
Abstract
Long non-coding RNAs (lncRNAs) are a prominent class of eukaryotic regulatory genes. Despite the numerous available transcriptomic datasets, the annotation of plant lncRNAs remains based on dated annotations that have been historically carried over. We present a substantially improved annotation of Arabidopsis thaliana lncRNAs, generated by integrating 224 transcriptomes in multiple tissues, conditions, and developmental stages. We annotate 6764 lncRNA genes, including 3772 that are novel. We characterize their tissue expression patterns and find 1425 lncRNAs are co-expressed with coding genes, with enriched functional categories such as chloroplast organization, photosynthesis, RNA regulation, transcription, and root development. This improved transcription-guided annotation constitutes a valuable resource for studying lncRNAs and the biological processes they may regulate.
Similar content being viewed by others

Introduction
Long non-coding RNAs (lncRNAs) are transcripts greater than 200 nt with little or no coding potential1,2,3,4. In contrast to the coding genes, they are smaller, have fewer exons, and have lower expression levels than their protein-coding counterparts1,3,5,6,7,8,9. In addition, they often have tissue- and cell-specific expression patterns1,4,7,8,9. lncRNAs have been widely studied in vertebrates. However, few plant lncRNAs have been experimentally characterized to date10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31.
The available studies on lncRNAs in plants reinforce functional similarities originally observed in animals, including modulation of chromatin topology, miRNA levels (miRNA sponges), precursors of small RNA, and acting as a scaffold for the formation of protein complexes11,13,32,33,34. Plant lncRNAs also participate in the response to biotic and abiotic stresses and environmental stimuli such as bacterial infection19, salinity20, drought25, cold10,31, nutrient stresses13,35,36, light11,18, and heat 26. They also play a role in reproductive development10,12,31, growth and development14,21, chromosome modification11,22 and the regulation of small RNA abundance via target mimicry13,18,37. All the functions mentioned above have in common the interaction of a lncRNA with some other biomolecule (RNA, DNA, or protein).
The search of lncRNAs in plants has resulted in numerous reference annotations. For example, in A. thaliana, lncRNAs have been identified and annotated multiple times in competing databases3,38,39,40,41,42,43. Two of the most popular long intergenic non-coding RNAs (lincRNAs) and natural antisense lncRNAs (NATs) reference annotations were generated using 200 A. thaliana tiling array datasets and four baseline transcriptomes to annotate all identifiable lincRNAs 4 and a reference annotation for NATs was generated using sense and antisense strand-specific RNA sequencing from 12 strand-specific root transcriptomes4,44 sequenced in the now discontinued SOLiD sequencing platform45. Both of these annotations are now outdated because first, tiling arrays only provide partial information on lncRNA position and expression and can only be used to annotate lincRNAs; second, the SOLiD platform had several problems with decoding when errors occurred during sequencing, as well as with palindromic regions45. Moreover, these studies used only four transcriptomes (in the case of lincRNAs), or transcriptomes exclusive to a single tissue (root in the case of the NATs) which limited their capacity to identify a complete suite of lncRNAs, particularly because most of these molecules are expressed in a tissue-specific fashion9,46.
Several databases store and classify plant lncRNAs3,38,39,41. Among these, we wish to highlight the CANTATAdb v2.0 database, which contains 4080 lncRNA genes41. The annotations in CANTATAdb are based on ten A. thaliana transcriptomes and a robust annotation methodology, including identifying lncRNAs using the Coding Potential Calculator (CPC)47. Another important database is GreeNC38,48, which also uses a predictive annotation through CPC to identify lncRNAs in different species based on transcripts available in Phytozome49 and ENSEMBL50, including 2752 genes in A. thaliana. In addition, it classifies lncRNAs that can function as miRNA precursors38. The most widely used lncRNA reference annotation is Araport1140. Araport11 has 3559 lncRNA genes (2444 lincRNAs and 1115 NATs)40. While coding gene annotations in Araport11 arise from the integrative annotation pipeline analysis of 113 RNA-seq experiments on different tissues from plants grown under various conditions, the lncRNAs annotated in Araport11 arise from various sources. In particular, it combines the annotations mentioned above of lincRNAs from4,44 and the NAT annotations from44 with lncRNAs well annotated in literature (e.g., FLINC and COOLAIR)12,31. Thus, the lncRNA annotation process in Araport11 was nowhere nearly as strict as their approach to annotating protein-coding genes.
Despite these multiple available sources of annotated plant lncRNAs, few of them have been experimentally characterized or assigned a possible function. A commonly used approach to assign a biological function to lncRNAs is the so-called “guilt-by-association” strategy51,52. This involves generating gene co-expression networks and their subsequent functional annotation to assign potential biological functions to lncRNA genes51,52. Co-expression networks represent the similarity between the expression patterns of different genes in a set of conditions, developmental stages, and tissues53. Genes co-regulated in a wide array of biological conditions are likely controlled by the same regulators or may participate in the same or related biological function or process52,54,55,56. This idea underlies “guilt-by-association” approaches, as lncRNAs can be assumed to work concurrently with the genes it is expressed with, and it is thus preemptively assigned the functions of the genes within its co-expression group. For this approach to work, multiple transcriptomes of the same organism in different stages of development, tissues, and various types of stress are required53,57,58. The more transcriptomes used, the better the statistical significance of the co-expression relationship between genes becomes. Furthermore, the diversity of transcriptomes makes it possible to identify specific networks for a condition or tissue and general networks54. In plants, co-expression networks have been successfully used for the identification of functions in both coding genes59,60,61,62,63 and, more recently, in lncRNAs6,64,65,66,67,68,69.
To address the need for a better annotation of lncRNAs in A. thaliana, we leverage the numerous publicly available RNA-Seq datasets to carry out a comprehensive reannotation of lncRNAs in A. thaliana. We reanalyzed 220 publicly available RNA-Seq datasets, in addition to four seedling transcriptomes generated in-house. Furthermore, we integrate these better annotated and expanded lncRNAs within gene co-expression networks, which enable us to identify potential functions.
Methods
Publicly available transcriptomes used
We selected 220 publicly available transcriptomes using the following criteria: (1) a minimum of 0.5 gigabases (GB) per transcriptome, and (2) generated in a condition, tissue, or developmental stage of wild-type Col-0 A. thaliana. These included: embryo, seed, hypocotyl, cotyledon, root tip, shoot apical meristem (SAM), seedling, root, plant callus, petiole, leaf, carpel, flower pedicel, petal, pollen, sepal, stamen, flower, stem internode, stem node, septum, valve, whole adult plant and conditions such as cold, heat, salinity, drought, blue light, red light, limited phosphate, limited iron and presence of abscisic acid (ABA). All transcriptomes were downloaded as raw reads from Gene Expression Atlas (GEA)70 and Gene Expression Omnibus (GEO)71. Each dataset is described in detail in Table 1. Additionally, we generated four transcriptomes from the aerial part and roots of A. thaliana 8 day post-germination seedlings (see details below), totaling 224 transcriptomes (Dataset S3).
In-house transcriptome generation
Seedlings were grown A. thaliana in Murashige and Skoog (MS) solid medium within growth chambers under conditions of long days (21 °C, 16/8 h photoperiod cycles), approximately for 8 days. The aerial part (shoot) and roots were collected separately, with two biological replicates for each organ (fully open cotyledons and 2 rosette leaves greater than 1 mm long). Total RNA was extracted using TRIzol (Invitrogen, 15,596,018), and according to company specifications, samples were DNase I treated using TURBO™ DNase (Invitrogen, AM2238). The quality and concentration of the samples were measured using the NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific Inc). The integrity of the RNA was verified using a 1.5% agarose gel, and the mRNA was enriched using the NEBNext Poly (A) mRNA Magnetic Isolation protocol (NEBNext, E7490S). The libraries were prepared using the NEBNext Ultra II Directional RNA library kits (NEBNext, E7760S) and NEBNext Multiplex oligos for Illumina (SET 1) (NEBNext, E7335). The libraries were sequenced using the Hi-Seq X from Illumina, using 2 × 150 nt (PE150). The depth and characteristics of these libraries are summarized in Table S1. All the experiments were performed in accordance with relevant guidelines and regulations.
