
Abstract
Proteus mirabilis (P. mirabilis) is a frequent cause of catheter-associated urinary tract infections. This study aims to investigate the anti-infective effect of Alhagi maurorum extract (AME), the traditional medicinal plant in the middle east, on the biofilm-forming P. mirabilis isolates. Hydroalcoholic extract and oil of A. maurorum were characterized by HPLC and GC–MS. The antiproliferative, anti-biofilm, and bactericidal activity of AME at various concentrations were assessed by turbidity, crystal violet binding, and agar well diffusion assays, respectively. The AME’s effect on adhesion and quorum sensing (QS) were investigated by in vitro adhesion assay on cell culture and agar overlay assay using Janthinobacterium lividum (ATCC 12472) as a biosensor strain. In addition, the expression level of selected genes involved in QS and biofilm regulation were determined by quantitative Real-Time PCR. Furthermore, the bladder phantom model was created to evaluate the assays and investigate the catheter’s calcium deposition. The most effective chemical compounds found in AME were tamarixetin, quercetin, and trans-anethole. Although AME did not inhibit swarming motility, it reduced biofilm production and exerted a concentration-dependent anti-adhesive and anti-QS activity against P. mirabilis. AME also downregulated the expression level of selected genes involved in biofilm formation and QS. This study showed that AME as a natural compound reduced biofilm formation of P. mirabilis by targeting virulence factor genes, quorum sensing, and other strategies that include preventing the adhesion of P. mirabilis to the cells. The results suggest that A. maurorum extract might have the potential to be considered for preventing UTIs caused by P. mirabilis.
Similar content being viewed by others

Introduction
Urinary tract infections (UTIs) are one of the most prevalent nosocomial infections. Most UTIs are caused by Enterobacteriaceae. E. coli and P. mirabilis are the most common causal bacteria in the family, accounting for about 90% of complex UTIs1. The increasing use of antibiotics to treat UTIs has led to the development of multi-drug resistant (MDR) bacteria2. P. mirabilis UTIs, particularly in the patients undergoing long-term catheterization, cause the formation of urinary stones (urolithiasis) and long-term catheterized infections3. Among many virulence factors used by P. mirabilis Catheter-Associated Urinary Tract Infections (CAUTIs), some virulence factors were linked to their ability to form biofilms, such as swarming motility, fimbriae, urease production, capsule polysaccharide, and efflux pumps4. The ability to form strong biofilm is one of the major virulence factors in P. mirabilis causing UTIs. The biofilm formed by P. mirabilis has a critical role in UTIs and makes MDRs and recurrent infections to current antibiotics. Biofilm formation enhances the resistance to antibiotics as well as the host immune system5. In the recent decade, efforts to develop new antibiotics have reduced, while drug-resistant strains have become challenging6. Therefore, new alternative strategies are needed to eradicate biofilm-forming pathogens.
Plant essential oils and extracts are considered a rich source of a wide range of bioactive compounds known as phytochemicals7. The possible antibiofilm activity of phytochemicals has made interest, particularly in applying the plants as alternatives to treat infectious diseases8. Phytochemicals could inhibit bacterial adherence to the surfaces, quorum sensing (QS), urease activity, and adhesion to the exopolysaccharide matrix4. The Alhagi maurorum (A. maurorum) is a traditional medicinal plant (known as camelthorn or manna plant) and is found in the middle east, including Iran9. Many studies showed the medicinal properties and frequent use of A. maurorum in rheumatic pains, bilharzias, liver disorders, and gastrointestinal discomfort disease treatment. Furthermore, A. maurorum has a potential effect on treating UTIs and acts as a powerful diuretic and antilithiastic10. Although the possible mechanisms of some phytochemicals assessed for P. mirabilis were studied4, the anti-biofilm activity and the molecular mechanisms caused by A. maurorum extract (AME) are unclear.
This study aimed to evaluate the effect of A. maurorum extract in biofilm degradation and QS genes expression of P. mirabilis isolated from the urinary catheters.
Results
Gas chromatography-mass spectrometry (GC–MS)
The analytic results of the bioactive compounds present in the aqueous extract of A. maurorum essence by GC–MS showed the presence of 8 bioactive phytochemical compounds. The highest percentage content of the compounds are as follows: Trans-Anethole (41.96%), 2-Propanone, 1–4-methoxyphenyl (20.46%), Benzene,1-methoxy-4-(2-propenyl) (9.54%). Other active compounds with their peak number, concentration (peak area%), and retention time (RT) are presented in (Table 1, Fig. 1).

Gas chromatography spectra of biologically active compounds of Alhagi marurum.
High-performance liquid chromatography (HPLC)
The dried extract was standardized using quercetin and tamarixetin as the bioactive marker for the standardization of the extract. Quercetin and tamarixetin peaks of extract appeared at a retention time of 8.730, 9.588 min, respectively. Using a calibration curve, the extract was standardized to contain 19 μg/100 mg of quercetin and 55 μg/100 mg of tamarixetin (Fig. 2).

HPLC results of crude extract of Alhagi maurorum. (A) Result of crude extract, (B) Calibration curve of quercetin using HPLC analysis method.
