOE and MOE showed outstanding result73,74,75. In this work, the protein file was prepared for docking by adding missing hydrogen atoms on protein by QuickPrep module of MOE. QuickPrep also calculates partial charges using Amber10:EHT force field. The two-dimensional structures of active CA-II inhibitors were drawn by ChemDraw and imported into MOE database. In MOE database, MOE-WASH module was used to convert 2D strictures into three-dimensional format by which all hydrogen atoms and partial charges on all the compounds and minimize the structure of each ligand with RMS gradient of 0.1RMS kcal/mol/Å. The docking was performed with Triangle Matcher docking algorithm and London dG scoring function. After docking, the best docked conformation of each ligand was selected based on good docking score and good binding interaction.
Results and discussion
Chemistry
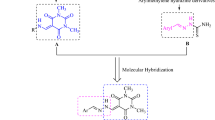
To discover the biological potential of 4-(dimethylamino)-cinnamaldehyde-based thiosemicarbazones (3a–q), a thiosemicarbazones (TSC) series bearing cycloalkyl and aryl substituents were synthesized. A classic method of condensation was adopted in which various thiosemicarbazides (2a–q) were reacted with 4-(dimethylamino)-cinnamaldehyde (1). The reaction was conducted in methanol using 1–2 drops of glacial acetic acid. The optimum conditions were set by the reaction of phenyl thiosemicarbazide (16) and 4-dimethylamino cinnamaldehyde (1) in a 1:1 ratio and by shifting the polarity of solvent from polar to non-polar i.e., methanol, DMSO, ethanol, THF, and DCM. The reaction conditions were optimized by using methanol as solvent and catalytic amount of glacial acetic acid (1-2 drops). The extent of the reaction was investigated and expanded by utilizing thiosemicarbazides (2a–q) with various cycloalkyl, heterocyclic, and aryl substituents at the N-4 position of thiosemicarbazides. The aimed compounds (3a–q) were obtained in good to outstanding yields (46–98%).
The structures of 4-(dimethylamino)-cinnamaldehyde conjugates of N-4 thiosemicarbazides (3a–q) were established by using spectral data i.e., IR, 1H-NMR, 13C-NMR (S.I. 1–34), microanalysis (CHN), and ESI spectrometry. In FTIR, the signal for C=S appeared in the 1177–1246 cm−1 range while the C=N band for several derivatives appeared ranging from 1484 to 1605 cm−1. N–H stretching appeared in the 3162–3469 cm−1 range. In the 1HNMR, the N=CH signal was seen as a doublet at δ 7.54–7.91 ppm, though the N–NH–C=S appeared as a singlet at δ 9.21–11.59 ppm; and CH3 linked with N-CH2 was seen as a triplet in the range of δ 1.08–1.11 ppm. The NH–CS proton displayed variable behavior and appeared as a singlet in the range of δ 9.04–9.49 ppm when directly attached to an aromatic ring. While, in the compounds 3c, 3k, and 3n, the NH–CS appeared as a multiplet in the range of δ: 7.59–7.63 ppm because of the presence of neighboring CH2-group while in TSC 3q bearing N-4 cyclohexyl substituent, a doublet appeared for NH-CS at δ: 7.20 ppm (J = 8.4 Hz). The spectral details of other aromatic and aliphatic protons were also following the proposed compounds. In 13C NMR spectra of 3n, owing to the presences of F, coupling of C-F was also observed.
In-vitro inhibition of bCA-II and structure activity relationship (SAR)
All thiosemicarbazones derivatives (3a–q) were tested against bovine carbonic anhydrase II (bCA-II). The assay was conducted at µM level and acetazolamide was used as a reference inhibitor. The novel derivatives displayed effective inhibition of bCA-II, with inhibition constant (IC50) ranging from 10.3 ± 0.62–46.6 ± 0.62 µM except for 3j, 3p, and 3q which showed activity less than 50% (Table 1). Additionally, the compounds of interest 3f, 3g, 3h, 3m, and 3n displayed higher inhibitory activity than the standard drug, acetazolamide (IC50 = 18.2 ± 0.43 µM). Among all the synthesized compounds, the best bCA-II inhibitor was 3n (IC50 = 10.3 ± 0.62 µM), followed by 3g (IC50 = 12.1 ± 1.01 µM), 3h (IC50 = 13.4 ± 0.52 µM), 3m (IC50 = 14.7 ± 1.42 µM) and 3f (IC50 = 17.9 ± 1.23 µM). While compounds 3l, 3e and 3o depicted inhibitory activities like acetazolamide with IC50 values in range of 18.3 ± 1.32–21.8 ± 0.85 µM. Moreover, 3i, 3d, 3c, 3b, 3a and 3k were found as least active inhibitors with IC50 in range of 33.6–49.6 µM.
