
Abstract
Pristine zirconia, ZrO2, possesses high premise as photocatalyst due to its conduction band energy edge. However, its high energy-gap is prohibitive for photoactivation by solar-light. Currently, it is unclear how solar-active zirconia can be designed to meet the requirements for high photocatalytic performance. Moreover, transferring this design to an industrial-scale process is a forward-looking route. Herein, we have developed a novel Flame Spray Pyrolysis process for generating solar-light active nano-ZrO2−x via engineering of lattice vacancies, Vo. Using solar photons, our optimal nano-ZrO2−x can achieve milestone H2-production yield, > 2400 μmolg−1 h−1 (closest thus, so far, to high photocatalytic water splitting performance benchmarks). Visible light can be also exploited by nano-ZrO2−x at a high yield via a two-photon process. Control of monomeric Vo versus clusters of Vo’s is the key parameter toward Highly-Performing-Photocatalytic ZrO2−x. Thus, the reusable and sustainable ZrO2−x catalyst achieves so far unattainable solar activated photocatalysis, under large scale production.
Similar content being viewed by others

Introduction
Sunlight energy storage to H2 via photocatalytic water splitting1,2 bears key advantages, such as high energy-storage capacity of H2 141.6 kJ g−1 versus 0.46–0.72 kJ g−1 on Li-batteries3. Water photocatalysis to produce H2 is an inherently green-technology, fully compliant with circular economy4. Moreover, maximization of photocatalytic efficiency in tandem with minimized costs for industrial-scale production is mandatory. So far, most literature reports on high-performance photocatalysts (HPP) refer to nanomaterials with focus on performance optimization5. Except TiO2, industrial-scale and cost-effective production of HPP’s remains at its infancy.
Metal oxides can be HPP’s as long as they fulfill certain key criteria: H2 production and CO2 reduction, are favored by high conduction band (CB) semiconductors. Pristine zirconia (ZrO2) has one of the highest CB-edge energies among metal oxides ECB = − 1100 mV versus NHE. However, its band-gap energy Eg > 5 eV requires highly energetic photons, i.e., λ < 225 nm, which is prohibitive for solar-light harvesting. In this context, pristine ZrO2 despite its established uses in technology, e.g. as refracting coating6, thermal coating7, gas-sensing8, fuel cells9, Water–Gas-Shift reaction10, so far, has not been established as a high-performance photocatalyst. Historically, Sayama and Arakawa11 were the first to observe photocatalytic performance of pristine ZrO2 reporting a rather symbolic H2-production yield 72 μmol g−1 h−1 under X-ray irradiation.
Strategies to ameliorate photocatalytic ZrO2 activity, can include: (i) heteroatom insertion12,13,14,15,16,17 into ZrO2 crystal, or (ii) Oxygen-defects’ creation18,19,20,21,22,23. The influence of heteroatoms in ZrO2 has been investigated12,13,14,15,16,17, in photocatalytic dye degradation or O2 evolution. Cerium-doped ZrO2 (Ce-ZrO2) can be photoactive in visible λ > 420 nm light12. Erbium-doped ZrO214 allowed band-gap tuning towards solar photons, which contributed to improved Methylene-Blue degradation13. Nitrogen15 and carbon17 2p-orbitals can enhance photocatalytic dye degradation via generation of intra-bandgap states. However, in all aforementioned cases, the reported photocatalytic performances, despite improvement, remain by far inferior versus benchmark photocatalysts, such as TiO2 which routinely achieves H2 photoproduction of the order of several (millimoles H2 g−1 h−1) in typical lab set ups24. So far, the only ZrO2-based photocatalyst passing the threshold of (millimoles g−1 h−1) is 2.12 mmol H2 g−1 h−1 by a N-doped ZrO225.
Without heteroatom doping, generation of reduced states in reducible metal-oxides, such as TiO2, are decisively beneficial for H2 photo yield. Examples include the work of Mao et al.23, Naldoni et al.26 and our Flame Spray Pyrolysis (FSP)-made black TiO2−x27. ZrO2 is a notoriously non-reducible oxide28,29, since introduction of Vo’s into the ZrO2 lattice is not favored energetically8,15,16,17,18,19,20, thus specific synthesis methods are needed to achieve reduction of the ZrO2 lattice. To this direction, the most significant performance has been reported by Sinhamahapatra et al.22 and Zu et al.30, where defect-rich ZrO2−x has achieved production of ~ 0.5 mmolg−1 h−1 H2 in both cases, however, this is still significantly below that of TiO226,31.
Regarding material synthesis, none of the so far reported ZrO2−x synthesis methods were designed to be scalable at industrial level. Specifically, previous ZrO2−x synthesis methods include sol-gel19, hydrothermal20, high pressure/temperature processing18,21. More efficient methods able to overcome the non-reducibility of ZrO2 are magnesiothermic22, titaniothermic23 and lithiothermic reduction30 where an elementary heterometal M0 atom, i.e. Li0, Ti0, Mg0, is used to reduce ZrO2 and create the desired Vo at high yield. However, all aforementioned synthesis routes include multiple steps and do not always allow facile/reproducible control or tailoring of Vo placement and populations. Particularly, the methods which require heterometal contact on ZrO2 surface e.g. magnesiothermic, rely on harsh acid washes for removing the leftover hetero-metal oxide, something that mounts questions on how this may impact the state of the catalyst itself22.