Filtering, assembly, and quantification of transcripts across all transcriptomes
We assessed the quality of all transcriptomes using FastQC v0.11.272 and MultiQC v1.073. Low-quality reads and adapters were removed using Trimmomatic v0.32 (HEADCROP:10–5 LEADING:5 SLIDINGWINDOW:4:15 MINLEN:30–60)74. All quality filter reads were aligned to the A. thaliana TAIR10 genome75, using STAR v2.7.2.b (–alignMatesGapMax 120,000)76. The resulting alignments were assembled using StringTie v1.3.4 (− f 0.3 − m 50 − a 10 − j 15 − c 2.5)77, using the Araport11 annotation as a reference40. The resulting transcripts were joined using the merge function (− c 2.5 − f 0.3) of the StringTie v1.3.4 program77. Transcript counts were obtained using Kallisto v0.44.0 (parameters for single-end transcriptomes: –single − t 8 − l (40, 67, 80) − s (5, 10, 20); parameters for paired-end transcriptomes: default)78.
lncRNA identification
To identify the lncRNAs, we first generated the amino acid sequence for all transcripts using TransDecoder v5.3.079. We then applied nine sequential filters based on previous studies5,9 (see Fig. S1). We refer to this process as the Strict Method (SM). First, (1) we selected all autosomal transcripts ≥ 200 nt using the infoseq program of EMBOSS v6.6.080. We eliminated sequences whose translated ORF or nucleotide sequence had homology to proteins in the Uniprot database 81 as measured by the (2) blastp (e-value ≤ 1e−6) or (3) blastx (e-value ≤ 1e−6, strand = ”plus”) program, respectively82. We subsequently removed sequences with (4) identifiable protein domains found in the base of Pfam (v33.0)83 using the HMMER v3.1b2 program84 (e-value ≤ 1e−6) or (5) with identifiable signal peptides using signalP v4.185 (D-cutoff: 0.45). For any reminder sequences, (6) we removed those that had an ORF > 100 aa using the program getorf of EMBOSS v6.6.0 80. We did an additional filtering step of all sequences with homology to non-redundant proteins (nr) annotated in the NCBI database85,86 using BLASTx82 (evalue ≤ 1e−6, strand = “plus”). For each remaining transcript, we identified the best blast hit against the ‘nr’ database with a percentage of identity above 70% (pident ≥ 70.000). For each best hit, we used the blastdbcmd function82 to obtain the information related to the ID. The transcripts annotated in NCBI as: “hypothetical protein” (in Refseq), “similar to” (NCBI’s annotation pipeline), “putative protein”, “unknown (unknown protein, unknown, partial, unknown)”, “predicted protein” and “unnamed protein product”87 were retained. tRNAs and rRNAs were identified using infernal v1.1.288 and the covariance models in the Rfam database89. We additionally compared sequences with tRNAs and rRNAs reported in A. thaliana using BLASTn82 (evalue ≤ 1e−6, strand = “plus”). All sequences identified as tRNAs or rRNAs were discarded. Finally, we eliminated transcripts with introns > 6000 bp.
After filtering, we manually reviewed transcripts classified in Araport1140 as coding proteins or genes and in our annotation as lncRNAs. This manual review consisted of verifying if these genes had annotation as functional proteins or annotated domains; in these cases, the lncRNA was discarded; if it was a hypothetical or not described protein, the lncRNA was retained. Thus, all sequences that passed this final review constituted the final set of SM lncRNAs.
Classification of lncRNAs by genomic position
LncRNAs are generally classified by their positional relationship to other genes. We used the following non-overlapping categories, based on the GENCODE annotation1:
-
(1)
Intergenic lncRNAs (lincRNAs) lncRNAs found in intergenic regions.
-
(2)
Natural antisense lncRNA (NAT) lncRNAs that totally or partially overlap an exon of another gene in the complementary chain.
-
(3)
Sense-exonic lncRNAs lncRNAs that totally or partially overlap the exon of another gene with the same direction of transcription (transcribed from the same DNA strand).
-
(4)
Intronic lncRNAs lncRNAs found within the intron of another gene without overlapping any of its exons, including those on the same chain or complementary to the superimposed gene.
We further classified lncRNAs by their expression level, considering all lncRNAs with an expression level of fewer than 3 transcripts per million (TPM) in one transcriptome as Low Confidence (SM LC). The remaining lncRNAs were classified as High Confidence (SM HC).
It is worth mentioning that all the isoforms of the overlapping gene are considered for all these categories. To know with which genes our lncRNAs overlap, we used the annotation of Araport1140 and BedTools (2.26.0) intersectBed (sense_exonic lncRNAs [− wo − f 0.1 − s], NAT [− wo − f 0.1 − S], intronic [− wo − f 1] and lincRNAs [− wo − v])90. Finally, all final annotations were inspected by visualizing them in the UCSC Genome Browser91.
Coding potential assessment
CPAT (3.04)92 was used to estimate the sequence-based coding potential of all transcripts as an additional validation method. CPAT is a framework designed for the alignment-free analysis of coding potential in a transcript context, using statistical analysis of relative kmer-frequencies as its basis. Transcripts with known annotation were used to create a Hexamer frequency model and a Logistic regression model using the make_hexamer_tab.py and make_logitModel.py scripts of the CPAT software framework. The relative reliability of the Regression model was estimated by analysis of the associated ROC curve, yielding an area under curve (ROC) score of 0.968.
Transcript coding potentials were predicted for each sequence using this model and results grouped in one of four categories: coding (coding potential > 0.5), non-coding (coding potential ≤ 0.5), high confidence coding (coding potential ≥ 0.9) and high confidence non-coding (coding potential ≤ 0.1).
Comparisons with other lncRNA databases
The 6764 genes annotated as lncRNAs by the SM were compared with the 2752 genes in GreeNC (v1.12)38, 4080 genes in CANTATAdb (v2.0)41 and 3559 genes in Araport1140. We compared the coordinates between these databases using the intersectBed program (− wo − s − f 1 − F 1) from the BedTools toolkit90. We visualized all lncRNA annotations in the UCSC Genome Browser and corroborated the gene assignment for each lncRNA transcript. We summarized these comparisons using the VennDiagram (v1.7.1)92 and UpSetR (v1.4.0) packages93 in R.
Quantification of lncRNAs by tissue and stage
The transcriptomes were divided into tissue and developmental stage categories based on their age and tissue of origin. Notably, some categories are not bona fide tissues (e.g. whole plant, seedlings). However, these were considered their own category as these transcriptomes can be readily differentiated from others. All the transcriptomes were classified into five developmental stages based on the classification by94 (Fig. 2b). The first two stages belong to the vegetative phase and include: seed germination (Stage 1, 3 to 5 days old) and leaf development (Stage 2, 6 to 25 days old); the rest of the stages are part of the reproductive phase, ranging from the presence of the first inflorescence (at 26 days old) (Stage 3, 26 to 29 days old), flower production (Stage 4, 30 to 47 days old), to the generation of siliques (Stage 5, 48 to 51 days old) (Table 1, Fig. 2b).
To identify lncRNAs specific to a tissue or stage of development, we calculated the value of the tissue specificity index Tau95. The calculated Tau values range from 0 to 1 where genes that are tissue or stage-specific have values close to 1 (Fig. S2, Dataset S4). Only genes with Tau values higher than the median Tau value of mRNAs (0.54) were considered tissue-specific or developmental stage-specific (Dataset S4).