The preliminary SAR suggested that the compounds showed inhibitory activity mainly due to the presence of thiosemicarbazide moiety. However, we further investigate the effect of various substitutions on the phenyl ring of the thiosemicarbazide. It can be seen in Scheme 1, the derivatives 3n (4-fluorobenzyl), 3g (4-bromophenyl) and 3h (4-fluorophenyl), showed superior activity, as compared to acetazolamide, and other derivatives of this series. It indicates that electron donating groups (i.e., flouro and bromo) at the 4-position of phenyl or benzyl rings are highly important for the potent activity of these compounds against bCA-II. It was further proved when the activity of 3n was compared with the biological activity of 3a (4-CH3). A significant decrease in the inhibitory potential of 3a indicates the importance of electronegative atom at para position of substituted phenyl ring. Whereas other derivatives including 3l (2, 4-CH3), 3e (2-F) and 3o (3-CN) substituted phenyl ring, respectively, showed comparable activity when compared to acetazolamide, however these compounds exhibited less potent inhibitory activity as compared to 3n, 3g and 3h. As the variation occurred only at “R” position, thus we could generate limited SAR. The substituents on phenyl ring reflects the importance of position with regards to the inhibitory activities of compounds. As we noted in derivatives 3e (2-F) and 3j (3-F) the position of fluorine atom is changed, thus significant variation in the activity was observed.

The synthetic route of compounds 3(a–q).
Whereas compounds 3a (4-CH3), 3b (2, 6-CH3), 3c (4-C2H5), 3d (2, 3-Cl), 3i (4-C3H7) and 3k (4-CH2Cl) on the phenyl ring showed inferior activity as compared to acetazolamide. The SAR suggests that the nature of atom (either electron donating or electron withdrawing), and their position on the phenyl ring is highly important for the potent inhibitory activity of the compounds.
Molecular docking studies
Computational docking was employed to study the binding behavior of compounds at the active site of CA-II. All the compounds were fitted at the entrance of the active site, thus block the access of the substrate, therefore inhibit the normal function of CA-II. The docked orientation of the most active compound 3n, reflect that the dimethylamino-cinnamaldehyde moiety of the compound interact at the entrance of the enzyme, while the thiosemicarbazide group interacted with the side chain of Thr200 through H-bond, and the thio group mediated H-bond with the amino group of Thr199 and ionic interaction with Zn ion at the core of the active. The docking score (DS) of 3n is highly negative (− 6.75 kcal/mol) suggesting the higher inhibitory potential of this compound against CA-II, which is correlated with our experimental result.
Subsequently, four compounds (3g, 3h, 3m and 3f) exhibited higher inhibitory potency as compared to the standard compound, acetazolamide. The biological activity of these compounds was in range of 12.1–17.9 µM. The binding mode of 3g depict that it adapted the same binding mode of 3n, however its para substituted Br atom did not interacted with the surrounding residues due to steric hindrance, however, its thiosemicarbazide group mediated H-bonding with the side chains of Asn62 and Gln92, thus this compound exhibited inhibitory activity comparable with the biological activity of 3n. The substitution of fluorine group at ortho-para position of compound 3h caused the compound to tilt from core of active site towards hydrophobic residues (Trp5, His64 and Ala65). However, the thio group of 3h was oriented towards the core where the side chain of Thr200 provided H-bond to the thio group, and its linked amino group formed a H-bond with the side chain of Gln92. These strong interactions are responsible for good inhibitory activity of this compound. Similar conformation was adapted by compound 3m, which indicates that the addition of bulky group at substituted phenyl ring creates steric hindrance at the core of active site, as a result, phenyl ring bend towards the loop region (Trp5, His64 and Ala65). The carbazide amino group of 3m retained H-bonding with the side chain of Gln92, however its thio group lost interaction with the Thr200. The binding orientation of compound 3f was similar with the docked orientation of 3g. The thio and the amino groups of 3f were stabilized by the side chains of Asn62 and Gln92, respectively by H-bonding, however its substituted methoxy group at meta position of phenyl ring did not interact with the active residues. The compound 3l exhibited the inhibitory activity comparable to the acetazolamide. The binding mode of 3l was found similar with the docked conformations of 3g and 3f. The addition of dimethyl group at meta para positions of phenyl ring caused the compound 3l to move more towards the entrance of the active site, however its thio group mediate bidentate interaction with the side chains of Asn62 and Asn67. The addition of fluorine at ortho position of phenyl ring of compound 3e produced similar binding effects like 3h. The fluorophenyl and cyanophenyl rings of 3e and 3o, respectively were tilted towards His64, and Ala65, however, the thio group of 3e accepted two H-bonds with the side chains of Thr199 and Thr200, and the thiosemicarbazone moiety of 3o formed strong H-bonding with the side chains of Gln92 and Thr200. In addition, the side chain of His94 provided hydrophobic interaction to the substituted phenyl rings of both the compounds.