Herein we have developed a one-step Flame Spray Pyrolysis (FSP) process for synthesis of solar-light active nano-ZrO2−x via engineering of lattice-vacancies, Vo. FSP is eminently suited for synthesis of high crystallinity nano-ZrO232,33, however, the synthesis of ZrO2−x has not been reported by FSP. In principle, ZrO2 can possess two families of reduced states: (i) reduced Zr3+ centers, and (ii) oxygen vacancies not-located on Zr atoms (Vo’s). Over last decades, Giamello’s group34 has provided valuable insights into the complexity of these reduced states. Using Electron Paramagnetic Resonance (EPR) spectroscopy, in combination with quantum chemical calculations34, they prove that Zr3+ centers can create extra energy states right below the ECB of pristine ZrO2, (~ 4.5–5.0 eV). Based on all existing evidence, these Zr3+ centers are expected to have little effect on the photocatalytic activity of zirconia35. On the other hand, Vo’s can create mid-gap states34 but their role in photocatalytic H2 evolution has not been explored systematically. Herein, using FSP we have produced libraries of ZrO2−x nano catalysts with varying concentrations of Vo’s and identified the optimal configuration, towards high photocatalytic H2-production efficiency from H2O. Specific aims of the present work were: (i) to develop a novel industrial-scale FSP method for one-step synthesis of nanosized ZrO2−x with controllable population and placement of the O-vacancies (Vo’s). No heteroatoms were used. (ii) to optimize the ZrO2−x for highly efficient solar light H2 production, well beyond the current state of the art, i.e., well-above the threshold mmol g−1 h−1, (iii) to provide a comprehensive understanding of the physicochemical role of Vo, with emphasis on monomeric versus clusters of Vo’s related to the photocatalytic properties. We present a novel anoxic-FSP process that allows in-situ formation of Vo’s during the primary particle formation step. Using solar photons, the optimal nano-ZrO2−x can achieve milestone H2-production yield, > 2400 μmolg−1 h−1 which is the closest so far to high photocatalytic performance benchmarks. We demonstrate that optimal nano-ZrO2−x can be achieved by controlling the monomeric Vo versus clusters of Vo’s by two routes either during the FSP synthesis or via a short post-FSP oxidation process.
Results
Synthesis of nano ZrO2−x by anoxic-flame spray pyrolysis
The concept of the novel anoxic-FSP process is outlined in Fig. 1. It consists of a single-nozzle FSP reactor with enclosed flame, where a mixture of Dispersion-gas [O2 and CH4] is used to create a reducing reaction atmosphere. In FSP-process35, ZrO2 particles are formed in three stages (Fig. 1). First, Zr-precursor droplets are sprayed by the FSP nozzle and combusted to generate the primary particles (PP)35. Then, primary particles evolve in the high-temperature area of the flame, i.e. up to 2800 K and form nanometric ZrO2 particles via sintering of PP’s35. In classical-FSP, used in the majority of lab studies and industry, pure O2 is used as dispersion gas through the spray nozzle, to form the droplets and primary particles. For example, by adjusting the combustion stoichiometry35 ratio P/D = [fuel/dispersion O2] = 3/3, we obtain fully oxidized, pristine ZrO2. In our anoxic-FSP, combustion of CH4 in the dispersion gas creates reducing agents which, as we show herein, can reduce the primary Zr-particle via formation of Oxygen vacancies (Vo). We also have considered the possibility of the formation of Zr-Hydride states36, however, none of our data support this, thus we do not discuss it further.

One-step FSP process for ZrO2−x production. The photo at right shows as-produced 500 mg of [1.3/1.7] ZrO2−x particles.
In this way, we have produced a library of ZrO2−x nanoparticles of varying Vo concentrations, see photos in Fig. 2. Herein we have codenamed the particles according to the dispersion [O2/CH4] ratio used, (see full details in Table S1 in SI). For example, the material codenamed [3/0.1] has been synthesized using a dispersion gas-mixture of [O2 flow = 3.0 lt min−1 and CH4 flow = 0.1 lt min−1]. Pristine ZrO2 is codenamed F.O. for Fully Oxidized, while material [1.3/1.7] was the most reduced. As shown in Fig. 2, going from pristine ZrO2 (F.O.) towards more reduced particles, i.e. [3/0.1] to [1.3/1.7], induced a change of particle color from crispy-white to pale grey/yellowish [3/0.1] and dark-gray for [1.3/1.7] material. XPS data (Fig. 2d) shows a progressive increase of Vo’s, detected by their characteristic signal at 532 eV37,38. We have verified that no-carbon deposition is evidenced by Raman data, (Supplementary Fig. S1), thus the observed color changes in the ZrO2−x materials (Fig. 2), are assigned exclusively to the formation of Vo’s via the anoxic-FSP process. According to XPS, Fig. 2d increased dispersion of the CH4 in the FSP process, promotes the formation of Vo’s. No Zr3+ states are resolved in Zr-XPS data, i.e., only the Zr4+ doublet was detected (181.9 eV, 184.2 eV) (Supplementary Fig. S2a–d).

(a) XRD of FSP-made ZrO2−x materials, (b) TEM images for [3/0.1] and [1.3/1.7] materials, (c) Kubelka–Munk plots DRS UV–Vis spectra. (d) XPS spectra of F.O., [3/0.1], [3/0.2], [2.3/0.7] and [1.3/1.7]. Top-Inset: Photos of the nanopowders.