Generation of coding and non-coding gene co-expression networks
To determine the possible functions of lncRNAs, we used a guilty-by-association approach. This approach identifies enriched functional annotations of protein-coding genes co-expressed with the lncRNAs, which allows inferring the biological processes in which these lncRNAs may be involved. The co-expression network was built using the WGCNA (1.69) package96 based on the table of raw counts for the full transcriptome normalized using the variance stabilizing transformation (VST), part of the DESeq2 (1.28.1) package97. The adjacency function was weighted by the power of correlation between the different genes, and the law of free-scale networks determined the parameter β. To ensure that the average connectivity of the network was continuous, we chose a value of β = 12, which is the lowest value for which the unscaled topology index curve remains stationary (Fig. S3). From this point on, we will refer to the groups of co-expressed genes as co-expression modules or simply modules, following the nomenclature used by the WGCNA program64. The network was of type signed with a bicor correlation (biweight midcorrelation) and the option of separate modules (unmerged) with a minimum module size of 50 genes. The expression profiles were represented by their main component (module eigengene). An eigengene is the first right-singular vector of the standardized gene expression98 that serves as a summarized representation of the expression of all genes in each module. To identify the functions associated with each co-expressed module, we performed an enrichment analysis of Gene Ontology (GO.db_3.11.4) categories using topGO (2.40.0)99 and the genome-wide annotation of Arabidopsis (org.At.tair.db) as background for the Biological Process (BP) ontology. Finally, we used a Fisher test correcting for multiple testing (Benjamini-Hochberg) (qval.bh < 0.01, FDR < 1%) to assess the significance of the enrichment of GO categories. ReviGO (rrvgo v 1.6.0) was used to summarize and remove redundant GO terms and visualized using treemap v2.4-6 R library.
Genome browser
All lncRNA annotations were uploaded to the UCSC Genome Browser as a track for visualization 91. The coordinates of all lncRNAs genes and their classification are available in Dataset S1.
All code used for these analyses is available in our Github repository: https://github.com/RegRNALab/Transcriptome-guided_lncRNA_annotation.
Results
Using the SM, we identify 6764 lncRNA genes (7070 transcripts). These included 4354 lincRNAs (4548 transcripts), 2060 NATs (2133 transcripts), 213 sense-exonic (248 transcripts), and 185 intronic (187 transcripts) (Fig. 1a, Dataset S1), 78 intronic lncRNAs had no transcriptional orientation (sense) as they were identified in single-end transcriptomes only. Furthermore, 33 lncRNA genes (46 transcripts) were categorized as both NATs and sense-exonic due to the position of the lncRNA flanked by both sense and antisense coding genes in the DNA strand. These were manually verified to ensure they were not extended 3′ UTRs of overlapping protein-coding genes. Additionally, 15 genes had isoforms belonging to different categories (Dataset S2). To provide a measure of the observed expression for lowly expressed lncRNAs, we classified those that had less than 3 TPMs in a single transcriptome as Low Confidence (SM LC) and the remaining lncRNAs as High Confidence (SM HC) (Dataset S2). The single transcriptome threshold was used as there are numerous tissues (carpel, flower pedicel, petal, petiole, pollen, sepal, septum, stamen, stem internode, stem node, and valve) for which we only have a single transcriptome (Dataset S1). Additionally, we assessed the coding potential of the lncRNAs identified by the SM using CPAT100. We found they had significantly lower coding potential scores than coding genes in Araport11 (Fig. S4a) and that the large majority of them were classified as either non-coding or high confidence non-coding by CPAT (Fig. S4b).

Annotation of lncRNA genes and comparisons with other plant lncRNA databases. (a) Distribution of the 6764 lncRNA genes predicted by the SM. (b) Venn diagram comparing the SM (green) with the databases GreeNC (purple), CANTATAdb (pink) and Araport11(yellow) where the lncRNAs have been annotated from A. thaliana. SM = Strict Method.
As expected, the identified lncRNAs have fewer exons per transcript (median 1; average 1.23) (Fig. S5a) than coding genes (median 4; average 6). Furthermore, their mature transcripts are smaller (average 437.3 nt) than that of their coding counterparts (average 1799 nt) (Fig. S5b). These characteristics coincide with what has been previously observed in animals5,7,8,101, flies102 and other plants68,103,104,105,106.
The total of lncRNAs annotated by the SM (6764 genes) outnumbers the most prominent databases in A. thaliana: GreenNC (v1.12) has 2752 genes (3008 transcripts)38, CANTATAdb (v2.0) 4080 genes (4373 transcripts)41 and Araport11, 3559 genes (3970 transcripts)40. A comparison with these databases revealed that 3772 lncRNAs genes in our annotation are novel and have not previously been reported in any of these databases (Fig. 1b); the new lncRNAs were categorized into 2326 lincRNAs (2454 transcripts), 1218 NATs (1227 transcripts), 111 sense-exonic (124 transcripts) and 145 intronic (146 transcripts). These new lncRNAs represent a 93.08% (2275 over 2444) increase in the number of lincRNAs and a 134.70% (1502 over 1115) increase in NATs, with respect to the Araport11 database. Additionally, we find that 398 lncRNA genes of lncRNAs are shared between our annotation and the GreeNC database38, 1485 with CANTATAdb41, and 2637 with Araport1140, being the Araport11 database the one with the best agreement with our data; our annotation contains approximately 74.09% (2637 over 3559) of the lncRNAs annotated in Araport11 (Fig. 1b).
Surprisingly, only 130 lncRNAs are shared between GreeNC, CANTATAdb, and Araport11 databases, and there are only 42 lncRNAs shared among the four annotations (Fig. 1b). It is important to note that there are likely other lncRNAs in A. thaliana that are not identified in our analysis, since not all conditions, tissues, and developmental stages have been surveyed using RNA-Seq. However, our annotation is the first to take advantage of most of the transcriptomic data available for this species, ensuring that the sequences obtained are only those of expressed lncRNAs. This, combined with a robust annotation method, avoids redundancy with other types of transcripts that are not lncRNAs.
Interestingly, when comparing our annotation to Araport11, we observe that our annotations were not always in the same biotype classification. The most concordant classification between both annotations was among lincRNAs, where 1747 lincRNA genes correspond to the same annotation (Fig. S6). However, several lncRNAs identified in our annotation are not classified as lncRNAs in Araport11: 288 lncRNA genes (265 lincRNAs, 12 NATs, 6 sense-exonic, 4 lincRNA-NAT, and 1 sense-exonic lincRNA) are annotated in Araport11 as “novel transcribed region”, and 388 as “coding genes” (217 lincRNAs, 110 NATs, 43 sense-exonic, 13 NAT-sense-exonic, 3 sense exonic lincRNAs, 1 lincRNA-NAT and 1 intronic) (Fig. S6).
We were particularly interested in these 388 lncRNAs classified as “coding genes” in Araport11. We manually reviewed these annotations and concluded these are, in fact, lncRNAs that are erroneously annotated as “coding genes” in Araport11. Among these, we found IPS1 (Induced by Phosphate Starvation 1, AT3G09922), a lncRNA with a mimicry target function for microRNA miR399 in the absence of phosphate13. Another erroneously classified lncRNA was IPS1’s paralog At4 (AT5G03545)37, which is functionally redundant to IPS1. Both of these lncRNAs have been previously experimentally validated and found to be conserved across several plant species107,108,109,110. Similarly, the lncRNA APOLO (AUXIN-REGULATED PROMOTER LOOP, AT2G34655)16 is annotated as a protein-coding gene. We also found multiple lncRNAs erroneously annotated as snoRNAs, novel transcribed regions, and other RNAs, including the experimentally validated lncRNAs: HID1 (HIDDEN TREASURE 1, AT2G35747)11, MARS (MARneral Silencing, AT5G00580)22, and DRIR (Drought-induced RNA, AT1G21529)20, respectively (Table 2).
In addition to these categories, we identified numerous lncRNAs that were annotated as transposable elements (92, reclassified as 91 lincRNAs and 1 NAT), other RNA (83: 77 lincRNAs, 3 lincRNA-NAT, 1 NAT, 1 sense-exonic and 1 sense-exonic lincRNA), pseudogenes (48: 43 lincRNAs, 2 NAT, 2 sense-exonic and 1 NAT lincRNA), snoRNA (5: 5 lincRNAs) and snRNA (1: 1 NAT) (Fig. S6). Finally, we found 3222 lncRNA genes that are not shared between Araport11 and our annotation. These lncRNAs comprise 1885 lincRNAs, 1117 NATs, 143 intronic, 67 sense-exonic, and 10 genes shared between NATs and sense-exonic (9 genes) and intronic and sense-exonic (1 gene) (Fig. S6). These last 10 genes had two annotations due to having isoforms belonging to two different categories. This comparison shows that the annotation of lncRNAs in Araport11, one of the most prominent reference databases for A. thaliana, has significant inaccuracies that are resolved in our annotations, resulting in an improvement in the classification of lncRNA genes.