The compounds 3i, 3d, and 3c exhibited inhibitory potential with IC50 values in range of 33.6–39.7 µM, while compounds 3b, 3a and 3k displayed lowest inhibitory activities with IC50 ranging from 40.4 to 49.6 µM. The observed binding modes of these compounds (3i, 3d, 3c, 3b, 3a and 3k) reflect that those compounds mediated only one H-bond interaction either with the side chain of Gln92, Asn62 or Asn67, thus the loss of higher number of H-bonds with surrounding residues or ionic interaction with Zn ion in the active site could be the possible reason of lower activity of those compounds. Moreover, the binding score of these compounds are also in range of − 5.73 to − 4.42 kcal/mol, which is lowest score among all the compounds. The docking scores and the binding interactions of each docked ligand with the active site residues of bCA-II are given in Table 2. The docking results match with our in vitro results. The docked conformations of all the active inhibitors are displayed in Fig. 2.

(A) The binding mode of the most active compound (3n) is shown. The ligand is depicted in cyan ball and stick model, interacting residues are demonstrated in magenta stick model, and hydrogen bonds are presented in black dotted lines. (B) The docked conformations of all the active ligands are displayed in the active site of CA-II. Ligands are presented in magenta color, the active site residues are shown in orange stick, H-bonds are depicted in black dotted lines. The protein in shown in surface model.
Conclusion
In summary, we have designed, synthesized, and evaluated a series of thiosemicarbazide derivatives as carbonic anhydrase-II inhibitors. These compounds could be further investigated for the treatment of carbonic anhydrase related biological disorders. Several compounds showed potent activity (i.e., 3f, 3g, 3h, 3l, 3m, and 3n) in in vitro biochemical assay. The active molecules were scrutinized in silico by molecular docking method to study their mode of binding within the active site of bCA-II. We observed that the high active leads bind specifically with Gln92, Thr199, Thr200, Asn62, and Asn67 via H-bonding, thus significantly inhibit the enzyme activity. Based on the promising in vitro and in silico results, these lead molecules can be a good choice for the in vivo studies. The identified compounds can be used as a good template drug for the diseases associated with the hyperactivity of CA-II. Moreover, these compounds will be also checked on CA-II from human source to know their selectivity.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
Mishra, C. B., Tiwari, M. & Supuran, C. T. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: Where are we today?. Med. Res. Rev. 40, 2485–2565. https://doi.org/10.1002/med.21713 (2020).
Yang, Y. et al. A novel homozygous nonsense mutation in the CA2 gene (c. 368G> A, p. W123X) linked to carbonic anhydrase II deficiency syndrome in a Chinese family. Metab. Brain Dis. 36, 589–599 (2021).
Khan, A. et al. Quinazolinones as competitive inhibitors of carbonic anhydrase-II (human and bovine): synthesis, in-vitro, in-silico, selectivity, and kinetics studies. Front. Chem. 8, 25 (2020).
Tinivella, A., Pinzi, L. & Rastelli, G. Prediction of activity and selectivity profiles of human Carbonic Anhydrase inhibitors using machine learning classification models. J. Cheminform. 13, 1–15 (2021).