The DRS-UV/Vis data, (Fig. 2c) show a progressive alteration of the band gaps as evidenced by the absorbance at intra-gap energies of 2.5 to 4.5 eV. All materials exhibit the primary bandgap 5.1 eV (243 nm) typical for monoclinic-ZrO218,20. Fully oxidized ZrO2 exhibited only the primary bandgap at 5.1 eV with no mid-gap states. Slightly-reduced [3/0.1] exhibited an additional absorbance tail band extending to ~ 3.1 eV. Low concentration of Vo’s, created by 0.1 Lmin−1 CH4, (see XPS data Fig. 2d) [3/0.1], allows absorbance of photons near the middle of the primary bandgap, and creates a pale-yellow color, see photo Fig. 2. Increasing dispersion CH4 (materials [3/0.2], [2.3/0.7], [1.3/1.7]) causes a monotonous enhancement of the intra-band absorbances and grey-color intensification (Fig. 2c). Experimentally18,20,22,23,30 and theoretically29,34 the intra-band absorbances in the range of 2.8–3.5 eV, can be assigned to electrons being injected from the VBmaximum to approximately the middle of the gap, where extra DOS are made available through the introduction of Vo’s. For example, by removing one oxygen atom, a doubly occupied (diamagnetic) F-center39 can be created inside the bandgap ZrO2−x at 3.3 eV39.
Structural characterization
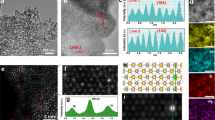
XRD (Fig. 2a) shows that our FSP-made Zr-particles consisted of monoclinic (m-ZrO2 in Fig. 2a) and tetragonal (t-ZrO2 in Fig. 2a) phases, at a ratio [t-ZrO2]: [m-ZrO2] = 4:1 and particle sizes 22–29 nm (monoclinic) to 16–20 nm (tetragonal phase) (Table S2). At ambient P, T, ZrO2 has monoclinic (m) structure with Zr4+ atoms sevenfold coordinated by O-anions (space group P21/c). TEM images, (Figs. 2b, 3), show that all ZrO2−X nanomaterials have quasi-spherical shapes, with highly crystalline Miller planes t-{101}. The more reduced ZrO2−X nanomaterials show some degree of surface distortion, see Fig. 2b. STEM images (Fig. 3a–c) indicate that the particles retain a high degree of crystal quality even at the more reduced ones. In some cases, formation of vacancies could be evidenced in the STEM images, (Fig. 3c). EDX data (Fig. 3) confirm a strong decrease of the Oxygen/Zr ratio in the more reduced [2.3/0.7] and [1.3./1.7] materials. BET analysis (Table S2) shows progressive SSA-decrease upon increase dispersion-CH4, attributed to increased aggregation of the particles at increased CH4, i.e. methane creates hotter flames i.e. methane heat of combustion = 50–55 MJ/kg (https://webbook.nist.gov/chemistry/).

STEM images and EDX spectra of (a) F.O. (b) 2.3/0.7 and (c) 1.3/1.7 materials.
Raman spectra (Fig. 4b) exhibit the vibrational modes from both monoclinic and tetragonal crystal phases40,41,42,43 (Fig. S4 and Table S3) and absence of carbon peaks (Fig. S1). In ZrO2−x materials, certain Raman modes are shifted (see Fig. 4b(I–III) and Table S4). More particularly, material [3/0.1] exhibits upshift of + 4 cm−1 at 313 cm−1 mode (Table S4). Material [2.3/0.7] exhibits downshifts of − 5 cm−1, − 2 cm−1, − 4 cm−1 at 313 472 cm−1, and 643 cm−1 respectively (Table S4). Material [1.3/1.7] exhibits downshift of up to 22 cm−1. Such downshifts can be attributed to tensile stress44 effectively lengthening the bonds, see Fig. 4a, i.e. due to loss of oxygen atoms from the lattice. Raman downshifts prevail in the t-ZrO2 phase; thus, the tetragonal phase is more responsive in the reductive atmosphere in anoxic-FSP. This can be explained by the existence of two different/non-equivalent Zr-O bond conformations in t-ZrO244 corresponding to 4-coordinated Zr(O4f), and 3-coordinated Zr atoms (O3f) respectively44. For comparison, the t-ZrO2 matrix consists solely of O4f Zr atoms while the m-ZrO2 matrix consists of both O4f and O3f44. Accordingly, the present Raman data indicate that removal of oxygen from the ZrO2 matrices is also non-equivalent, thus is easier to extract oxygen from an O4f site rather than an O3f site by 0.1 eV22, therefore it is easier to reduce t-ZrO2.

(a) Unit cell of ZrO2with two indicative t-ZrO2 lattice vibrations and (b) Raman downshifts in reduced ZrO2−x.
Photocatalytic H2 production at (millimole gr−1 h−1)
Figure 5a presents the photocatalytic H2 production from H2O, for all our ZrO2−x materials, under Xenon-illumination. In each panel in Fig. 5a the as-prepared photocatalysts are marked as “a.p”. The time indication in each bar, refers to the post-FSP oxidation-time at 400 °C (see also XRD data in Fig. S3 in S.I.). First, we discuss the as-prepared ZrO2−x materials i.e., see the first bar in each column group in Fig. 5a. Pristine, (F.O.) ZrO2 was practically non-photoactive in H2 production, with a yield of 20 μmol g−1 h−1.

