
Abstract
Within the microbial rhodopsin family, heliorhodopsins (HeRs) form a phylogenetically distinct group of light-harvesting retinal proteins with largely unknown functions. We have determined the 1.97 Å resolution X-ray crystal structure of Thermoplasmatales archaeon SG8-52-1 heliorhodopsin (TaHeR) in the presence of NaCl under acidic conditions (pH 4.5), which complements the known 2.4 Å TaHeR structure acquired at pH 8.0. The low pH structure revealed that the hydrophilic Schiff base cavity (SBC) accommodates a chloride anion to stabilize the protonated retinal Schiff base when its primary counterion (Glu-108) is neutralized. Comparison of the two structures at different pH revealed conformational changes connecting the SBC and the extracellular loop linking helices A–B. We corroborated this intramolecular signaling transduction pathway with computational studies, which revealed allosteric network changes propagating from the perturbed SBC to the intracellular and extracellular space, suggesting TaHeR may function as a sensory rhodopsin. This intramolecular signaling mechanism may be conserved among HeRs, as similar changes were observed for HeR 48C12 between its pH 8.8 and pH 4.3 structures. We additionally performed DEER experiments, which suggests that TaHeR forms possible dimer-of-dimer associations which may be integral to its putative functionality as a light sensor in binding a transducer protein.
Similar content being viewed by others

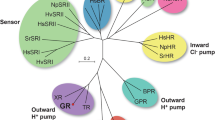
Heliorhodopsins (HeRs) are a newly discovered category of retinal-binding microbial rhodopsins1,2. Its first member, HeR 48C12, was discovered through functional metagenomics when the marine fosmid KIN48C12 yielded red E. coli colonies on retinal-containing culture plates1. This fosmid had low sequence similarity to known rhodopsins, which attributed to the lack of detection of HeRs in typical bioinformatic searches1. Now, 22 more HeRs have been discovered through pigmented E. coli screens3,4, and over 400 unique HeR sequences have been found from homology searches of whole genomes from microorganisms5,6. It turns out that HeRs comprise a large and diverse group of rhodopsins. HeRs are found in bacteria, archaea, eukaryota, and algal viruses1,5,7,8 (Fig. 1). They are globally distributed in marine, hypersaline, freshwater, and soil environments1,7. Moreover, HeRs can be found in psychrophiles, mesophiles, and hyperthermophiles that live in various temperature environments1,8. Interestingly, bioinformatic analysis revealed that HeRs are found in gram positive bacteria and are largely absent in gram negative bacteria6,8, although this taxonomic distribution in monoderms and diderms was somewhat debated5.

Phylogenetic tree of microbial rhodopsins with representative HeRs and type I rhodopsins. HeRs and type I rhodopsins form distinct branches arising from < 15% sequence identity. Microbial rhodopsins are widely distributed among archaea (orange squares), bacteria (blue circles), eukaryota (green triangles), and viruses (black stars). Type I rhodopsins have numerous diverse functions including outward H+ pumps (BRs, XRs, PRs, VirRs), inward H+ pumps (XeRs, SzRs), outward Cl– pumps (HRs, ClRs), outward Na+ pumps (NaRs), anion and cation channels (ChRs), sensors, and enzymerhodopsins (including PDEs). A small group of viral HeRs show proton-transport activity (gold). Many HeRs have no ion transport activity (grey). Some HeRs that have been spectroscopically characterized only (brown) or are uncharacterized (white) are also shown. The tree scale bar shows the average number of amino acid substitutions per site. Red colored circles indicate bootstrap values > 80% for 100 replicates. See Supplementary Table S1 for the full names of proteins and sequence (NCBI searchable codes).
Despite their global abundance and growing scientific interest, the functions of HeRs remain elusive, although light-gated proton channel activity has recently been reported for a few HeRs9. This motivated further research to uncover the similarities and differences between HeRs and other rhodopsins. Rhodopsins are light-harvesting membrane proteins that are classified as either microbial rhodopsins (type I) or animal rhodopsins (type II)10. HeRs fall within the microbial rhodopsins category, but they are phylogenetically distinct from previously discovered type I rhodopsins (Fig. 1).