Microbial isolation and identification
Of the 40 P. mirabilis isolates, 34 (82.5%) were MDR, twelve isolates (30%) had strong biofilm ability (OD590nm ≥ 2.5), twenty of them (50%) were moderate biofilm producer (1.5 ≤ OD 590 nm < 2.5), and eight (20%) of them had weak biofilm (0.7 ≤ OD590 nm < 1.5).
Determination of antimicrobial activity, growth inhibition analysis, and minimum inhibitory concentration (MIC) of AME and the bioactive compounds of AME (quercetin, tamarixetin, and trans-anethole)
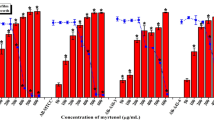
The bactericidal assay of AME was evaluated by the agar well diffusion method. None of the AME concentrations (125–1000 μg/mL) showed a bacteriocidic effect on the P. mirabilis over an 18 h incubation time. The bacterial cultures treated with AME at different concentrations did not show any significant inhibitory effect on the growth of P. mirabilis compared with the control (Fig. 3). The AME didn’t show any inhibitory effect on P. mirabilis, but MIC50 of trans-anethole was at 1 mg/mL. The inhibitory effect of tamarixetin and quercetin was 23% and 12% at 500 μg/mL compared to the control.

The effect of Alhagi maurorum extract at different concentrations (1000–125 µg/ml) on the growth of P. mirabilis in TSB media (solid line) or supplemented with the optimal value of Alhagi maurorum aqueous extract (dashed line). The data represent the absorbance of the mean ± SD values of experiments performed in triplicates.
Motility inhibition assay
The swarming motility of P. mirabilis ATCC7002 was clearly visible in control and in AME and tamarixetin supplements at concentrations from 0.125 to 1 mg/ml. The bacteria exhibited a total swarming diameter of 20 mm in the treated and untreated experiments (P > 0.05). The concentration of quercetin and trans-anethole with 1 and 2 mg/mL, respectively, showed swarming inhibition at 25% and 50% of swarming plates.
Qualitative QS inhibition assay
J. lividum synthesizes the violet pigment violacein as a result of QS. Loss of purple pigmentation of J. lividum in the vicinity of the plant extracts indicated QS inhibition by the plant extract, which was seen in 62.5, 125, 250, and 500 μg/mL (Fig. 4).

The qualitative anti-QS activity of different concentrations of AME.
Quantitative anti-QS activity: violacein inhibition
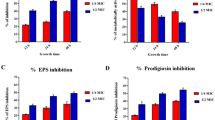
The inhibitory effect of all Alhagi crude extract on the violacein pigment production at different concentrations (62.5–1000 μg/mL) was measured spectrophotometrically and quantified. The violacein pigment production decreased as the AME concentration increased (Fig. 5). This experiment was performed at different times (18, 48, and 72 h), and the optimum concentration of 125 μg/ml AME was considered for the following experiments.

The percentage reduction of quantitative QS in different concentrations of AME and different times of exposure (18 h, 48 h, and 72 h).
Eradication of biofilm
As shown in Fig. 6, AME significantly inhibited biofilm formation. AME and its bioactive compound (quercetin, tamarixetin, and trans-anethole) inhibited the biofilm formation concentration-dependent. The bacteria were treated with AME at 125, 250, 500, and 1000 μg/mL; the biofilm inhibition rate was approximately 74%, 71%, 60%, and 54%, respectively. Quercetin had the most effective compound in AME with 80%, 95%, and 96% biofilm eradication at 125, 250, and 500 μg/mL. The second effective compound of AME was trans-anethole with 44%, 62% and 79% biofilm eradication at 500 μg/mL, 1 mg/mL, and 2 mg/mL. tamarixetin showed a lower reduction in biofilm with 45% and 49% at 62.5 and 125 μg/mL of concentration.

The effectiveness of AME with a different concentration in preventing biofilm formation of P. mirabilis.
Cell viability assay
The cytotoxicity of the AME was examined in HeLa cells. Cells treated with the AME at different concentrations (62.5–1000 μg/mL) survived as well as the control cells (P > 0.05), indicating that a high dose of AME did not affect HeLa survival (Fig. 7).

Survival assay with MTT. The absorbance of different concentrations of AME at OD570 nm.
Adhesion assay
The quantitative binding of P. mirabilis was investigated on the HeLa cell line by enumeration by plating on TSA. AME at different concentrations (125–1000 μg/mL) decreased the adherence of P. mirabilis to the HeLa cell line in a concentration-dependent manner. Results showed that at the concentration of 0.125 mg/ml of AME, P. mirabilis presents a 40% reduction in adhesion to HeLa cells (Fig. 8). Although, at the higher concentration of the extract (0.5 and 1 mg/ml), no significant reduction in the adhesion of P. mirabilis to HeLa cells was seen compared to the control (P > 0.05).

The effect of the different concentrations of AME on adhesion of P. mirabilis to HeLa cell.
Bladder phantom model
To precisely evaluate the impact of AME on crystalline biofilm formation, models of late-stage infection were deactivated after 18 h, and calcium levels on catheter sections were quantified. As demonstrated in Fig. 9, AME significantly reduced the levels of encrustation at the concentration of 0.125 mg/ml. The urine pH was measured after treatment, and there was no significant difference in urine pH after treatment of extract in comparison with control (P > 0.05).