(a) Photocatalytic H2 production from H2O, by as-prepared and oxidized ZrO2−x catalysts under full-solar Xenon-light irradiation. (b) H2 production by [2.3/0.7] ZrO2−x catalyst under various oxidation times (400 °C). (c) Comparison of photocatalytic H2 production by the best performing ZrO2−x catalysts under full-solar Xenon-light versus visible 405 nm LED irradiation. (d) XPS spectra, (e) EPR spectra, indicating the evolution of the Vo’s under oxidation of [2.3./0.7] ZrO2−x. (f) Schematic DOS configuration and excitation under visible 405 nm, see full DFT data in S.I. Fig. S10.
In contrast, a slightly reducing FSP atmosphere, i.e., as-prepared [a.p. 3/0.1], enables an impressive amelioration of H2 evolution of 1700 μmol g−1 h−1. This demonstrates that anoxic-FSP can provide as-prepared ZrO2−x material exhibiting millimoles per gram per hour H2 production. Further increase of O2/CH4 ratio impacted negatively the H2 photogeneration with a tendency towards a steady production near 500 μmol g−1 h−1 of H2 for the highly reduced as-prepared materials (see the first bar in each group in Fig. 5a). Soft oxidation by calcination at 400 °C under ambient O2, exerted a dramatic influence on the H2-photoproduction: a characteristic bell-shaped dependence was observed for the [3.0/0.2] and [2.3/0.7] materials. The optimum oxidation time was 90 min for [2.3/0.7] ZrO2−x which, achieved a remarkable yield of 2428 μmol H2 g−1 h−1. The best performance of the [3.0/0.2] ZrO2−x material was 1500 μmol H2 g−1 h−1 (Fig. 5a). Table S5 summarizes a comparison of FSP-ZrO2−x versus other pertinent ZrO2−x materials reported in the literature (see also Fig. S7).
The catalyst with the higher H2 yield, [2.3/0.7]-90, is highly recyclable, (Fig. S5a), retaining 100% of its activity after two reuses and > 96% after four reuses. XRD (Fig. S5b), shows that the [2.3/0.7]-90 crystal remains intact after 4-uses. Concurrently, DRS-UV/Vis (Fig. S5c), demonstrates that its light-absorbance profile remains also intact. As we discuss hereafter, optimization of monomeric Vo-concentration is determinant for photocatalytic activity. In [2.3/0.7]-90, the monomeric Vo’s is optimized, see EPR and XPS data in Figs. 5d,e, and S6a,b see also the trends in XPS, EPR for [2.3/0.7] in Fig. 5d,e. After photocatalytic use of [2.3/0.7]-90 material, the monomeric vacancies are not altered, neither the ZrO2−x crystal. Thus, the ZrO2−x provide a robust reusable photocatalyst.
Discussion
The data in Fig. 5a demonstrate that there are two options to achieve high-photocatalytic performance ZrO2−x materials: either (i) to be prepared at low O2/CH4 ratio e.g. [3.0/0.1], or (ii) after a soft post-FSP oxidation of more reduced materials e.g. [2.3/0.7]. Very-high O2/CH4 ratio [1.3/1.7] results in highly-reduced ZrO2−x which cannot be improved by post-FSP oxidation.
The origins of these trends can be understood by considering the types and populations of the Vo’s in ZrO2−x. Figure 5b,d,e indicate a correlation of the H2 production rates for the [2.3/0.7] ZrO2−x catalysts, with the Vo’s detected by XPS, Fig. 5d, and EPR Fig. 5e. The XPS and EPR data show a clear decrease in the population of Vo’s of the [2.3/0.7] material upon oxidation at 400 °C. Importantly, theoretical simulation of the EPR signals, Fig. 6, allows distinction between Vo-clusters versus monomeric Vo’s. For example, in [a.p. 2.3/0.7] Vo clusters prevail, while in [3.0/0.1], monomeric Vo’s prevail, as we can see from percentages in Fig. 6c. Oxidation progressively eliminates Vo clusters towards monomeric Vo’s. This is also evident from the progressive elimination of the deep-grey color, and the changes in the UV–Vis spectra (Fig. S8 in SI). Taking this information into account, the bell-shaped H2 production trend in Fig. 5b, indicates that a high concentration of O-vacancies, forming Vo clusters, is detrimental to the photocatalytic activity of ZrO2−x. Fewer Vo’s are better suited for optimal photocatalytic activity (see trend in Fig. 5a, for [2.3/0.7]). However, further oxidation of Vo’s tends to delimit the photoactivity. This teaches us that a quantitative control of the Vo’s clusters versus monomeric Vo is necessary, to achieve highly-performance photocatalytic ZrO2−x, see full trend in Fig. 6e. We consider that this factor was also of pertinence in the magnesiothermically reduced ZrO2−x materials22. Although not noticed by these authors22, inspection of their EPR spectra, shows that these correspond to Vo clusters, which concurs with their limited H2 production of 506 μmol H2 g−1 h−1, resembling our as-prepared [2.3/0.7] material.

(a) Experimental (solid-lines) and theoretical EPR (dashed-lines) spectra of Vo’s formed in as-prepared [3/0.1] ZrO2−x (blue, monomeric Vo’s) or [1.3/1.7] ZrO2−x (red, Vo clusters) (b) Experimental EPR spectra, showing the progressive transition from pure-monomeric Vo’s in [3/0.1] ZrO2−x towards Vo-clusters in [1.3/1.7] ZrO2−x (c) Ratio of EPR detectable oxygen vacancies VOEPR over total Zr-atoms present in each ZrO2−x material (d) Ratio of VoXPS determined by XPS over total oxygen atoms in the lattice. (e) Control of (Monomeric Vo/Vo Cluster) ratio in as-prepared ZrO2−x or via post-FSP oxidation, allows optimization of the photocatalytic H2 production. The size of the sun-symbol exemplifies the H2 production efficiency.