To date, several HeRs have been studied including HeR 48C121,5,7,11,12,13,14,15, Thermoplasmatales archaeon SG8-S2-1 heliorhodopsin (TaHeR)12,13,16, Bellilinea caldifistulae heliorhodopsin (BcHeR)3, and more than 30 others that have been spectroscopically characterized or functionally investigated1,4,9,16,17. From experimental studies of these HeRs, it has become apparent that they share numerous structural and photochemical properties with other microbial rhodopsins. Similar to type I rhodopsins, HeRs possess seven-transmembrane α-helices (helix A to G) and covalently bind the all-trans-retinal chromophore at a conserved lysine on helix G through a Schiff base linkage5,11,12,16. In the ground state, the retinal Schiff base (RSB) is protonated (RSBH+).
HeRs and type I rhodopsins also have similar photoreaction dynamics13. In HeRs, the retinal chromophore isomerizes from all-trans to 13-cis with comparable ultrafast time scales (τisom = 0.42 ps for HeR 48C12 and τisom = 0.22 ps for TaHeR) and slightly lower isomerization quantum yields as type I rhodopsins13, which triggers a photocycle in the millisecond to second time frame. Both type I rhodopsins and HeRs pass through similar photocycle intermediates including K, M, and O1,3,4,5,9, although the M-intermediate was not observed for BcHeR3. The M-intermediate, a state in which the RSB is deprotonated, is also not observed in chloride-pumping microbial rhodopsins10. Thus, the lack of an M-intermediate for BcHeR may speak towards the functional diversity of HeRs. Further, as known for microbial rhodopsins, retinal adaptation depends on the protein. HeRs can either exhibit retinal adaptation like HeR 48C12 (dark-adaption: 97% all-trans-retinal; light-adaption: 40% all-trans-retinal)1, or no retinal adaptation like BcHeR (88% all-trans-retinal in both light and dark adapted states)3.
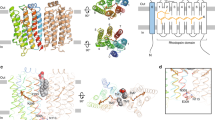
Despite these similarities, HeRs have unique sequences and topologies. First of all, HeRs share less than 15% sequence identity with other type I and type II rhodopsins1,5,7,8. More notably, HeRs have an inverted membrane topology compared to known microbial and animal rhodopsins1,16 (Fig. 2). HeRs expose their N-terminal tail towards the cytoplasm, while the N-terminus of type I and type II rhodopsins orient towards the outside of the cell.

Cartoon structure representation of HeRs (yellow) and type I rhodopsins (pink) with major structural differences emphasized in red. Both HeRs and type I rhodopsins have seven transmembrane α-helices labelled A–G (except enzymerhodopsin, which has eight helices), with a covalently linked retinal chromophore (red sticks) bound to a lysine (pink or yellow sticks) by a Schiff base linkage (blue sticks) on helix G. HeRs have an inverted orientation in the membrane with the N-terminus (red N) facing inward and C-terminus (red C) facing outward, which consequently flips the retinal orientation. HeRs possess a long extracellular A–B loop (AB) composed of a twisted β-sheet (red arrows), and an intracellular B–C loop forming an α-helix (red cylinder labelled BC). In contrast, type I rhodopsins typically have an extracellular B–C loop forming a β-sheet.
There may be as many as 10 different sub-families of HeRs with potentially different functions5. A natural hypothesis is that HeRs may function like other microbial rhodopsins—as light-activated ion pumps, ion channels, light sensors, or enzymes. Very recently, three proton-transporting HeRs were discovered in marine giant viruses, including Emiliania huxleyi virus 202 (V2HeR3) and Emiliania huxleyi virus PS401 (VPS401HeR)9 as shown in Fig. 1. However, most known HeRs do not function as ion pumps or channels because they do not show light-dependent pH changes, including HeR 48C121, BcHeR3, TaHeR16, ten HeRs discovered through functional metagenomics (EINA and HULA variants shown in Fig. 1)4, and other HeRs1,9,16. Interestingly, the photocycles for these HeRs (lacking ion-transport function) often exhibit a Schiff base deprotonation step (M-intermediate), but the proton does not exit the protein, not even transiently1.