It is worth noting that within our annotation, 48 lncRNA genes (120 transcripts) have an ambiguous annotation, as they are simultaneously annotated as NAT and sense-exonic (33 lncRNA genes; 93 transcripts), lincRNAs and sense-exonic (5 genes; 12 transcripts), lincRNAs and NAT (9 genes; 13 transcripts), and intronic and sense-exonic (1 gene; 2 transcripts) (Dataset S2). Specifically, in the case of lncRNAs annotated as NAT sense-exonic, they overlap two different protein-coding genes, thereby acquiring a separate annotation for each gene. Similarly, other lncRNA genes had isoforms in different categories, depending on the genomic location of each isoform.
Expression patterns of lncRNAs
In addition to annotating lncRNAs, we leveraged the transcriptomic information to explore how many lncRNAs were expressed amongst A. thaliana tissues, developmental stages, and conditions (Fig. 2). We found more lncRNAs expressed in flower, root, seedling, and silique (Fig. 2a). Organs with higher cell-type diversity, such as flowers, silique, roots, seedlings and leaves had a higher number of lncRNAs (Fig. 2a). This tendency has been previously observed in animals, where organs with more diversity of cell types, such as the brain, express more lncRNAs112,113,114. Reproductive tissues are also known to host a greater diversity of lncRNAs. Similarly, in our data, flowers have more lncRNAs than other organs (Fig. 2a). Interestingly, the number of lncRNAs expressed in the flower is much higher than in its individual parts (stamen, sepal, petal, carpel, and pedicel), further suggesting the high tissue and cell-type diversity of this organ may be due to the multiple tissues that make up this organ. An enrichment of lncRNAs in reproductive tissues has been previously reported in multiple plant species such as soy, corn, and rice34,115,116 and animal testis9,57,117,118. Another category that stands out for its number of expressed lncRNAs is seedlings (Fig. 2a), composed of a mixture of tissues in a particular developmental stage. As most of the transcriptomes from abiotic stress conditions used in this study were from seedlings, many lncRNAs expressed in response to these stresses are expressed in and thus assigned to seedlings (Fig. 2a). Also, the number of transcriptomes and the sequencing depth in each category correlates positively with the number of lncRNAs found (Fig. 2a).

Expression of lncRNAs by tissue and life stage. (a) Histograms of the number from left to right of (x-axis): lncRNAs per tissue, unique lncRNAs, unique coding RNAs, followed by box-plots displaying the number of lncRNAs per transcriptome, GB per transcriptome and number of transcriptomes found in each tissue and life stage studied (y-axis), ordered by the mean number of days after germination. (b) The tissues were grouped into five life stages of A. thaliana using their position within the A. thaliana’s developmental progression. (c) Number of lncRNAs per life stage. The histogram contains the number of lncRNAs identified in each of these stages, while the second histogram contains the number of unique lncRNAs for each of these stages.
In terms of development, the germination phase (stages 1) has the highest number of lncRNAs (Fig. 2c), followed by stage 4 (flower development) (Fig. 2), and does not appear to correlate with the number of transcriptomes in each developmental stage (Fig. 2c). Developmental stages where tissue differentiation or organ formation occur tend to express multiple lncRNAs in both plants6,119,120,121 and animals8,46,57,113. Unfortunately, the early stages of tissue differentiation are not represented in our data set, which could help us identify lncRNAs that participate in tissue formation.
Tissue and stage-specific lncRNA expression
Genes specifically expressed in a particular tissue or stage of development may be important for establishing the identity of that tissue or stage. We found that lncRNAs in A. thaliana, as in most organisms, are expressed in a more tissue-specific manner compared to coding genes (Fig. S2b). The embryo and the whole adult plant had the highest amount of unique lncRNAs, while the root and the embryo expressed more unique coding genes (Fig. 2a). Interestingly, despite not being the most abundant in lncRNAs (Fig. 2a), the embryo has the highest number of unique lncRNAs. Also, the root expressed most of the unique coding and lncRNAs genes (Fig. 2a). Interestingly, there were many more unique coding genes in the root and almost no unique lncRNAs expressed (Fig. 2a). We did not observe an increase in unique genes in tissues with various stress conditions. Also, most unique lncRNAs are expressed in the reproductive phase of the plant rather than in the vegetative phase (Fig. 2c). Dividing the lncRNAs by biotype, we find that 67.1% (143 of 213) sense-exonic, 64% (1318 of 2060) of NATs, 59.8% (2604 of 4354) of lincRNAs , and 48.1% (89 of 185) of intronic lncRNAs belong to a single tissue or stage. These results indicate a high specificity of lncRNAs in the different tissues and stages.
LncRNAs with known tissue-specific functions
Some lncRNAs with known functions display a high tissue-specificity measured by Tau that agrees with their reported functional tissue (Table S2). Among these, we find lncRNAs IPS1 and At4, which have functions related to phosphate starvation13,37, and the lncRNA MARS, which is involved in changes of the chromatin conformation in response to ABA22. As expected, these three lncRNAs have high tissue-specificity values in root tissues (Dataset S4). In addition, the lncRNA FLINC, related to the regulation of flowering12, is specifically enriched in the SAM. On the other hand, the tissue-specific expression of some known functional lncRNAs does correspond to the tissue where they are reported to function. Such is the case of HID1, a lncRNA involved in hypocotyl elongation11, which has high tissue-specificity in the SAM (Table S2), despite being previously found to be ubiquitously expressed11. Similarly, APOLO, which participates in lateral root development in response to auxin16,120, has high tissue-specificity in the petiole (Table S2). This discrepancy is likely due to the lack of auxin-treated roots in our dataset, which is where we expected to see the highest APOLO Tau values.
Co-expression of lncRNAs with coding genes
To infer a possible function for all annotated lncRNAs, we used a so-called guilty-by-association approach. To this aim, we constructed a co-expression network including all coding and non-coding genes using WGCNA96. A total of 224 transcriptomes with 34,937 genes were analyzed to construct this network.
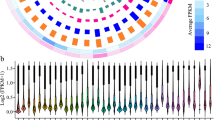
We obtained a total of 45 co-expression modules (Fig. 3). 1425 (21%) lncRNA genes were found in 44 of the 45 co-expression modules. Overall, 516 lincRNAs, 746 NAT, 104 sense-exonic, and 59 intronic were co-expressed. Module 1 harbored the most lncRNAs, with 383 of them, primarily NATs (290), followed by lincRNAs (79), 13 sense, and one intronic lncRNA (Fig. 3). According to the functional enrichment for biological processes, this module stood out for processes related to photosynthesis, the organization of chloroplasts, and response to light. The next modules with the highest number of lncRNAs are modules 4 and 3; these modules are related to the processing and transcription of RNA. In total, 91% (40/44) of modules that housed lncRNAs presented functional enrichment for biological processes. Interestingly, 746 novel lncRNAs were co-expressed with coding genes and distributed amongst 40 modules. Modules 1, 3, 4, and 6 had the most newly annotated lncRNAs, most of them NAT lncRNAs (Fig. S7). It is worth noting that most novel lncRNAs (3026, 80.2%) in our annotation were not co-expressed with coding genes.

LncRNAs per module. Number of lncRNAs in each co-expression module and classified by biotype (Intergenic (lincRNA)—blue, Intronic—purple, NAT—pink, Sense-exonic—yellow).
We found that the 40 co-expression modules that housed lncRNAs and had functional enrichment could be grouped into 9 categories (Fig. S8 to S16) mainly by their function followed by their eigengene values (representative gene expression) in the different tissues or developmental stages96. These functional categories are chloroplast organization and photosynthesis (4 modules with 409 lncRNAs) (Fig. S8), RNA regulation and transcription (4 modules with 375 lncRNAs) (Fig. S9), root development and response to root-related stress (5 modules with 125 lncRNAs) (Fig. S10), protein labeling and transport with (5 modules with 117 lncRNAs) (Fig. S11), cell division (5 modules with 112 lncRNAs) (Fig. S12), lipids and membranes (3 modules with 97 lncRNAs) (Fig. S13), response to pathogens (4 modules with 72 lncRNAs) (Fig. S14), DNA repair (2 modules with 61 lncRNAs) (Fig. S15) and response to stress (5 modules with 17 lncRNAs) (Fig. S16).