The data under visible 405 nm LED irradiation (Fig. 5c), indicate that significant part of photocatalytic H2 production, at least 70% versus the solar-light photons, can be excited by visible 405 nm photons (3.1 eV). Taking into account the DRS-UV/Vis data, (Fig. 2c) this can be attributed to the occurrence of the mid-gap states, (Fig. 5f). Theoretical DFT calculations (Fig. S10) show that in ZrO2−x, few oxygen vacancies can create mid-gap states, located at energy distances around 3.0 eV12,45 from both the VB-top and ~ 2.0 eV from the CB-bottom. Thus, in ZrO2−x the 3.1 eV photons (405 nm LED) are able to excite two consecutive electron transitions, (Fig. 5f). Increased anoxicity, i.e., as in material [2.3/0.7], enhances the DOS band-tailing (see S.I. Figs. S10 and 5f). This would increase the probability of two-photon electron photoexcitation via VB → Vo, and Vo → CB. These electrons are favorably transferred to the Pt particles, which act as electron collectors i.e. work function of Pt, φ = + 0.9 eV versus NHE46, is favorable for acceptance of electrons from the highly-excited electrons in the CB.
Quantitate analysis of VO-clusters versus monomeric Vo’s, by EPR and XPS
Numerical EPR simulations, dashed lines Fig. 6a, show that monomeric Vo’s were characterized by an inhomogeneous line-shape with linewidth \(\Delta H_{monomer} = \left( {9.4 \pm 0.1} \right){\text{Gauss}}\) and a rhombic gmonomer-tensor, (Table S5). Vo clusters are characterized by a Lorenz line-shape and \(\Delta H_{cluster} = \left( {4.6 \pm 0.1} \right){\text{ Gauss}}\) and isotropic gcluster-tensor, (Table S6).
The structural significance of this is: for isotropic EPR signals with low g-anisotropy, a Gaussian line-shape is the fingerprint of the so-called inhomogeneously-broadened S = 1/2 states47,48 which is indicative of isolated Vo’s, with no-interactions47,48. Physically, this indicates that in the ZrO2−x particles produced under low CH4-flow e.g. [3/0.1], monomeric isolated VoEPR centers. The Lorentz line-shape of the Vo clusters indicates that it originates from Vo centers with spin-exchange and/or fast dipolar interactions. In ZrO2−x, this Lorentz line-shape indicates formation of Vo-clusters upon increasing dispersion-CH4.
Comparing the XPS data for surface-Vo’s (Fig. 6d) versus the quantitative data for total-paramagnetic Vo’s, (Fig. 6c and Table S7 in SI), derived from the EPR spectra (Fig. 6b) and their deconvolution in monometer/clusters (see example for [1.3/1.7] in Fig. S9 in SI), we notice a correlation: highly reduced ZrO2−x, have higher surface-Vo’s, and total paramagnetic Vo’s. Figure 6e provides an overview plot, which shows that optimization of H2 photocatalysis can be achieved via optimization of VO-Clusters versus monomeric Vo’s by two routes: (i) control of FSP anoxicity, or (ii) by soft post-FSP oxidation. This demonstrates that our novel anoxic-FSP process allows facile synthesis of solar-light active nano-ZrO2−x via engineering of lattice-vacancies, Vo. Control of monomeric Vo versus clusters of Vo’s is the key-parameter toward Highly-Performing-Photocatalytic ZrO2−x. The anoxic-FSP process presented here, should be easily adaptable to existing industrial-scale FSP reactors. This offers an efficient technology that can be adopted in the future and provide new tools for the design of other families of photoactive nanomaterials via control of oxygen vacancies.
Methods
Synthesis of nanomaterials by FSP
Precursor solution was prepared by dissolving Zirconium (IV) Propoxide (70 wt% in 1-propanol) in xylene and acetonitrile in a 2.2/1.0 ratio at a concentration of 0.25 M. Then, the solution was fed through a capillary at 3 ml min−1 and dispersed to a self-sustained oxygen/methane (4–2 L min−1) pilot flame to initiate combustion. An important distinction must be made which leads to the innovation of the presented work. ZrO2−x materials were prepared by modifying the dispersion feed. While keeping the 3 ml min−1 dispersion constant, methane gas (CH4) was fed along with the traditional dispersion gas (O2). The resulting high temperatures and hydride formation through the decomposition of methane lead to the formation of ZrO2−x. Furthermore, the protocol of methane injection ensures the formation of bulk defects as the particle is influenced at its early stages of flight, at the primary particle stage. Finally, the pressure drop was fixed at 1.5–2.0 bar, and an additional 10 L min−1 O2 sheath was used to aid in particle collection which was made possible by a vacuum pump (Busch V40) and by a glass microfiber filter (GF 6 257, Hahnemühle, Dassel, Germany).