Impact of AME with an optimal concentration on crystalline biofilm formation on the catheter. ***P < 0.001.
Effect of AME on gene expression
We used a qRT-PCR assay to examine the effect of AME at an optimal concentration of 125 μg/ml on the adhesion and quorum sensing gene expression levels. Results showed that all of the mrpA, pmfA, luxS, rsmA, and rsbA genes were significantly downregulated, and their expression levels were reduced approximately by 2−3.9, 2−5.6, 2−1.6, 2−4.5, and 2−1.4-fold, respectively. Among examined different time intervals (4, 16, and 48 h), a significant reduction in the expression of these genes was seen after 16 h treatment (P < 0.05) (Fig. 10).

Relative expression and fold change according to the exposure time with Alhagi extract. A significant reduction was demonstrated especially in 16 h treatment.
Discussion
Antibiotic resistance in bacterial biofilms piqued researchers' interest in looking for additional anti-biofilm drugs and alternative therapeutics. Plants have long been thought to be a rich source of phytochemicals, which are bioactive compounds. Medicinal plants are a good substitute for commonly used antimicrobial drugs11. Among their various applications, phytochemicals have attracted particular interest to their antibiofilm activity, which was attributed to the inhibition of virulence factors, including microbial adherence, quorum sensing, urease activity, and exopolysaccharide matrix production4.
A. maurorum extract is composed of chemical compounds that show good antibacterial activity against different bacterial pathogens, including Escherichia. coli, Staphylococcus. aureus, Bacillus. subtilis, Pseudomonas. aeruginosa and Salmonella. typhimurium12. The phytochemical analysis of AME by GC–MS and HPLC revealed the presence of Trans-Anethole (p-methoxy propenyl benzene), tamarixetin, and quercetin. Trans-Anethole (tA), as a significant component of many essential oils, is an organic compound and a by-product of terpene synthesis13. Kwiatkowski et al. reported the significant antibacterial activity of tA on S. aureus. They showed that tA increased 2–3 times the inhibition zone of bacterial lawn and reduced 60%- 80% the biofilm formation of S. aureus14,15. In another study, Wieczyńska et al. showed the antimicrobial activities of some bio-based compounds, including eugenol, carvacrol, and trans-anethole (tA), against coliforms16. We examined the antimicrobial activity of A. maurorum extract, which showed the extract has neither a bactericidal nor bacteriostatic direct role against P. mirabilis isolates. These results were in line with those reported by Sosa17, who showed that I. lutea extract had no antibacterial and bactericidal activity against Uropathogenic P. mirabilis strains. In a study by Hadadi et al., the methanolic crude extract of A. maurorum had no antibacterial activity on some bacteria, including B. subtilis, E. coli, Pseudomonas. syringae, Pseudomonas. viridiflava and Xanthomonas. campestris18. Similarly, Ahmed et al. showed that the crude extract of A. maurorum was not active against E. coli19. Wang et al. study was assessed the inhibition effect of resveratrol compound on some virulence factor genes expression in P. mirabilis, and the results were similar to our findings20. We further investigated the swarming motility of P. mirabilis ATCC7002 phenotypically and observed clearly visible swarming in AME-treated bacteria and the control. Although, Aygul et al. showed that quercetin (as an active component of AME) inhibited the swarming motility of P. mirabilis. They supposed that the inhibitory effect of quercetin on P. mirabilis swarming was possibly in terms of regulating the expression level of polyamine enzymes which trigger the swarming differentiation or active pump proteins21.
The virulence factors such as swarming, biofilm formation, and the presence of an efflux pumping system are involved in the pathogenesis of P. mirabilis in UTIs4. We indicated that AME inhibits QS in a dose-dependent manner. The anti-QS activity of different plant extracts was investigated, and surprisingly, a wide range of natural products and traditional medicinal herbs showed significant anti-QS ability against Gram-positive and Gram-negative bacteria22,23. Another important virulence factor is biofilm formation. Several reports assessed the anti-biofilm activity of phytochemicals24,25,26. We evaluated the effect of AME on biofilm formation by crystal violet with the microtiter plate method. AME achieved up to 76% inhibition of biofilm formation at a concentration of 0.125 mg/mL. It is possible that the phytochemical compounds could inhibit bacterial growth in different pathways, including weakening the virulence of bacteria without showing bactericidal activity.
The inhibition of quorum sensing (QS) and initial attachment to cells may be related to the biofilm inhibition and decrease in adhesion to cells, for instance, the study which was done by Šimunović et al. showed that some of the natural extracts such as oregano, nettle, winter savory, roseroot, yarrow, and rosemary could reduce the motility and adhesion of C. jejuni via the modulation of LuxS (QS) system27. However, the molecular mechanisms of Alhagi hydroalcoholic crude extract have not been studied yet. To analyze the mechanism of the biofilm inhibition, the difference in expression levels of selected genes involved in biofilm formation and QS were evaluated by RT-qPCR. Gene expression analysis showed an altered pattern (2.6–20.7-fold) downregulation of the genes that affected the virulence properties of P. mirabilis, such as motility, biofilm formation, and QS activity. The data obtained from genotypic analysis confirmed the phenotypic results and showed AME could interact with biofilm and QS regulators in a dose- and time-dependent manner. Our data were in agreement with the data achieved from qualitative and quantitative QS inhibition assay performed by J. lividum. In agreement with our results, several reports illustrated the anti-QS,—biofilm formation, and -luxS expression of natural compounds28,29,30. Two existing genes, rsmA and rsbA in P. mirabilis, regulate swarming and virulence factor expression31. Our results demonstrated that treatment results in the downregulation of these genes, which might be led to swarming inhibition.