Kose, L. P. et al. The effects of some avermectins on bovine carbonic anhydrase enzyme. J. Enzyme Inhibit. Med. Chem. 31, 773–778 (2016).
Kusian, B., Sültemeyer, D. & Bowien, B. Carbonic anhydrase is essential for growth of Ralstonia eutropha at ambient CO2 concentrations. J. Bacteriol. 184, 5018–5026 (2002).
Krishnamurthy, V. M. et al. Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein−ligand binding. Chem. Rev. 108, 946–1051 (2008).
Burger, A. Medicinal Chemistry and Drug Discovery: Cardiovascular Agents and Endocrines, Vol*** 3 (Wiley, 2003).
Supuran, C. T. & Scozzafava, A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg. Med. Chem. 15, 4336–4350 (2007).
Pastorekova, S. & Supuran, C. Carbonic Anhydrase IX: From Biology to Therapy In Hypoxia and Cancer, Cancer Drug Discovery and Development (Springer, 2014).
Ruusuvuori, E. & Kaila, K. Carbonic anhydrases and brain pH in the control of neuronal excitability. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications, Subcellular Biochemistry, Vol*** 75 (eds Frost, S. & McKenna, R.) 271–290 (Springer, 2014).
Zaraei, S.-O. et al. Sulfonate and sulfamate derivatives possessing benzofuran or benzothiophene nucleus as potent carbonic anhydrase II/IX/XII inhibitors. Bioorg. Med. Chem. 27, 3889–3901 (2019).
Frazier, M. L., Lilly, B. J., Wu, E. F., Ota, T. & Hewett-Emmett, D. Carbonic anhydrase II gene expression in cell lines from human pancreatic adenocarcinoma. Pancreas 5, 507–514 (1990).
Pastorekova, S. et al. Carbonic anhydrase IX, MN/CA IX: analysis of stomach complementary DNA sequence and expression in human and rat alimentary tracts. Gastroenterology 112, 398–408 (1997).
Parkkila, S. et al. Carbonic anhydrase inhibitor suppresses invasion of renal cancer cells in vitro. Proc. Natl. Acad. Sci. 97, 2220–2224 (2000).
Malikowski, T., Duffey, M. & Patel, S. Different roles of carbonic anhydrase in human vs bovine corneal endothelial transport. Investig. Ophthalmol. Vis. Sci. 54, 5409–5409 (2013).
Malikowski, T. M., Bosch, J. B., Min, S., Duffey, M. E. & Patel, S. P. Carbonic anhydrase inhibitors in corneal endothelial transport. Invest. Ophthalmol. Vis. Sci. 55, 2652–2658 (2014).
Matsa, R., Makam, P., Kaushik, M., Hoti, S. & Kannan, T. Thiosemicarbazone derivatives: Design, synthesis and in vitro antimalarial activity studies. Eur. J. Pharm. Sci. 137, 104986 (2019).
Prajapati, N. P. & Patel, H. D. Novel thiosemicarbazone derivatives and their metal complexes: Recent development. Synth. Commun. 49, 2767–2804 (2019).
Afrasiabi, Z. et al. Transition metal complexes of phenanthrenequinone thiosemicarbazone as potential anticancer agents: synthesis, structure, spectroscopy, electrochemistry and in vitro anticancer activity against human breast cancer cell-line, T47D. J. Inorg. Biochem. 95, 306–314 (2003).
Byrnes, R. W., Mohan, M., Antholine, W. E., Xu, R. X. & Petering, D. H. Oxidative stress induced by a copper-thiosemicarbazone complex. Biochemistry 29, 7046–7053 (1990).
Kolocouris, A. et al. New 2-(1-adamantylcarbonyl) pyridine and 1-acetyladamantane thiosemicarbazones–thiocarbonohydrazones: cell growth inhibitory, antiviral and antimicrobial activity evaluation. Bioorg. Med. Chem. letters 12, 723–727 (2002).
Baldini, M. et al. Copper (II) complexes with substituted thiosemicarbazones of α-ketoglutaric acid: synthesis, X-ray structures, DNA binding studies, and nuclease and biological activity. Inorg. Chem. 43, 7170–7179 (2004).
Klayman, D. L., Bartosevich, J. F., Griffin, T. S., Mason, C. J. & Scovill, J. P. 2-Acetylpyridine thiosemicarbazones. 1. A new class of potential antimalarial agents. J. Med. Chem. 22, 855–862 (1979).