Characterization techniques
Powder X-Ray Diffraction (XRD) data were collected at room temperature using a Bruker D8 Advance 2theta diffractometer with copper radiation (Cu Ka, λ = 1.5406 Å) and a secondary monochromator operating at 36 kV and 36 mA. Crystal size is calculated by the Scherrer formula. X-Ray photoelectron spectroscopy (XPS) data were collected by a surface analysis ultrahigh vacuum system (SPECS GmbH) equipped with a twin Al–Mg anode X-ray source and a multichannel hemispherical sector electron analyzer (HSA Phoibos 100). The base pressure was 2 − 5 × 10−9 mbar. A monochromatized Mg Kα line at 1253.6 eV and analyzer pass energy of 20 eV were used in all XPS measurements. The binding energies were calculated with reference to the energy of C1s peak of contaminant carbon at 284.5 eV. The peak deconvolution was calculated using a Shirley background.
Raman HORIBA-Xplora Plus spectrometer, equipped with an Olympus BX41 microscope. A 785 nm diode laser was used as an excitation source, and the laser beam was focused on the sample with the aid of the microscope. Before measurement, each powder material was softly pressed between two glass plates to form a pellet-like structure. Brunauer–Emmett–Teller (BET) adsorption–desorption isotherms were recorded at 77 K using a Quantachrome NOVA touch LX2. Outgassing was performed at 80 °C for 5 h under vacuum, before the measurements. The absorption data points in the relative pressure P/Po range of 0.1–0.3 was used to calculate the specific surface area (SSA).
Electron paramagnetic resonance spectroscopy
The X-band electron paramagnetic resonance (EPR) spectra of ZrO2/ZrO2−x materials were recorded with a Bruker ER200D spectrometer at 77 K, equipped with an Agilent 5310A frequency counter. The spectrometer was running under a custom-made software based on LabView. Adequate signal-to-noise ratio was obtained after 15–20 scans, with a microwave power fixed at 20 mW. The EPR instrumental conditions were as follows: microwave frequency = 9.53 GHz and modulation amplitude = 10 Gpp.
Theoretical analysis of the EPR spectra
The experimental EPR spectra were simulated using the EasySpin software. A S = 1/2 Spin Hamiltonian was used \(\hat{H} = \beta \vec{B} \cdot g \cdot \vec{S}\) where β is the Bohr magneton, \(\vec{B}\) is the applied magnetic field, g is the spectroscopic g-tensor and \(\vec{S}\) the spin angular momentum. The X-band electron paramagnetic resonance (EPR) spectra were recorded with a Bruker ER200D spectrometer at 77 K, equipped with an Agilent 5310A frequency counter. The spectrometer was running under a home-made software based on LabView. Adequate signal-to-noise ratio was obtained after 30–50 scans. The EPR instrumental conditions were as follows: microwave frequency = 9.55 GHz, modulation frequency = 50.00 kHz, and modulation amplitude = 10 Gauss peak-to-peak.
Photocatalytic H2 evolution procedure
The photocatalytic hydrogen reactions were realized into a double wall Pyrex reactor, cooled with tap circulation (T = 25 °C). Light source was a Solar Simulator, (Sciencetech, Class AAA, model SciSun-150) with average irradiation intensity of 180 W m−2 equipped with a xenon lamp of 150 W and Air Mass filter (1 sun, AM1.5G). As Visible light source was used a Led lamp FireEdge™ FE410 (λ = 405 nm) supplied by Phoseon company, which power intensity was set to be 180 W m−2, using a power meter (Thorlabs Inc., USA). In each experiment, 50 mg of the catalyst was suspended into 150 ml water/methanol mixture 20% v/v (final concentration of the catalyst 330 mg L−1). Atmospheric O2 from the suspension was removed, fulfilling the content of reactor with Ar gas (99.9997%) at least 1 h. As Pt source were used the dihydrogen hexachloroplatinate (IV) hydrate complex (H2Pt4Cl6. 6H2O, 99.99%, Αlfa Αesar) which was photodeposited in situ, at the reaction mixture. Qualitative and quantitative monitoring of produced H2 and CO2 gases was done via a continuous online GasChromatography System combined with a Thermo-conductive Detector (GC-TCD- Shimadzu GC-2014, carboxen 1000 column, Ar carrier gas49.
Post-FSP oxidation
A ThermaWatt furnace was used, equipped with a tubular Quartz compartment50. Oxidations were performed under atmospheric O2 at temperature 400 °C and calcination time was varied from 30 to 120 min through intervals of 30 min.
Data availability
All data generated or analysed during this study are included in this published article (and its supplementary information files).
References
Hwang, J. et al. Perovskites in catalysis and electrocatalysis. Science 358, 751–756 (2017).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, 4998 (2017).
Fang, H. Challenges with the ultimate energy density with Li-ion batteries. IOP Conf. Ser. Earth Environ. Sci. 781, 042023 (2021).
Villa, K., Galán-Mascarós, J. R., López, N. & Palomares, E. Photocatalytic water splitting: Advantages and challenges. Sustain. Energy Fuels 5, 4560–4569 (2021).
Singh, N., Prakash, J. & Gupta, R. K. Design and engineering of high-performance photocatalytic systems based on metal oxide–graphene–noble metal nanocomposites. Mol. Syst. Des. Eng. 2, 422–439 (2017).
Aksel, C. The microstructural features of an alumina–mullite–zirconia refractory material corroded by molten glass. Ceram. Int. 29, 305–309 (2003).
Clarke, D. R., Oechsner, M. & Padture, N. P. Thermal-barrier coatings for more efficient gas-turbine engines. MRS Bull. 37, 891–898 (2012).