An optimized antiadhesive compound should interact with the adhesins of the pathogen, leading to significant inhibition of the docking process between bacteria and eukaryotic cells32. As a result, AME reduces the adhesion of P. mirabilis on our constructed bladder phantom model and consequently affects the calcium deposition in the catheter.
There are limited data about the molecular investigation for the ability of AME in the prevention of UTIs. As a result, AME, a widely used medicinal plant in folk medicine, might strongly regulate QS and biofilm formation of P. mirabilis and could decrease the amount of calcium deposited on the catheter. Moreover, the concentrations used do not show cytotoxicity suggests that this extract has the potential to be considered for further studies on the topics, including the prevention of UTIs caused by P. mirabilis. This study showed that AME as a natural compound reduced biofilm formation of P. mirabilis by targeting virulence factor genes, quorum sensing, and other strategies that include preventing the adhesion of P. mirabilis to the cells. The results suggest that A. maurorum extract might have the potential to be considered for preventing UTIs caused by P. mirabilis.
Materials and methods
Preparation of Alhagi crude extract
The whole part of the A. maurorum plant was collected during the flowering stage in July 2020 from the desert areas around Isfahan province (Gaz, Isfahan, Iran). The plant samples were authenticated by a specialist. The material was identified by J.B and M.G. A voucher specimen of the material is retained in the archives of the Department of Pharmacognosy, Isfahan Pharmaceutical Sciences Research Center under the designation 38,330 (FUMH). Ten grams of freshly powdered plant material were extracted with 100 mL of 50% ethanol for 15 min (3 × 5 min) under ice-cooling by rotor–stator extractor (Ultraturrax®) at maximum rotor speed. The extraction step was repeated 3 times. Then, the suspension was centrifuged at 5.000 × g for 15 min, and the clear supernatant was dried by a rotary vacuum evaporator to yield 2.0 g of dry extract (herbal material: extract ratio = 5:1). The A. maurorum extract (AME) was stored at − 20 °C in sealed containers under a vacuum12.
Essential oil (EO) isolation
The powdered A. maurorum (100 g) was subjected to hydro distillation for 4 h using the Clevenger apparatus (Clevenger, 1928). Then, the EOs were dehydrated by olive oil and stored in tightly sealed glass vials at − 20 °C for further analysis.
Gas chromatography-mass spectrometry (GC–MS) analysis
The gas chromatograph was equipped with a programmable split/spitless injector, a capillary column, and a programmable oven. A sample volume of 2 μL was injected at 271 °C, in spitless mode, in a baffle Siltek-deactivated liner (2 mm × 2.75 mm × 120 mm) provided by Thermo Fisher Scientific. Samples were analyzed via gas chromatography (Agilent USB-393752) equipped with an FID detector and capillary column33.
High-performance liquid chromatographic (HPLC)
Active phytochemical compounds were determined in the aqueous extract of the leaves by HPLC. A 100 mg of dried extract was hydrolyzed in HCl: Tetrahydrofuran (2.5 M) for 1 h. Flavonoid analytes were extracted into a water-soluble solvent (HCL (2 N) and diethyl ether), followed by partitioning of the analyte molecules in an organic solvent in the presence of a salt mixture (salting-out effect). The binary mobile phase consisted of solvent A (water: H3PO4 10 mM; 99:1; v/v) and solvent B (acetonitrile). NUCLEOSIL® 100–5 RP-18 (Thermo scientific column, 150 mm × 4.6 mm) was used to separate phenolic compounds with isocratic elution: 75% A to 25% B at a flow rate of 1.2 ml/min, the time rum was over 10 min. A UV detector detected the phenolic acids and flavonoids at 200–500 nm wavelength. A standard calibration curve in the range of 0.005 to 0.1 mg/ml was prepared for quantitative analysis using different concentrations of standards (0.005, 0.025, 0.01, 0.1 mg/ml). The chromatographic peaks were identified by comparing the retention time of analytics with that of the reference compounds. The relationship between the concentration and peak area of the standard was measured using the minimum square method (R2 value).
Determination of cell viability (MTT assay)
Cytotoxic assays were done in the HeLa cell line (ATCC CCL-2) obtained from the National Cell Bank of Iran, Pasteur Institute of Iran (Tehran, I.R. Iran). HeLa cells (0.5 × 104 cells/ well) were seeded in 96 well-microtiter plates in the presence of Dulbecco’s Modified Eagle Medium (DMEM, Gibco, USA), supplemented with 5% FBS (Gibco, USA), and incubated for 12 h in a humidified atmosphere with 5% CO2 at 37 °C. AME was solubilized in water to give a stock solution with a 2 mg/mL final concentration. Serial ten-fold dilutions of AME in DMEM were prepared to reach 62.5–1000 μg/mL concentrations. Then, 100 µl of each dilution was added to each well. Hela cells with the growth medium were used as control. After incubation for 24 h, the viability of the cells were assessed by MTT assay as described previously34.