Bermejo, E. et al. Complexes of group 12 metals with 2-acetylpyridine 4N-dimethylthiosemicarbazone and with 2-acetylpyridine-N-oxide 4N-dimethylthiosemicarbazone: synthesis, structure and antifungal activity. Z. Nat. B 54, 777–787 (1999).
Iakovidou, Z. et al. Platinum (II) and palladium (II) complexes with 2-acetylpyridine thiosemicarbazone: cytogenetic and antineoplastic effects. Anticancer Drugs 12, 65–70 (2001).
Pham, V. H., Phan, T. P. D., Phan, D. C. & Vu, B. D. Synthesis and bioactivity of thiosemicarbazones containing adamantane skeletons. Molecules 25, 324 (2020).
Arooj, M. et al. Coumarin based thiosemicarbazones as effective chemosensors for fluoride ion detection. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 261, 120011 (2021).
Hashmi, S. et al. Probing 4-(diethylamino)-salicylaldehyde-based thiosemicarbazones as multi-target directed ligands against cholinesterases, carbonic anhydrases and α-glycosidase enzymes. Bioorg. Chem. 107, 104554 (2021).
Haribabu, J. et al. Isatin based thiosemicarbazone derivatives as potential bioactive agents: Anti-oxidant and molecular docking studies. J. Mol. Struct. 1110, 185–195 (2016).
Pingaew, R., Prachayasittikul, S. & Ruchirawat, S. Synthesis, cytotoxic and antimalarial activities of benzoyl thiosemicarbazone analogs of isoquinoline and related compounds. Molecules 15, 988–996 (2010).
de Oliveira, R. B. et al. Synthesis and antimalarial activity of semicarbazone and thiosemicarbazone derivatives. Eur. J. Med. Chem. 43, 1983–1988 (2008).
Sheikhy, M., Jalilian, A. R., Novinrooz, A. & Motamedi-Sedeh, F. Synthesis and in vitro antibacterial evaluation of some thiosemicarbazides and thiosemicarbazones. J. Biomed. Sci. Eng. 5, 17616 (2012).
El-Sharief, M. A. S., Abbas, S. Y., El-Bayouki, K. A. & El-Gammal, E. W. Synthesis of thiosemicarbazones derived from N-(4-hippuric acid) thiosemicarbazide and different carbonyl compounds as antimicrobial agents. Eur. J. Med. Chem. 67, 263–268 (2013).
Opletalovà, V. et al. Identification and characterization of thiosemicarbazones with antifungal and antitumor effects: cellular iron chelation mediating cytotoxic activity. Chem. Res. Toxicol. 21, 1878–1889 (2008).
Siwek, A., Stefańska, J., Dzitko, K. & Ruszczak, A. Antifungal effect of 4-arylthiosemicarbazides against Candida species. Search for molecular basis of antifungal activity of thiosemicarbazide derivatives. J. Mol. Model. 18, 4159–4170 (2012).
Kulandaivelu, U. et al. Synthesis, antimicrobial and anticancer activity of new thiosemicarbazone derivatives. Arch. Pharm. 344, 84–90 (2011).
Altıntop, M. D. et al. Synthesis and biological evaluation of new naphthalene substituted thiosemicarbazone derivatives as potent antifungal and anticancer agents. Eur. J. Med. Chem. 108, 406–414 (2016).
Wang, Y. et al. Design, synthesis and anticancer activity of novel nopinone-based thiosemicarbazone derivatives. Bioorg. Med. Chem. letters 27, 2360–2363 (2017).
de Oliveira, J. F. et al. Thiosemicarbazones and 4-thiazolidinones indole-based derivatives: synthesis, evaluation of antiproliferative activity, cell death mechanisms and topoisomerase inhibition assay. Eur. J. Med. Chem. 136, 305–314 (2017).
Hu, K., Yang, Z.-H., Pan, S.-S., Xu, H.-J. & Ren, J. Synthesis and antitumor activity of liquiritigenin thiosemicarbazone derivatives. Eur. J. Med. Chem. 45, 3453–3458 (2010).