Radhakrishnan, J. K., Kamble, S. S., Krishnapur, P. P., Padaki, V. C. & Gnanasekaran, T. Zirconia Oxygen Sensor for aerospace applications. In 2012 Sixth International Conference on Sensing Technology (ICST) 714–717 (IEEE, 2012). https://doi.org/10.1109/ICSensT.2012.6461771
Dhanasekaran, P., Williams, S. R., Kalpana, D. & Bhat, S. D. Boosting efficiency and stability using zirconia nanosphere-held carbon for proton exchange membrane fuel cells. RSC Adv. 8, 472–480 (2018).
Jeong, D.-W., Na, H.-S., Shim, J.-O., Jang, W.-J. & Roh, H.-S. A crucial role for the CeO2–ZrO2 support for the low temperature water gas shift reaction over Cu–CeO2–ZrO2 catalysts. Catal. Sci. Technol. 5, 3706–3713 (2015).
Sayama, K. & Arakawa, H. Photocatalytic decomposition of water and photocatalytic reduction of carbon dioxide over zirconia catalyst. J. Phys. Chem. 97, 531–533 (1993).
Gionco, C. et al. Cerium-doped zirconium dioxide, a visible-light-sensitive photoactive material of third generation. J. Phys. Chem. Lett. 5, 447–451 (2014).
Hernández, S. et al. Insights into the sunlight-driven water oxidation by Ce and Er-doped ZrO2. Front. Chem. 6, 368 (2018).
Gionco, C. et al. Rare earth oxides in zirconium dioxide: How to turn a wide band gap metal oxide into a visible light active photocatalyst. J. Energy Chem. 26, 270–276 (2017).
Sudrajat, H., Babel, S., Sakai, H. & Takizawa, S. Rapid enhanced photocatalytic degradation of dyes using novel N-doped ZrO2. J. Environ. Manag. 165, 224–234 (2016).
Bugrov, A. N., Rodionov, I. A., Zvereva, I. A., Smyslov, RYu. & Almjasheva, O. V. Photocatalytic activity and luminescent properties of Y, Eu, Tb, Sm and Er-doped ZrO2 nanoparticles obtained by hydrothermal method. Int. J. Nanotechnol. 13, 147 (2016).
Poungchan, G., Ksapabutr, B. & Panapoy, M. One-step synthesis of flower-like carbon-doped ZrO2 for visible-light-responsive photocatalyst. Mater. Des. 89, 137–145 (2016).
Wang, Q. et al. Photocatalytic hydrogen generation on low-bandgap black zirconia (ZrO2) produced by high-pressure torsion. J. Mater. Chem. A 8, 3643–3650 (2020).
Imparato, C. et al. Unraveling the charge state of oxygen vacancies in ZrO2–x on the basis of synergistic computational and experimental evidence. J. Phys. Chem. C 123, 11581–11590 (2019).
Song, L., Cao, X. & Li, L. Engineering stable surface oxygen vacancies on ZrO2 by hydrogen-etching technology: An efficient support of gold catalysts for water–gas shift reaction. ACS Appl. Mater. Interfaces 10, 31249–31259 (2018).
Teeparthi, S. R., Awin, E. W. & Kumar, R. Dominating role of crystal structure over defect chemistry in black and white zirconia on visible light photocatalytic activity. Sci. Rep. 8, 5541 (2018).
Sinhamahapatra, A., Jeon, J.-P., Kang, J., Han, B. & Yu, J.-S. Oxygen-deficient zirconia (ZrO2−x): A new material for solar light absorption. Sci. Rep. 6, 27218 (2016).
Qi, F. et al. Interfacial reaction-induced defect engineering: Enhanced visible and near-infrared absorption of wide band gap metal oxides with abundant oxygen vacancies. ACS Appl. Mater. Interfaces 12, 55417–55425 (2020).
Solakidou, M., Georgiou, Y. & Deligiannakis, Y. Double-nozzle flame spray pyrolysis as a potent technology to engineer noble metal-TiO2 nanophotocatalysts for efficient H2 production. Energies 14, 817 (2021).
Wang, Y. et al. Novel N-doped ZrO2 with enhanced visible-light photocatalytic activity for hydrogen production and degradation of organic dyes. RSC Adv. 8, 6752–6758 (2018).
Naldoni, A. et al. Photocatalysis with reduced TiO2: From black TiO2 to cocatalyst-free hydrogen production. ACS Catal. 9, 345–364 (2019).
Fujiwara, K., Deligiannakis, Y., Skoutelis, C. G. & Pratsinis, S. E. Visible-light active black TiO2–Ag/TiOx particles. Appl. Catal. B Environ. 154–155, 9–15 (2014).
Ruiz Puigdollers, A., Schlexer, P., Tosoni, S. & Pacchioni, G. Increasing oxide reducibility: The role of metal/oxide interfaces in the formation of oxygen vacancies. ACS Catal. 7, 6493–6513 (2017).
Pacchioni, G. Numerical simulations of defective structures: The nature of oxygen vacancy in non-reducible (MgO, SiO2, ZrO2) and reducible (TiO2, NiO, WO3) oxides. In Defects at Oxide Surfaces Vol. 58 (eds Jupille, J. & Thornton, G.) 1–28 (Springer International Publishing, 2015).
Zu, D. et al. Black ZrO2 synthesized by molten lithium reduction strategy for photocatalytic hydrogen generation. J. Am. Ceram. Soc. 103, 4035–4042 (2020).