Microbial isolation and identification
In this study, 40 P. mirabilis were isolated from the catheters collected from intensive care unit (ICU) patients of various hospitals in Isfahan and confirmed with conventional biochemical and genetic tests35. The P. mirabilis ATCC7002 was used as a standard control.
Antimicrobial activity of AME, growth measurement and MIC of AME and the bioactive compounds of AME (quercetin, tamarixetin, and trans-anethole)
The antimicrobial activity of AME was initially tested against P. mirabilis strain (ATCC7002) by the agar well diffusion method36. A freshly prepared culture of P. mirabilis was adjusted to OD620 of 0.2 and suspended in sterile PBS. 100 μL of the bacterial suspension were swabbed on the Muller-Hinton plate and spread homogeneously. Then, 6 mm of the wells were cut into the agar plate, followed by adding 50 µL of AME dissolved in PBS at different concentrations (62.5–2000 μg/mL). Plates were incubated at 37℃ for 24 h. The inhibition zones around the tested wells were measured to detect the AME range of effect against P. mirabilis. The disc antibiotic model of ofloxacin (5 μg/mL) was put on an agar surface as a positive control, and PBS was added in well served as a negative control.
For growth measurement, the overnight culture of P. mirabilis 7002 (108 CFU/ml) was inoculated into 10 mL of Luria Bertani Broth, and the OD620 value was adjusted to 0.1. Then, 50 µL of the culture was transferred into each well of a 96-well polystyrene microtiter plate that contained 100 µL of LB broth. Subsequently, AME at different final concentrations (125–1000 μg/mL) was added to the wells, and the cultures were incubated at 37 °C for 24 h while shaking (180 rpm). Gentamicin (100 μg/mL) and liquid medium served as positive and negative controls, respectively. The bacterial growth was monitored at 30 min intervals, and the OD620 nm was recorded by a microplate reader (Infinite F50, Tecan)37. The test was done in triplicate for each concentration. MIC values of crude extract and its essential oils were determined using the microdilution broth method described by Wiegand et al.28.
Adhesion assay
HeLa cells (0.5 × 105 cells/ well) were seeded in 24-well plates with/without different extract concentrations (125–1000 μg/mL) and infected with 106 CFU/mL of P. mirabilis and incubated at 37 °C under 5% CO2 for two hours. The wells were washed three times with PBS to remove non-adherent bacteria. To detect adherent bacteria, cell cultures were treated with 500 μl 0.025% Triton X-100 for 5 min at 37 °C in 5% CO2 to detach and lyse the cell monolayer. After that, the cell lysates were diluted in ten serial dilutions. Bacterial colonies were counted after the cell lysates were inoculated on Trypticase Soy Agar (TSA) and incubated at 37 °C for 24 h. The number of bacterial colonies in treated plates was compared to the control29.
Swarming motility assay on agar
Fifty microliters of the series of AME (125–1000 μg/mL), quercetin (1 mg/mL), tamarixetin (1 mg/mL), trans-anethole (1 mg/mL) were mixed with 10 ml of molten Mueller–Hinton agar medium and poured immediately over the surface of the plate as an overlay. The plate was point-inoculated with an overnight culture of P. mirabilis (ATCC7002) once the overlaid agar had solidified and incubated at 37 °C for 3 days. The extent of swarming was determined by measuring the area of the colony30. The test was done in triplicate for each concentration.
Static biofilm assay
In this study, twelve MDR P. mirabilis strains were isolated from CAUTIs of patients attending reference AL-Zahra hospital (Isfahan, Iran) and identified, as described previously35. The P. mirabilis clinical isolates were assessed for their biofilm activity in a microtiter plate according to the previously described method35. The clinical isolates which had strong biofilm formation were chosen for further investigation. To study the extract's antibiofilm activity, 100 µl (OD620 = 0.1) of each isolate culture were plated into a 96-well polystyrene microtiter plate and incubated for 72 h at 37 °C. Then, the media were discarded, and the biofilms were washed with PBS (pH 7.2). The biofilms were supplemented with 100 µl of the AME (125–1000 μg/mL), quercetin (62.5–1000 μg/mL), tamarixetin (62.5 μg/mL–1 mg/mL), trans-anethole (62.5 μg/mL–2 mg/mL), individually and incubated for 18 h at 37 °C. Then, the media were removed, and the wells were fixed with 96% ethanol, followed by staining with 0.1% crystal violet for 15 min. The wells were consequently washed 5 times with H2O, solubilized in acetone 33% and ethanol 80% (1:1). The amount of biomass was quantified by measuring the OD620 using an ELISA- microtiter plate reader (Infinite F50, Tecan). Each treatment was done in triplicates. As a control, 100 µl of nutrient broth was added to the original biofilm of the isolated P. mirabilis. The percentage of biofilm reduction is calculated with this formula: (control untreated OD590 nm—the mean of three replicants test OD590 nm/control untreated OD590 nm) × 10038. All of the OD of tests were normalized by subtracting the OD590 of stained treated and untreated (bacteria only) from the OD590 of stained control wells containing bacteria-free medium only.