We found that the largest number of coexpressed lncRNAs are in the functional category enriched for chloroplast organization and photosynthesis, with positive expression eigengene values in organs related to photosynthesis such as leaves, cotyledons, and hypocotyls. These lncRNAs are divided into four modules (Fig. S8) related to more specific functions such as response to radiation (response to red light, high-intensity light) (module 1), chloroplast and plastid organization (modules 1, 32, and 33), response to cold (modules 1 and 36) and seed, embryo development (modules 32, 33 and 36). The following functional category where we find numerous lncRNAs is related to RNA regulation and its transcription. This category comprises four modules (Fig. S9) with functions such as mRNA metabolic process (module 4), RNA processing (module 3), and regulation of gene expression (modules 16, 17). Genes in these functional categories are most highly expressed in embryos, SAM, and plant callus. The expression profile in this functional category is very similar to the function category of cell division (5 modules) (Fig. S12), which has positive expression values in embryos, seeds, SAM, and roots. The group of modules with fewer lncRNAs is enriched in genes that participate in the response to abiotic conditions (Fig. S16). However, many modules (modules 1, 2, 5, 6, 14, 19, 23, 36, and 45) are enriched in genes that participate in other stress responses (such as drought and cold). Still, they were classified in other functional groups, such as root development (Fig. S10).
For example, in Module 7, we identify 15 lncRNAs highly expressed in root, root tip, plant callus, and seedlings (which include root tissues) and appear to be upregulated in response to limited phosphate conditions (Fig. 4). Amongst the genes in this module we find ERF71, a transcriptional activator involved in root development122; FRO2, which is involved in root growth in response to lack of iron123; NRT21, a repressor of lateral root initiation in response to low nitrate or high sucrose conditions124; MYB93, a transcription factor that acts as a negative regulator of lateral root formation125, Aux/IAA proteins, which function as repressors of early auxin response genes126; MiZ1, which plays a role in lateral root development by maintaining auxin levels and negatively regulates sensitivity to cytokinins127 among several others (Dataset S5). Indeed, this module is highly enriched in transmembrane transport and root system development processes.

Module 7 expression and functions. (a) Barplot of representative expression (eigengene) by transcriptome in module 7, which is enriched in genes expressed and participating in root development. Each bar represents the expression in one transcriptome, organized by tissue and ordered by age from younger (left) to older (right). The lower bar describes which tissue belongs to each color, while the upper one the different experimental treatments of each transcriptome. (b) Treemap of enriched biological functions in module 7.
Several functionally characterized lncRNAs belong to specific functional categories. For example, the DRIR, At4, and APOLO lncRNAs are found in the group of modules related to root function and stress response. It is known that DRIR regulates the closure of stomata in drought20, At4 is associated with the response to phosphate deficiency128, and APOLO is a regulatory lncRNA that directly controls its neighboring gene PID and a many of independent genes by DNA association in response to auxin16,129. The functions of these lncRNAs fit what we observed in the functional enrichment of the modules where they are found. In addition to these examples, we have some others in the group of chloroplasts and photosynthesis, such as FLORE24. This lncRNA has been identified as an important factor in the photoperiod. The lncRNA FLINC, identified as a mediator of flowering in response to temperature12, is found in the group of RNA regulation and transcription functions (Dataset S5).
Discussion
Here, we generated a new and improved annotation of lncRNAs in A. thaliana, supported by 224 transcriptome datasets (Dataset S3) obtained from 24 organs (parts of the plant), 11 conditions, and 5 developmental stages (20 timepoints) (Dataset S3). We found 6764 lncRNAs genes (7070 transcripts), including 3772 novel lncRNAs (Fig. 1b). Among our annotated lncRNAs, we identified 58 genes (86 transcripts) of lncRNAs experimentally validated in A. thaliana from the EvlncRNAs database130, which supports our ability to identify functionally relevant lncRNAs by leveraging existing publicly available transcriptomes.
Given our much cohort of transcriptomic evidence, we find few lncRNAs shared with databases such as GreeNC (398 lncRNAs genes shared)38 and CANTATAdb (1485 lncRNAs genes shared)41, and about 74.09% of the lncRNAs in Araport11 are found in our annotation40 Importantly, our curation approach helped us identify several lncRNAs that were erroneously annotated as coding genes including the lncRNA IPS1, an experimentally validated lncRNA with multiple target sites for miR399, induced in the absence of phosphate13. Another example is its paralog At4, which presents functional redundancy with IPS137. Although these two lncRNAs are functionally conserved in tomato (Lycopersicon esculentum L.) (lncRNA TPSI1)107, Medicago truncatula (lncRNA Mt4)108, rice (lncRNA OsIPS1)109 and barley (HvIPS1)110. Similarly, the well-characterized lncRNA APOLO, which regulates lateral root development16, is annotated as a protein-coding gene. The experimentally characterized lncRNAs HID111, MARS22, and DRIR20 were also erroneously classified (Table 2). Given the wide usage of the Araport11 database, we recommend a revision of their annotations based on our results.
The hundreds of transcriptomics datasets we used allowed us to analyze the abundance of lncRNAs in the different tissues and the development stages. Our analysis revealed that organs with more cell-type diversity display the highest number of lncRNAs in A. thaliana (Fig. 2a). This pattern is particularly prominent in organs related to reproduction (flower & silique), similarly to previous reports in multiple animals9,57,117,118 and plant species34,115,116.
We find that the depth and the number of the transcriptomes are the experimental factors that most affect our capacity to identify novel lncRNAs in any given sample, similarly to previous annotation efforts in various species116,131,132. Thus, we recommend having higher sequencing depth to expedite the discovery of lncRNAs. One limitation of our study is the lack of data from stages where tissue differentiation occurs in the plant, including the flower formation and embryonic stages and the formation of the gametes—surveying these biological conditions would be essential to help complete the catalog of A. thaliana lncRNAs and further our understanding of the role of lncRNAs in the formation of plant structures.
In animals, organ formation and differentiation primarily occur in the embryonic stage, while in plants, it occurs not only in the embryonic phase but also in germination and flower development. It has been shown that widely expressed and conserved lncRNAs are expressed during tissue development, which have the highest probability of being functional. As the tissue matures, an increasing number of species and organ-specific lncRNAs are more likely to be non-functional46.
We find that the expression of lncRNAs is significantly more specific than the expression of coding genes. Nearly 62% (4188) of lncRNAs have expression profiles restricted to a specific tissue or stage, while only 45.6% (12,638) of proteins are specific to a particular tissue or stage. This finding agrees with previous reports in A. thaliana4 and other species1,9,133,134. Moreover, most sense-exonic, NATs and lincRNAs displayed high tissue specificity, while intronic lncRNAs had the lowest tissue specificity, overall very similar to the tissue specificity of protein coding genes.
We found 1241 co-expressed lncRNAs, which we could associate with our broad functional categories (Figs. 3, S8–S16). Using this approach, we find functional categories involving lncRNAs similar to those previously reported in both A. thaliana and other plant species. For example, we find 70 A. thaliana lncRNAs distributed in modules 5, 21, 18, and 39, all functionally enriched in coding genes associated with drought. Numerous lncRNAs are involved in this response in plants135, including 664 lncRNAs in maize136, 51 in cassava, 1096 and 126 in a drought-resistant variety of Brassica napus137. Similarly, we identified five modules with 72 lncRNAs related to response to pathogens (Fig. S14). lncRNAs have been previously found to be differentially expressed in response to infection in tomato138 and maize139. However, the functions that we can assign to lncRNAs are limited by our set of transcriptomes; we can only identify enriched biological functions in the tissues and conditions available in our panel. This analysis could be improved by including more transcriptomes in the future.