Sinhamahapatra, A., Jeon, J.-P. & Yu, J.-S. A new approach to prepare highly active and stable black titania for visible light-assisted hydrogen production. Energy Environ. Sci. 8, 3539–3544 (2015).
Mädler, L., Kammler, H. K., Mueller, R. & Pratsinis, S. E. Controlled synthesis of nanostructured particles by flame spray pyrolysis. J. Aerosol Sci. 33, 369–389 (2002).
Stahl, J. et al. Comparing co-catalytic effects of ZrOx, SmOx, and Pt on COx methanation over Co-based catalysts prepared by double flame spray pyrolysis. ChemCatChem 13, 2815–2831 (2021).
Gionco, C. et al. Paramagnetic defects in polycrystalline zirconia: An EPR and DFT study. Chem. Mater. 25, 2243–2253 (2013).
Koirala, R., Pratsinis, S. E. & Baiker, A. Synthesis of catalytic materials in flames: Opportunities and challenges. Chem. Soc. Rev. 45, 3053–3068 (2016).
Weaver, J. H., Peterman, D. J., Peterson, D. T. & Franciosi, A. Electronic structure of metal hydrides. IV. TiHx, ZrHx, HfHx, and the fcc-fct lattice distortion. Phys. Rev. B 23, 1692–1698 (1981).
Lackner, P., Zou, Z., Mayr, S., Diebold, U. & Schmid, M. Using photoelectron spectroscopy to observe oxygen spillover to zirconia. Phys. Chem. Chem. Phys. 21, 17613–17620 (2019).
Ma, W., Herbert, F. W., Senanayake, S. D. & Yildiz, B. Non-equilibrium oxidation states of zirconium during early stages of metal oxidation. Appl. Phys. Lett. 106, 101603 (2015).
Ricca, C., Ringuedé, A., Cassir, M., Adamo, C. & Labat, F. A comprehensive DFT investigation of bulk and low-index surfaces of ZrO2 polymorphs. J. Comput. Chem. 36, 9–21 (2015).
Quintard, P. E., Barbéris, P., Mirgorodsky, A. P. & Merle-Méjean, T. Comparative lattice-dynamical study of the Raman spectra of monoclinic and tetragonal phases of zirconia and hafnia. J. Am. Ceram. Soc. 85, 1745–1749 (2004).
Naumenko, A. P., Berezovska, N. I., Biliy, M. M. & Shevchenko, O. V. Vibrational analysis and Raman spectra of tetragonal zirconia. Phys. Chem. Solid State 9, 121–125 (2008).
Kim, B.-K. & Hamaguchi, H. Mode assignments of the Raman spectrum of monoclinic zirconia by isotopic exchange technique. Phys. Status Solidi B 203, 557–563 (1997).
Hirata, T., Asari, E. & Kitajima, M. Infrared and Raman spectroscopic studies of ZrO2 polymorphs doped with Y2O3 or CeO2. J. Solid State Chem. 110, 201–207 (1994).
Nye, J. F. & Lindsay, R. B. Physical properties of crystals: Their representation by tensors and matrices. Phys. Today 10, 26–26 (1957).
Perevalov, T. V. & Islamov, D. R. Atomic and electronic structure of oxygen polyvacancies in ZrO2. Microelectron. Eng. 178, 275–278 (2017).
Tisdale, W. A. et al. Hot-electron transfer from semiconductor nanocrystals. Science 328, 1543–1547 (2010).
Portis, A. M. Electronic structure of F centers: Saturation of the electron spin resonance. Phys. Rev. 91, 1071–1078 (1953).
Castner, T. G. Saturation of the paramagnetic resonance of a V center. Phys. Rev. 115, 1506–1515 (1959).
Solakidou, M. et al. Efficient photocatalytic water-splitting performance by ternary CdS/Pt-N-TiO2 and CdS/Pt-N, F-TiO2: Interplay between CdS photo corrosion and TiO2-dopping. Appl. Catal. B Environ. 254, 194–205 (2019).
Stathi, P., Solakidou, M. & Deligiannakis, Y. Lattice defects engineering in W-, Zr-doped BiVO4 by flame spray pyrolysis: Enhancing photocatalytic O2 evolution. Nanomaterials 11, 501 (2021).
Acknowledgements
This research was funded by the Hellenic Foundation for Research and Innovation (H.F.R.I) under the “First Call for H.F.R.I Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment grant” (Grant ΗFRI-1888).
Author information
Authors and Affiliations
Contributions
A.M.; Particle synthesis and characterization, writing-original draft, A.Z.; Particle synthesis, Writing-original draft, M.S.; Photocatalytic experiments, Writing—review and editing, S.S. TEM analysis, Y.D.; Conceptualization, Supervision, Funding acquisition, Writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deligiannakis, Y., Mantzanis, A., Zindrou, A. et al. Control of monomeric Vo’s versus Vo clusters in ZrO2−x for solar-light H2 production from H2O at high-yield (millimoles gr−1 h−1). Sci Rep 12, 15132 (2022). https://doi.org/10.1038/s41598-022-19382-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-19382-3
This article is cited by
-
A homogeneous Zr based polyoxometalate coupled with Ppy/PTA for efficient photocatalytic degradation of organic pollutants
Journal of Materials Science: Materials in Electronics (2024)
-
Non-graphitized carbon/Cu2O/Cu0 nanohybrids with improved stability and enhanced photocatalytic H2 production
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