Qualitative screening of anti-QS activity
We used pigmented biosensor strain of Janthinobacterium lividum (ATCC 12472) as a reporter to study the anti-QS potential of the four crude A. maurorum extracts39. Agar overlay assay was done using 5 ml of molten soft Luria–Bertani (LB) agar (0.3% agar, 45℃), and 50 μL of the freshly prepared culture of the J. lividum (OD620 = 0.7) was then added before plating the supernatants on the media. The agar-culture solution was immediately poured over the surface of pre-warmed LB agar plates. Then, 20μL of the AME (125–4000 μg/mL) was pipetted on sterile paper discs and let to dry. The discs were placed on the solidified agar. The plates were incubated overnight at 30 °C. Antibacterial activity was revealed through a zone of clearance at the center, and QS inhibition was observed around a colorless, opaque zone with intact bacteria. DMSO was used as a control40. This assay was performed in triplicate.
Quantitative anti-QS assay
Quantitative evaluation of QS inhibitory activity of the AME was carried out based on their ability to inhibit the production of purple pigment violacein by J. lividum ATCC 12472. The strain was cultured aerobically in LB at 30 °C supplemented with the optimal concentrations determined by a qualitative anti-QS test (125–1000 μg/mL). Eugenol (0.625 mg/mL; Sigma, St. Louis, MO, USA) was used as QSI-positive control. One milliliter of an overnight culture of the J. lividum was centrifuged (13,000 rpm, 10 min) to precipitate the insoluble violacein, and the pellet was evenly resuspended in 1 mL of DMSO. The solution was centrifuged (13,000 rpm, 10 min) to remove the cells, and the violacein was quantified at OD620 nm using a UV spectrophotometer (UV-1800, Shimadzu, Kyoto, Japan). The percentage of violacein inhibition was calculated by the following formula: Percentage of violacein inhibition = (control OD620 nm − test OD620 nm/control OD620 nm) × 10040.
Bladder phantom model for treating the biofilm with crude extract
In vitro bladder models, originally described by Stickler et al.41, were set up and operated. Size-18 French all-silicone Foley catheters (Chenkang, China) were used in all experiments and were inserted into the bladder via an outlet before retention balloons were inflated with 10 ml of sterile water. To form a sterile and closed drainage system, catheters were attached to a drainage bag. P. mirabilis 7002 suspensions (1010 CFU, representing late-stage infection) were inoculated directly into the residual bladder urine, and flow was suspended for 1 h to permit cells to establish within the system. At 45 min after bacterial inoculation, test models were treated with an optimal concentration of AME in a volume of 1 ml; the flow was restored 15 min later. The amount of deposited calcium on the primary 2 cm of the catheter was measured and compared with the control. pH was also measured at the start and end of experiments by sampling the medium in the bladder42.
Quantification of crystalline biofilm formation on catheter sections
To measure the levels of crystalline biofilm formation and catheter encrustation in control and extract-treated models, the amount of calcium present on catheter sections removed from bladder models run for a set time (18 h) was quantified by flame photometry42.
Quantitative real-time PCR analysis
The quantitative real-time PCR (qRT-PCR) assay was carried out to study the effect of AME on the expression of QS and adhesion genes (mrpA, pmfA, luxS, rsmA, and rsbA) of P. mirabilis. An overnight inoculated pooled urine with P. mirabilis7002 was transferred to fresh urine, treated with an optimal concentration of AME, and incubated for different time intervals (4, 16, and 48 h) at 37 °C. Then, cells were washed with sterile PBS (pH 7.2) three times and collected after 10 min centrifugation at 4 °C. Total RNA was extracted from bacterial cells using an RNA extraction kit (Jena Bioscience, Germany) following the manufacturer’s instructions. Reverse transcription PCR was conducted, and cDNA was synthesized according to the Jena bioscience kit (Germany) protocol. A qRT-PCR was performed on an ABI system (Applied Biosystems StepOne PlusTM, USA). Each 20 µL reaction contained 2 × Master Mix (SYBR® Green Ampliqon, Denmark), diluted cDNA (5 ng/μL), primers (10 pM of each forward and reverse primers), and RNase-free ddH2O. The thermocycling conditions were as follows: denaturation for 10 min at 95 °C, followed by 40 cycles of denaturation for 15 s at 95 °C, annealing, and extension at 54 °C for 60 s. 16 s rRNA was used as an internal control. The primers used in this study were designed by the online tool Primer 3 web version 4.0.0 and listed in Table 2. All samples were run in triplicate. The relative expression of target genes was calculated by the conventional 2−ΔΔCT method43.
Statistical analysis
Statistical analysis was performed by the SPSS software package (Version v16, IBM Corporation, Armonk, NY, USA). All results were presented as mean ± standard deviation (SD). One-way ANOVA plus post-hoc Tukey test or two-tailed paired t-test was used to evaluate statistical significance between samples. Statistical significance was regarded as p values < 0.05.