Previous works have already established the relationship between lncRNAs and processes related to photosynthesis in A. thaliana and rice140, as well as in the response to different types of light18,141. Photosynthesis is arguably the most important biological pathway in plants. Our results show that the function with the highest number of lncRNAs is related to chloroplast organization and response to light (Fig. S8); this indicates that a large number of lncRNAs may be involved in these processes. It is worth noting that most of our data were obtained from photosynthetic tissues and seedlings, which may explain why our largest modules, comprising the majority of lncRNAs, are associated with photosynthetic processes.
We also identified lncRNAs co-expressed with genes involved in root development and root response to multiple environmental stimuli (Fig. 4). lncRNAs have previously been shown to participate in root differentiation and response to different stress conditions in A. thaliana16,129,142,143,144, Medicago truncatula, where 5561 lncRNAs change their expression in the root due to osmotic stress145, and in Populus, where 295 lncRNAs change their expression during root development119.
Surprisingly, several previously characterized lncRNAs, including ELENA1, MARS, COOLAIR, IPS1, and HID1, are not associated with any particular module. This might be partly because some of these lncRNAs perform their regulatory function in specific environmental conditions (e.g., prolonged cold in the case of COOLAIR), poorly represented in our transcriptomic panel31. Furthermore, the functional association via co-expression is not a fail-proof method; it only identifies lncRNAs expressed in most tissues sampled and that have a strong expression association with genes with similar functions. Thus, many other novel lncRNAs reported in our annotation with no functional association may have important functions that this approach could not identify.
We hope this highly curated, transcriptomic informed lncRNA annotation with functional associations via co-expression in A. thaliana becomes a valuable resource to the A. thaliana and the plant lncRNA community. In the future, we want to assess if the functional association relationships between lncRNAs and other RNAs are conserved in different species and how their loss or gain might be associated with the loss or gain of particular traits in this and other plant families.
Data availability
Raw datasets, software, and documents are available under a CC-BY license at Github146 and FigShare (see Supplementary Information) and NCBI (PRJNA765039).
References
Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789 (2012).
Mattick, J. S. & Rinn, J. L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 22, 5–7 (2015).
Zhao, Y. et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucl. Acids Res. 44, D203–D208 (2016).
Liu, J. et al. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 24, 4333–4345 (2012).
Gaiti, F. et al. Dynamic and Widespread lncRNA expression in a sponge and the origin of animal complexity. Mol. Biol. Evol. 32, 2367–2382 (2015).
Liu, S., Sun, Z. & Xu, M. Identification and characterization of long non-coding RNAs involved in the formation and development of poplar adventitious roots. Ind. Crops Prod. 118, 334–346 (2018).
Hezroni, H. et al. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 11, 1110–1122 (2015).
Darbellay, F. & Necsulea, A. Comparative transcriptomics analyses across species, organs, and developmental stages reveal functionally constrained lncRNAs. Mol. Biol. Evol. 37, 240–259 (2020).
Cabili, M. N. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes. Dev. 25, 1915–1927 (2011).
Heo, J. B. & Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 331, 76–79 (2011).
Wang, Y. et al. Arabidopsis noncoding RNA mediates control of photomorphogenesis by red light. Proc. Natl. Acad. Sci. U. S. A. 111, 10359–10364 (2014).
Severing, E. et al. Arabidopsis thaliana ambient temperature responsive lncRNAs. BMC Plant Biol. 18, 145 (2018).
Franco-Zorrilla, J. M. et al. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 39, 1033–1037 (2007).
Ding, J. et al. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. U. S. A. 109, 2654–2659 (2012).
Gultyaev, A. P. & Roussis, A. Identification of conserved secondary structures and expansion segments in enod40 RNAs reveals new enod40 homologues in plants. Nucl. Acids Res. 35, 3144–3152 (2007).
Ariel, F. et al. Noncoding transcription by alternative RNA polymerases dynamically regulates an auxin-driven chromatin loop. Mol. Cell. 55, 383–396 (2014).
Bardou, F. et al. Long noncoding RNA modulates alternative splicing regulators in Arabidopsis. Dev. Cell. 30, 166–176 (2014).
Sun, Z., Huang, K., Han, Z., Wang, P. & Fang, Y. Genome-wide identification of Arabidopsis long noncoding RNAs in response to the blue light. Sci. Rep. 10, 6229 (2020).
Seo, J. S. et al. ELF18-INDUCED LONG-NONCODING RNA associates with mediator to enhance expression of innate immune response genes in arabidopsis. Plant Cell 29, 1024–1038 (2017).
Qin, T., Zhao, H., Cui, P., Albesher, N. & Xiong, L. A nucleus-localized long non-coding RNA enhances drought and salt stress tolerance. Plant Physiol. 175, 1321–1336 (2017).
Fedak, H. et al. Control of seed dormancy in Arabidopsis by a cis-acting noncoding antisense transcript. Proc. Natl. Acad. Sci. U. S. A. 113, E7846–E7855 (2016).
Roulé, T. et al. The lncRNA MARS modulates the epigenetic reprogramming of the marneral cluster in response to ABA. Mol. Plant. https://doi.org/10.1101/2020.08.10.236562 (2022).
Xu, S. et al. The vernalization-induced long non-coding RNA VAS functions with the transcription factor TaRF2b to promote TaVRN1 expression for flowering in hexaploid wheat. Mol. Plant. 14, 1525–1538 (2021).
Henriques, R. et al. The antiphasic regulatory module comprising CDF5 and its antisense RNA FLORE links the circadian clock to photoperiodic flowering. New Phytol. 216, 854–867 (2017).
Zhao, X. et al. Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 9, 5056 (2018).
Wunderlich, M., Groß-Hardt, R. & Schöffl, F. Heat shock factor HSFB2a involved in gametophyte development of Arabidopsis thaliana and its expression is controlled by a heat-inducible long non-coding antisense RNA. Plant Mol. Biol. 85, 541–550 (2014).
Di, C. et al. Characterization of stress-responsive lncRNAs in Arabidopsis thaliana by integrating expression, epigenetic and structural features. Plant J. 80, 848–861 (2014).
MacIntosh, G. C., Wilkerson, C. & Green, P. J. Identification and analysis of Arabidopsis expressed sequence tags characteristic of non-coding RNAs. Plant Physiol. 127, 765–776 (2001).
Wu, J. et al. A novel hypoxic stress-responsive long non-coding RNA transcribed by RNA polymerase III in Arabidopsis. RNA Biol. 9, 302–313 (2012).
Kindgren, P., Ard, R., Ivanov, M. & Marquardt, S. Transcriptional read-through of the long non-coding RNA SVALKA governs plant cold acclimation. Nat. Commun. https://doi.org/10.1101/287946 (2018).
Csorba, T., Questa, J. I., Sun, Q. & Dean, C. Antisense COOLAIR mediates the coordinated switching of chromatin states at FLC during vernalization. Proc. Natl. Acad. Sci. 111, 16160–16165 (2014).
Rai, M. I., Alam, M., Lightfoot, D. A., Gurha, P. & Afzal, A. J. Classification and experimental identification of plant long non-coding RNAs. Genomics 111, 997–1005 (2019).
Kim, D.-H., Xi, Y. & Sung, S. Modular function of long noncoding RNA, COLDAIR, in the vernalization response. PLoS Genet. 13, e1006939 (2017).
Zhang, Y.-C. et al. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 15, 512 (2014).
Hirsch, J. et al. Characterization of 43 non-protein-coding mRNA genes in Arabidopsis, including the MIR162a-derived transcripts. Plant Physiol. 140, 1192–1204 (2006).
Yuan, J. et al. Systematic characterization of novel lncRNAs responding to phosphate starvation in Arabidopsis thaliana. BMC Genomics 17, 655 (2016).
Shin, H., Shin, H.-S., Chen, R. & Harrison, M. J. Loss of At4 function impacts phosphate distribution between the roots and the shoots during phosphate starvation. Plant J. 45, 712–726 (2006).
Paytuví Gallart, A. et al. GREENC: A Wiki-based database of plant lncRNAs. Nucleic Acids Res. 44, D1161–D1166 (2016).