Muñoz-Ruiz, P. et al. Design, synthesis, and biological evaluation of dual binding site acetylcholinesterase inhibitors: new disease-modifying agents for Alzheimer’s disease. J. Med. Chem. 48, 7223–7233. https://doi.org/10.1021/jm0503289 (2005).
Pacca, C. C. et al. Thiosemicarbazones and Phthalyl–Thiazoles compounds exert antiviral activity against yellow fever virus and Saint Louis encephalitis virus. Biomed. Pharmacother. 87, 381–387 (2017).
Yanardag, R. et al. Synthesis, characterization and antidiabetic properties of N1–2, 4-dihydroxybenzylidene-N4-2-hydroxybenzylidene-S-methyl-thiosemicarbazidato-oxovanadium (IV). Eur. J. Med. Chem. 44, 818–826 (2009).
Shehzad, M. T. et al. Benzoxazinone-thiosemicarbazones as antidiabetic leads via aldose reductase inhibition: Synthesis, biological screening and molecular docking study. Bioorg. Chem. 87, 857–866 (2019).
Shehzad, M. T. et al. Exploring antidiabetic potential of adamantyl-thiosemicarbazones via aldose reductase (ALR2) inhibition. Bioorg. Chem. 92, 103244 (2019).
Basri, R. et al. Exploration of chromone-based thiosemicarbazone derivatives: SC-XRD/DFT, spectral (IR, UV–Vis) characterization, and quantum chemical analysis. ACS Omega 5, 30176–30188 (2020).
Heffeter, P. et al. Anticancer thiosemicarbazones: chemical properties, interaction with iron metabolism, and resistance development. Antioxid. Redox Signal. 30, 1062–1082 (2019).
Gou, Y. et al. α-N-heterocyclic thiosemicarbazone Fe (III) complex: Characterization of its antitumor activity and identification of anticancer mechanism. Eur. J. Med. Chem. 123, 354–364 (2016).
Islam, M. et al. Synthesis and characterization of new thiosemicarbazones, as potent urease inhibitors: In vitro and in silico studies. Bioorg. Chem. 87, 155–162 (2019).
Letizia, C. S., Cocchiara, J., Lapczynski, A., Lalko, J. & Api, A. M. Fragrance material review on cinnamic acid. Food Chem. Toxicol. Int. J. Publ. Brit. Ind. Biol. Res. Assoc. 43, 925–943. https://doi.org/10.1016/j.fct.2004.09.015 (2005).
Chen, B. J. et al. Cinnamaldehyde analogues as potential therapeutic agents. Mini. Rev. Med. Chem. 17, 33–43. https://doi.org/10.2174/1389557516666160121120744 (2017).
Mohamed Jawed, A. et al. Synthesis, antiproliferative, and antioxidant activities of substituted N-[(1,3,4-oxadiazol-2-yl) methyl] benzamines. Lett. Drug Des. Discov. 17, 145–154. https://doi.org/10.2174/1570180816666181113110033 (2020).
Glomb, T., Szymankiewicz, K. & Świątek, P. Anti-cancer activity of derivatives of 1,3,4-oxadiazole. Molecules 23, 3361 (2018).
Barygina, V. et al. Treatment with low-dose cytokines reduces oxidative-mediated injury in perilesional keratinocytes from vitiligo skin. J. Dermatol. Sci. 79, 163–170. https://doi.org/10.1016/j.jdermsci.2015.05.003 (2015).
Basak, P. Y., Gultekin, F. & Kilinc, I. The role of the antioxidative defense system in papulopustular acne. J. Dermatol. 28, 123–127 (2001).
Wei, Q.-Y., Xiong, J.-J., Jiang, H., Zhang, C. & Ye, W. The antimicrobial activities of the cinnamaldehyde adducts with amino acids. Int. J. Food Microbiol. 150, 164–170 (2011).
Han, Y. et al. Mesenchymal stem cells for regenerative medicine. Cells 8, 886 (2019).
Bulut, N. et al. Synthesis of some novel pyridine compounds containing bis-1, 2, 4-triazole/thiosemicarbazide moiety and investigation of their antioxidant properties, carbonic anhydrase, and acetylcholinesterase enzymes inhibition profiles. J. Biochem. Mol. Toxicol. 32, e22006 (2018).
Akocak, S., Lolak, N., Bua, S. & Supuran, C. T. Discovery of novel 1, 3-diaryltriazene sulfonamides as carbonic anhydrase I, II, VII, and IX inhibitors. J. Enzyme Inhib. Med. Chem. 33, 1575–1580 (2018).