Ethics approval and collection permission
Ethics approval was obtained from the Ethics Committee of Isfahan University of Medical Sciences (IR. MUI. MED.REC.1398.259). All methods were performed in accordance with the relevant guidelines and regulations.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Jacobsen, S. M., Stickler, D. J., Mobley, H. L. T. & Shirtliff, M. E. Complicated catheter-associated urinary tract infections due to Escherichia coli and Proteus mirabilis. Clin. Microbiol. Rev. 21, 26–59 (2008).
Dasgupta, C., Rafi, M. A. & Salam, M. A. High prevalence of multidrug resistant uropathogens: A recent audit of antimicrobial susceptibility testing from a tertiary care hospital in Bangladesh. Pak. J. Med. Sci. 36, 1297 (2020).
Mirzaei, A., Habibi, M., Bouzari, S. & Karam, M. R. A. Characterization of antibiotic-susceptibility patterns, virulence factor profiles and clonal relatedness in proteus mirabilis isolates from patients with urinary tract infection in Iran. Infect. Drug Resist. 12, 3967 (2019).
Wasfi, R., Hamed, S. M., Abd Allah, M. A. W. & Ismail, L. Proteus mirabilis biofilm: development and therapeutic strategies. Front. Cell. Infect. Microbiol. 10, 414 (2020).
Høiby, N., Bjarnsholt, T., Givskov, M., Molin, S. & Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 35, 322–332 (2010).
Gutiérrez-Barranquero, J. A., Reen, F. J., McCarthy, R. R. & O’Gara, F. Deciphering the role of coumarin as a novel quorum sensing inhibitor suppressing virulence phenotypes in bacterial pathogens. Appl. Microbiol. Biotechnol. 99, 3303–3316 (2015).
Ahmed, F. & Urooj, A. Glucose-lowering, hepatoprotective and hypolipidemic activities of stem bark of Ficus racemosa in streptozotocin-induced diabetic rats. J. young Pharm. 1, 160 (2009).
Barros, C. H. N. & Casey, E. A review of nanomaterials and technologies for enhancing the antibiofilm activity of natural products and phytochemicals. ACS Appl. Nano Mater. 3, 8537–8556 (2020).
Tavassoli, A. P. et al. Phytochemistry and therapeutic effects of Alhagi spp. and tarangabin in the traditional and modern medicine: a review. J. Herbmed. Pharmacol. 9, 86–104 (2020).
Muhammad, G., Hussain, M. A., Anwar, F., Ashraf, M. & Gilani, A. Alhagi: A plant genus rich in bioactives for pharmaceuticals. Phyther. Res. 29, 1–13 (2015).
Khalate, S. et al. Biofilm inhibition of UTI pathogens using Terminalia arjuna and Ipomea carnea plant extract. Indian J. Sci. Technol. 13, 2452–2462 (2020).
Hassanshahian, M., Saadatfar, A. & Masoumipour, F. Formulation and characterization of nanoemulsion from Alhagi maurorum essential oil and study of its antimicrobial, antibiofilm, and plasmid curing activity against antibiotic-resistant pathogenic bacteria. J. Environ. Health Sci. Eng. 18, 1015–1027 (2020).
Shimoni, E., Baasov, T., Ravid, U. & Shoham, Y. The trans-anethole degradation pathway in an Arthrobacter sp. J. Biol. Chem. 277, 11866–11872 (2002).
Kwiatkowski, P. et al. The effect of fennel essential oil and trans-anethole on antibacterial activity of mupirocin against Staphylococcus aureus isolated from asymptomatic carriers. Adv. Dermatology Allergol. Dermatologii i Alergol. 36, 308 (2019).
Kwiatkowski, P. et al. The effect of subinhibitory concentrations of trans-anethole on antibacterial and antibiofilm activity of mupirocin against mupirocin-resistant Staphylococcus aureus strains. Microb. Drug Resist. 25, 1424–1429 (2019).
Wieczyńska, J. & Cavoski, I. Antimicrobial, antioxidant and sensory features of eugenol, carvacrol and trans-anethole in active packaging for organic ready-to-eat iceberg lettuce. Food Chem. 259, 251–260 (2018).
Zhu, H. & Sun, S. J. Inhibition of bacterial quorum sensing-regulated behaviors by Tremella fuciformis extract. Curr. Microbiol. 57, 418–422 (2008).
Hadadi, Z., Nematzadeh, G. A. & Ghahari, S. A study on the antioxidant and antimicrobial activities in the chloroformic and methanolic extracts of 6 important medicinal plants collected from North of Iran. BMC Chem. 14, 1–11 (2020).
Ahmad, N., Shinwari, Z. K., Hussain, J. & Perveen, R. Phytochemicals, antibacterial and antioxidative investigations of Alhagi maurorum medik. Pak J Bot 47, 121–124 (2015).
Wang, W.-B. et al. Inhibition of swarming and virulence factor expression in Proteus mirabilis by resveratrol. J. Med. Microbiol. 55, 1313–1321 (2006).
Aygül, A., Öztürk, İ, Çilli, F. F. & Ermertcan, Ş. Quercetin inhibits swarming motility and activates biofilm production of Proteus mirabilis possibly by interacting with central regulators, metabolic status or active pump proteins. Phytomedicine 57, 65–71 (2019).
Vattem, D. A., Mihalik, K., Crixell, S. H. & McLean, R. J. C. Dietary phytochemicals as quorum sensing inhibitors. Fitoterapia 78, 302–310 (2007).