Jin, J., Liu, J., Wang, H., Wong, L. & Chua, N.-H. PLncDB: Plant long non-coding RNA database. Bioinformatics 29, 1068–1071 (2013).
Cheng, C.-Y. et al. Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 89, 789–804 (2017).
Szcześniak, M. W., Bryzghalov, O., Ciomborowska-Basheer, J., Makałowska, I. CANTATAdb 2.0: Expanding the collection of plant long noncoding RNAs. In Methods in Molecular Biology 415–29 (2019).
Yi, X., Zhang, Z., Ling, Y., Xu, W. & Su, Z. PNRD: A plant non-coding RNA database. Nucl. Acids Res. 43, D982–D989 (2015).
Xuan, H. et al. PLNlncRbase: A resource for experimentally identified lncRNAs in plants. Gene 573, 328–332 (2015).
Li, S., Liberman, L. M., Mukherjee, N., Benfey, P. N. & Ohler, U. Integrated detection of natural antisense transcripts using strand-specific RNA sequencing data. Genome Res. 23, 1730–1739 (2013).
El Bairi, K., Azzam, F. & Amrani, M. The arrival of next-generation sequencing: An overview of current technologies. In Illuminating Colorectal Cancer Genomics by Next-Generation Sequencing: A Big Chapter in the Tale (ed. El Bairi, K.) 73–89 (Springer International Publishing, 2020).
Sarropoulos, I., Marin, R., Cardoso-Moreira, M. & Kaessmann, H. Developmental dynamics of lncRNAs across mammalian organs and species. Nature 571, 510–514 (2019).
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucl. Acids Res. 35, W345–W349 (2007).
Di Marsico, M., Paytuvi Gallart, A., Sanseverino, W. & Aiese, C. R. GreeNC 2.0: A comprehensive database of plant long non-coding RNAs. Nucl. Acids Res. 50, D1442–D14427 (2021).
Goodstein, D. M. et al. Phytozome: a comparative platform for green plant genomics. Nucl. Acids Res. 40, D1178–D1186 (2012).
Hubbard, T. The Ensembl genome database project. Nucl. Acids Res. 30, 38–41 (2002).
Guttman, M. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227 (2009).
Rinn, J. L. & Chang, H. Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166 (2012).
Stuart, J. M., Segal, E., Koller, D. & Kim, S. K. A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255 (2003).
He, F. & Maslov, S. Pan- and core- network analysis of co-expression genes in a model plant. Sci. Rep. https://doi.org/10.1038/srep38956 (2016).
Dinger, M. E. et al. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 18, 1433–1445 (2008).
Gupta, R. A. et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076 (2010).
Necsulea, A. et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505, 635–640 (2014).
You, Q. et al. Co-expression network analyses identify functional modules associated with development and stress response in Gossypium arboreum. Sci. Rep. 6, 38436 (2016).
Usadel, B. et al. Co-expression tools for plant biology: Opportunities for hypothesis generation and caveats. Plant Cell Environ. 32, 1633–1651 (2009).
Hofmann, F., Schon, M. A. & Nodine, M. D. The embryonic transcriptome of Arabidopsis thaliana. Plant Reprod. https://doi.org/10.1101/479584 (2019).
Tong, C. et al. Comprehensive analysis of RNA-seq data reveals the complexity of the transcriptome in Brassica rapa. BMC Genomics 14, 689 (2013).
Greenham, K. et al. Temporal network analysis identifies early physiological and transcriptomic indicators of mild drought in Brassica rapa. Elife https://doi.org/10.7554/elife.29655 (2017).
Sharma, R., Singh, G., Bhattacharya, S. & Singh, A. Comparative transcriptome meta-analysis of Arabidopsis thaliana under drought and cold stress. PLoS ONE 13, e0203266 (2018).
Wang, J. et al. Genome-wide analysis of long non-coding RNAs responsive to multiple nutrient stresses in Arabidopsis thaliana. Funct. Integr. Genomics 21, 17–30 (2021).
Yan, Q. et al. Differential co-expression networks of long non-coding RNAs and mRNAs in Cleistogenes songorica under water stress and during recovery. BMC Plant Biol. 19, 23 (2019).
Deforges, J., Reis, R. S., Jacquet, P., Vuarambon, D. J. & Poirier, Y. Prediction of regulatory long intergenic non-coding RNAs acting in trans through base-pairing interactions. BMC Genomics 20, 601 (2019).
Simopoulos, C. M. A. et al. Coding and long non-coding RNAs provide evidence of distinct transcriptional reprogramming for two ecotypes of the extremophile plant Eutrema salsugineum undergoing water deficit stress. BMC Genomics 21, 396 (2020).
Zhao, X. et al. Genome-wide identification and characterization of long non-coding RNAs in peanut. Genes https://doi.org/10.3390/genes10070536 (2019).
Palos, K. et al. Identification and functional annotation of long intergenic non-coding RNAs in Brassicaceae. Plant Cell https://doi.org/10.1093/plcell/koac166 (2022).
Petryszak, R. et al. Expression Atlas update–an integrated database of gene and protein expression in humans, animals and plants. Nucl. Acids Res. 44, D746–D752 (2016).
Edgar, R. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucl. Acids Res. 30, 207–210 (2002).
Andrews S. FastQC: A quality control tool for high throughput sequence data. (2015).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Berardini, T. Z. et al. The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 53, 474–485 (2015).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 16, 276–277 (2000).
UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucl. Acids Res. 49, D480–D489 (2021).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Finn, R. D. et al. Pfam: The protein families database. Nucl. Acids Res. 42, D222–D230 (2014).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: Interactive sequence similarity searching. Nucl. Acids Res. 39, W29–W37 (2011).
Petersen, T. N., Brunak, S., von Heijne, G. & Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 (2011).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI reference sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucl. Acids Res. 33, D501–D504 (2005).
Gene Frequently Asked Questions. National Center for Biotechnology Information (US). (2018).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Nawrocki, E. P. et al. Rfam 120: Updates to the RNA families database. Nucl. Acids Res. 43, D130–D137 (2015).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Chen, H. & Boutros, P. C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 12, 35 (2011).
Conway, J. R., Lex, A. & Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940 (2017).
Boyes, D. C. et al. Growth stage-based phenotypic analysis of Arabidopsis: A model for high throughput functional genomics in plants. Plant Cell 13, 1499 (2001).
Yanai, I. et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21, 650–659 (2005).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Langfelder, P. & Horvath, S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 1, 54 (2007).
Alexa, A., Rahnenführer, J. & Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607 (2006).
Wang, L. et al. CPAT: Coding-potential assessment tool using an alignment-free logistic regression model. Nucl. Acids Res. 41, e74 (2013).
Kern, C. et al. Genome-wide identification of tissue-specific long non-coding RNA in three farm animal species. BMC Genomics 19, 684 (2018).
Chen, M.-J. M. et al. Integrating RNA-seq and ChIP-seq data to characterize long non-coding RNAs in Drosophila melanogaster. BMC Genomics 17, 220 (2016).
Deng, P., Liu, S., Nie, X., Weining, S. & Wu, L. Conservation analysis of long non-coding RNAs in plants. Sci. China Life Sci. 61, 190–198 (2018).
Zhu, Y. et al. Global transcriptome analysis reveals extensive gene remodeling, alternative splicing and differential transcription profiles in non-seed vascular plant Selaginella moellendorffii. BMC Genomics 18, 1042 (2017).
He, H. et al. Genome-wide analysis identified a set of conserved lncRNAs associated with domestication-related traits in rice. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22094742 (2021).
Wang, P. et al. Global profiling of lncRNAs expression responsive to allopolyploidization in cucumis. Genes 11, 1500 (2020).
Liu, C., Muchhal, U. S., Uthappa, M., Kononowicz, A. K. & Raghothama, K. G. Tomato phosphate transporter genes are differentially regulated in plant tissues by phosphorus. Plant Physiol. 116, 91–99 (1998).
Burleigh, S. H. & Harrison, M. J. The down-regulation of Mt4-like genes by phosphate fertilization occurs systemically and involves phosphate translocation to the shoots. Plant Physiol. 119, 241–248 (1999).