Askin, S. et al. Design, synthesis, characterization, in vitro and in silico evaluation of novel imidazo [2, 1-b][1, 3, 4] thiadiazoles as highly potent acetylcholinesterase and non-classical carbonic anhydrase inhibitors. Bioorg. Chem. 113, 105009 (2021).
Hewitt, C. S. et al. Structure–activity relationship studies of acetazolamide-based carbonic anhydrase inhibitors with activity against Neisseria gonorrhoeae. ACS Infect. Dis. 7, 1969–1984 (2021).
Dizdaroglu, Y. et al. Design, synthesis and molecular modelling studies of some pyrazole derivatives as carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 35, 289–297 (2020).
Eldehna, W. M. et al. Benzofuran-based carboxylic acids as carbonic anhydrase inhibitors and antiproliferative agents against breast cancer. ACS Med. Chem. Lett. 11, 1022–1027 (2020).
Nocentini, A. et al. Discovery of new sulfonamide carbonic anhydrase IX inhibitors incorporating nitrogenous bases. ACS Med. Chem. Lett. 8, 1314–1319 (2017).
Sever, B., Türkeş, C., Altıntop, M. D., Demir, Y. & Beydemir, Ş. Thiazolyl-pyrazoline derivatives: In vitro and in silico evaluation as potential acetylcholinesterase and carbonic anhydrase inhibitors. Int. J. Biol. Macromol. 163, 1970–1988 (2020).
Pedrood, K. et al. Design, synthesis, characterization, enzymatic inhibition evaluations, and docking study of novel quinazolinone derivatives. Int. J. Biol. Macromol. 170, 1–12 (2021).
Khan, M. et al. Inhibitory efficacy of thiosemicarbazones for carbonic Anhydrase II (bovine and human) as a target of calcification and tumorigenicity. Curr. Pharm. Des. 20, 25 (2022).
Pocker, Y. & Meany, J. The catalytic versatility of erythrocyte carbonic anhydrase. II. Kinetic studies of the enzyme-catalyzed hydration of pyridine aldehydes. Biochemistry 6, 239–246 (1967).
Ur Rehman, N. et al. Antiproliferative and carbonic anhydrase II inhibitory potential of chemical constituents from Lycium shawii and aloe vera: evidence from in silico target fishing and in vitro testing. Pharmaceuticals 13, 94 (2020).
Sippel, K. H. et al. High-resolution structure of human carbonic anhydrase II complexed with acetazolamide reveals insights into inhibitor drug design. Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun. 65, 992–995 (2009).
Molecular Operating Environment (MOE), C. C. G. U., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2022.
Khan, I. et al. Utilization of the common functional groups in bioactive molecules: Exploring dual inhibitory potential and computational analysis of keto esters against α-glucosidase and carbonic anhydrase-II enzymes. Int. J. Biol. Macromol. 167, 233–244 (2021).
Avula, S. K. et al. Synthesis of new 1H–1, 2, 3-triazole analogs in aqueous medium via “Click” chemistry: A novel class of potential carbonic anhydrase-II inhibitors. Front. Chem. 9, 642614 (2021).
Rehman, N. U. et al. Commikuanoids AC: New cycloartane triterpenoids with exploration of carbonic anhydrase-II inhibition from the resins of Commiphora kua by in vitro and in silico molecular docking. Fitoterapia 20, 105125 (2022).
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through under Grant number (RGP. 2/106/43). Z. S is thankful to the Alexander von Humboldt Foundation for the award of Georg Forster Research Fellowship for Experienced Researchers.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.R., Z.B. and M. K. carried out synthesis and interpretation of spectral data; S.A.H and A.K did biological analysis and molecular docking; A.T. did SAR analysis; Z.S. A.K and A. Al.H. developed the concept and wrote the main manuscript. M.A.S and T.E.A repeated some spectra and interpreted the results / revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rasool, A., Batool, Z., Khan, M. et al. Bis-pharmacophore of cinnamaldehyde-clubbed thiosemicarbazones as potent carbonic anhydrase-II inhibitors. Sci Rep 12, 16095 (2022). https://doi.org/10.1038/s41598-022-19975-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-19975-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