Koh, C.-L. et al. Plant-derived natural products as sources of anti-quorum sensing compounds. Sensors 13, 6217–6228 (2013).
Silva, A. J. & Benitez, J. A. Vibrio cholerae biofilms and cholera pathogenesis. PLoS Negl. Trop. Dis. 10, e0004330 (2016).
Kim, H.-S. & Park, H.-D. Ginger extract inhibits biofilm formation by Pseudomonas aeruginosa PA14. PLoS ONE 8, e76106 (2013).
Sánchez, E. et al. Antibacterial and antibiofilm activity of methanolic plant extracts against nosocomial microorganisms. Evid. Based Complement. Altern. Med. eCAM 2016, (2016).
Šimunović, K., Ramić, D., Xu, C. & Smole Možina, S. Modulation of Campylobacter jejuni motility, adhesion to polystyrene surfaces, and invasion of INT407 cells by quorum-sensing inhibition. Microorganisms 8, 104 (2020).
Wiegand, I., Hilpert, K. & Hancock, R. E. W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175 (2008).
Rocha, S. P. D. et al. Aggregative adherence of uropathogenic Proteus mirabilis to cultured epithelial cells. FEMS Immunol. Med. Microbiol. 51, 319–326 (2007).
Kazemian, H. et al. Antibacterial, anti-swarming and anti-biofilm formation activities of Chamaemelum nobile against Pseudomonas aeruginosa. Rev. Soc. Bras. Med. Trop. 48, 432–436 (2015).
Rather, P. N. Swarmer cell differentiation in Proteus mirabilis. Environ. Microbiol. 7, 1065–1073 (2005).
González de Llano, D. et al. Anti-adhesive activity of cranberry phenolic compounds and their microbial-derived metabolites against uropathogenic Escherichia coli in bladder epithelial cell cultures. Int. J. Mol. Sci. 16, 12119–12130 (2015).
Ahmad, S. et al. Chemical composition, antioxidant and anticholinesterase potentials of essential oil of Rumex hastatus D. Don collected from the North West of Pakistan. BMC Complement. Altern. Med. 16, 1–11 (2016).
Van Meerloo, J., Kaspers, G. J. L. & Cloos, J. Cell sensitivity assays: the MTT assay. in Cancer Cell Culture 237–245 (Springer, 2011).
Mirzaei, A., Nasr Esfahani, B., Raz, A., Ghanadian, M. & Moghim, S. From the urinary catheter to the prevalence of three classes of integrons, β-lactamase genes, and differences in antimicrobial susceptibility of proteus mirabilis and clonal relatedness with rep-PCR. Biomed. Res. Int. 2021 (2021).
Augustine, S. K., Bhavsar, S. P. & Kapadnis, B. P. A non-polyene antifungal antibiotic from Streptomyces albidoflavus PU 23. J. Biosci. 30, 201–211 (2005).
Mohammadi, S. et al. A label-free, non-intrusive, and rapid monitoring of bacterial growth on solid medium using microwave biosensor. IEEE Trans. Biomed. Circuits Syst. 14, 2–11 (2019).
Burt, S. A., Ojo-Fakunle, V. T. A., Woertman, J. & Veldhuizen, E. J. A. The natural antimicrobial carvacrol inhibits quorum sensing in Chromobacterium violaceum and reduces bacterial biofilm formation at sub-lethal concentrations. PLoS ONE 9, e93414 (2014).
O’Brien, K., Perron, G. G. & Jude, B. A. Draft genome sequence of a red-pigmented Janthinobacterium sp. native to the Hudson Valley watershed. Genome Announc. 6, e01429-e1517 (2018).
Chenia, H. Y. Anti-quorum sensing potential of crude Kigelia africana fruit extracts. Sensors 13, 2802–2817 (2013).
Stickler, D. J., Morris, N. S. & Winters, C. [35] Simple physical model to study formation and physiology of biofilms on urethral catheters. Methods Enzymol. 310, 494–501 (1999).
Nzakizwanayo, J. et al. Bacteriophage can prevent encrustation and blockage of urinary catheters by Proteus mirabilis. Antimicrob. Agents Chemother. 60, 1530–1536 (2015).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Aboutalebian, S. et al. Direct detection and identification of the most common bacteria and fungi causing otitis externa by a stepwise multiplex PCR. Front. Cell. Infect. Microbiol. 11, 210 (2021).
Acknowledgements
This research benefited from a grant from the Isfahan University of Medical Sciences. This work was supported by Isfahan University of Medical Sciences, Isfahan, I.R. Iran, through Grant No. 398353.
Author information
Authors and Affiliations
Contributions
S.M. contributed to organizing and supervising the whole study and was responsible for the funding acquisition. A.M. and M.G. conducted the experiments. A.M. drafted the manuscript. S.M. and B.N.E. mainly contributed to designing the experiments and data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mirzaei, A., Nasr Esfahani, B., Ghanadian, M. et al. Alhagi maurorum extract modulates quorum sensing genes and biofilm formation in Proteus mirabilis. Sci Rep 12, 13992 (2022). https://doi.org/10.1038/s41598-022-18362-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-18362-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