Ajmera, I. et al. Regulatory feedback response mechanisms to phosphate starvation in rice. NPJ. Syst. Biol. Appl. 4, 4 (2018).
Huang, C. Y., Shirley, N., Genc, Y., Shi, B. & Langridge, P. Phosphate utilization efficiency correlates with expression of low-affinity phosphate transporters and noncoding RNA, IPS1, in barley. Plant Physiol. 156, 1217–1229 (2011).
Calixto, C. P. G. et al. Cold-dependent expression and alternative splicing of arabidopsis long non-coding RNAs. Front Plant Sci. 10, 235 (2019).
Mattick, J. S. The central role of RNA in human development and cognition. FEBS Lett. 585, 1600–1616 (2011).
Grote, P. & Herrmann, B. G. Long noncoding RNAs in organogenesis: Making the difference. Trends Genet. 31, 329–335 (2015).
Mercer, T. R., Dinger, M. E., Sunkin, S. M., Mehler, M. F. & Mattick, J. S. Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. U. S. A. 105, 716–721 (2008).
Li, L. et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. BioMed. Central 15, 1–15 (2014).
Golicz, A. A., Singh, M. B. & Bhalla, P. L. The long intergenic noncoding RNA (LincRNA) landscape of the soybean genome. Plant Physiol. 176, 2133–2147 (2018).
Golicz, A. A., Bhalla, P. L. & Singh, M. B. lncRNAs in plant and animal sexual reproduction. Trends Plant Sci. 23, 195–205 (2018).
Brown, J. B. et al. Diversity and dynamics of the Drosophila transcriptome. Nature 512, 393–399 (2014).
Sian, L., Ling, W., Haoran, Q. & Meng, X. LncRNA/circRNA–miRNA–mRNA networks regulate the development of root and shoot meristems of Populus. Ind. Crops Prod. Elsevier 133, 333–347 (2019).
Bhatia, G., Sharma, S., Upadhyay, S. K. & Singh, K. Long non-coding RNAs coordinate developmental transitions and other key biological processes in grapevine. Sci. Rep. 9, 3552 (2019).
Zhu, M. et al. Transcriptomic analysis of long non-coding RNAs and coding genes uncovers a complex regulatory network that is involved in maize seed development. Genes 8, 274 (2017).
Lee, S.-Y. et al. Arabidopsis AtERF71/HRE2 functions as transcriptional activator via cis-acting GCC box or DRE/CRT element and is involved in root development through regulation of root cell expansion. Plant Cell Rep. 34, 223–231 (2015).
Satbhai, S. B. et al. Natural allelic variation of FRO2 modulates Arabidopsis root growth under iron deficiency. Nat. Commun. 8, 15603 (2017).
Little, D. Y. et al. The putative high-affinity nitrate transporter NRT2.1 represses lateral root initiation in response to nutritional cues. Proc. Natl. Acad. Sci. U. S. A. 102, 13693–13698 (2005).
Gibbs, D. J. et al. AtMYB93 is a novel negative regulator of lateral root development in Arabidopsis. New Phytol. 203, 1194–1207 (2014).
Liscum, E. & Reed, J. W. Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol. Biol. 49, 387–400 (2002).
Moriwaki, T. et al. Hormonal regulation of lateral root development in Arabidopsis modulated by MIZ1 and requirement of GNOM activity for MIZ1 function. Plant Physiol. 157, 1209–1220 (2011).
Sánchez-Calderón, L. et al. Characterization of low phosphorus insensitive mutants reveals a crosstalk between low phosphorus-induced determinate root development and the activation of genes involved in the adaptation of Arabidopsis to phosphorus deficiency. Plant Physiol. 140, 879–889 (2006).
Ariel, F. et al. R-loop mediated trans action of the APOLO long noncoding RNA. Mol Cell. 77, 1055–65.e4 (2020).
Zhou, B. et al. EVLncRNAs: A manually curated database for long non-coding RNAs validated by low-throughput experiments. Nucl. Acids Res. 46, D100–D105 (2018).
Iyer, M. K. et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 47, 199–208 (2015).
Hon, C.-C. et al. An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 543, 199–204 (2017).
Ma, L., Bajic, V. B. & Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 10, 925–933 (2013).
Guttman, M. et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 28, 503–510 (2010).
Gelaw, T. A. & Sanan-Mishra, N. Non-coding RNAs in response to drought stress. Int. J. Mol. Sci. https://doi.org/10.3390/ijms222212519 (2021).
Zhang, W. et al. Identification of maize long non-coding RNAs responsive to drought stress. PLoS ONE 9, e98958 (2014).
Tan, X., Li, S., Hu, L. & Zhang, C. Genome-wide analysis of long non-coding RNAs (lncRNAs) in two contrasting rapeseed (Brassica napus L.) genotypes subjected to drought stress and re-watering. BMC Plant Biol. 20, 81 (2020).
Cui, J., Luan, Y., Jiang, N., Bao, H. & Meng, J. Comparative transcriptome analysis between resistant and susceptible tomato allows the identification of lncRNA16397 conferring resistance to Phytophthora infestans by co-expressing glutaredoxin. Plant J. 89, 577–589 (2017).
Han, G. et al. Identification of long non-coding RNAs and the regulatory network responsive to arbuscular mycorrhizal fungi colonization in maize roots. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20184491 (2019).
Shi, Y., Chen, J. & Hou, X. Similarities and differences of photosynthesis establishment related mRNAs and novel lncRNAs in early seedlings (Coleoptile/Cotyledon vs. True Leaf) of rice and Arabidopsis. Front Genet. 11, 565006 (2020).
Kohnen, M. V. et al. Neighbor detection induces organ-specific transcriptomes, revealing patterns underlying hypocotyl-specific growth. Plant Cell 28, 2889–2904 (2016).
Ben Amor, B. et al. Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 19, 57–69 (2009).
Bazin, J. & Bailey-Serres, J. Emerging roles of long non-coding RNA in root developmental plasticity and regulation of phosphate homeostasis. Front Plant Sci. 6, 400 (2015).
Stauffer, E. & Maizel, A. Post-transcriptional regulation in root development. Wiley Interdiscip. Rev RNA. 5, 679–696 (2014).
Wang, T.-Z., Liu, M., Zhao, M.-G., Chen, R. & Zhang, W.-H. Identification and characterization of long non-coding RNAs involved in osmotic and salt stress in Medicago truncatula using genome-wide high-throughput sequencing. BMC Plant Biol. 15, 131 (2015).
Functional and Evolutionary Genomics of Regulatory RNA (RegRNALab). Transcriptome-guided_lncRNA_annotation [Internet]. [cited 2022 Apr 18]. Available from: https://github.com/RegRNALab/Transcriptome-guided_lncRNA_annotation
Acknowledgements
We acknowledge Dr. Katarzyna Oktaba for her library quality and preparation advice.
Funding
This work was funded in part by Consejo Nacional de Ciencia y Tecnología (CONACYT Ph.D. Scholarships 338379 (JAC-G), 781634 (ELC-N), and 780678 (IJG-L) and by a Royal Society Newton Advanced Fellowship (NAF\R1\180303) awarded to SLF-V.
Author information
Authors and Affiliations
Contributions
J.A.C.-G. and S.L.F.-V. conceived and coordinated the study. J.A.C.-G. made assembly and annotation of lncRNAs, as well as tissues specific analysis and co-expression analysis interpretation. E.L.C.-N. performed RNA-seq experiments and the identification and classification of lncRNAs. C.K. calculated coding potential scores. J.A.P.-P. helped with batch processing of RNA-seq data. I.J.G.-L. did the co-expression and functional enrichment analysis. S.L.F.-V. obtained the funding. S.L.F.-V., J.A.C.-G., E.L.C.-N., C.K. and I.J.G.-L. drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Corona-Gomez, J.A., Coss-Navarrete, E.L., Garcia-Lopez, I.J. et al. Transcriptome-guided annotation and functional classification of long non-coding RNAs in Arabidopsis thaliana. Sci Rep 12, 14063 (2022). https://doi.org/10.1038/s41598-022-18254-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-18254-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