The current theory is that many HeRs function as light-driven sensors. There is a plethora of evidence to support sensory function. First, long photocycles of greater than 1 s are observed for HeR 48C121, TaHeR16, BcHeR3 and others4, which are characteristic for sensors like sensory rhodopsin I (SRI) and sensory rhodopsin II (SRII)18,19,20. In particular, the O-intermediate is long-lived in HeRs and a good candidate for a signalling state1,3,4. Secondly, HeRs are largely constrained to the photic zone—the surface layer of aquatic habitats that still receives enough sunlight for photosynthesis—which suggests that HeRs serve a light-harvesting function1,4. However, one exception is BcHeR which was discovered in thermophilic digester sludge which is an inherently low irradiance environment3. Thirdly, a resonance Raman study revealed that the vibrational mode of the RSB is like that of photosensory microbial rhodopsins12. Specifically, the RSBH+ of both TaHeR and HeR 48C12 forms a strong hydrogen bond to a glutamate residue12 with comparable strength to SRII from Halobacterium salinarum21 and Natronobacterium pharaonis22,23. If HeRs are indeed sensors, the transducer proteins have not yet been identified, because HeRs lack a consistent set of gene neighbours4. A wide genomic and metagenomic search provided compelling evidence that some HeRs are linked to histidine kinases, while other HeRs may be N-terminally fused to a MORN-repeat (Membrane Occupation and Recognition Nexus) or a Zinc-finger functional domain6. Another analysis of HeR-coding DNA segments revealed two protein families that were unique gene neighbors among some HeRs: Blh (β-carotene 15,15′-dioxygenase), required for the last step in the retinal biosynthetic pathway in prokaryotes; and the DegV family involved in activating fatty acids4. In order to resolve the biological role of HeRs, further functional and structural studies will be necessary.
The crystal structures of HeR 48C125,11 and TaHeR16 have provided insight into the unique architecture and potential functions of HeRs (Fig. 2). The structures revealed that HeRs assemble as dimers with the most extensive dimerization interface seen in microbial rhodopsins (2260 Å2 for HeR 48C12)11. The most distinguishing feature of HeRs is the very long, extracellular loop linking helices A and B (~ 30 residues, ~ 40 Å in length) that forms two antiparallel β-strands, which is unique to HeRs5,11,16. This A − B loop is essential for dimer formation and HeR function, as its deletion causes protein aggregation and a complete loss of retinal binding11. On the intracellular side, an unusual B–C loop (~ 14 residues, ~ 18 Å in length) forms a structured α-helix5. Another striking element observed in both HeRs is a fenestration (an opening between helices towards the membrane) above the β-ionone ring of retinal, which has also not been observed in other rhodopsin structures5,11,16. For TaHeR, it was postulated that since its host organism lacks a means of producing retinal, the fenestration could facilitate binding exogenous retinal from the environment16.
Kovalev et al.5 crystallized HeR 48C12 in two different states: (1) a violet, basic form at pH 8.8 and (2) a blue, acidic form at pH 4.3, postulating that the low pH structure could give insight into conformational changes due to the protonation of the RSBH+ proton acceptor. The starkest difference between the pH 4.3 and pH 8.8 structures was observed in the large hydrophilic Schiff base cavity (SBC). Whereas the SBC was filled with seven water molecules at pH 8.8, it contained three water molecules and one acetate anion at pH 4.35. Furthermore, key residues underwent pH-induced rearrangement which may indicate functionally relevant intramolecular signalling. In order to determine whether the observed structural changes between low and high pH forms are conserved among HeRs, we investigated the pH-induced structural changes of TaHeR. Using bicelle crystallization we determined the high resolution structure of TaHeR at pH 4.5, which revealed a chloride ion in the SBC and differences to the previously published pH 8 TaHeR structure16, arguing for a conserved intramolecular signalling behavior similar to that in HeR 48C12. This suggests that different sub-families of HeRs may potentially function as light sensors using a common mechanism.
Furthermore, we investigated the pH- and light-induced conformational changes of TaHeR in detergent using electron paramagnetic resonance (EPR) spectroscopy. EPR can provide course-grained site-specific structural information about the population of dynamic states of a spin-labelled protein in solution24,25,26. We explored the conformational dynamics of the spin-labelled A–B loop using continuous wave-EPR (CW-EPR) and double electron–electron resonance (DEER). CW-EPR detects the stabilized unpaired electron from a covalently attached spin label at a specific topological region of the protein and therefore provides local information on whether the spin label is mobile or restricted in motion25. DEER is an EPR technique in pulsed dipolar spectroscopy (PDS) that can be applied on a sample containing two or more spin labels. This method facilitates direct measurements of dipolar interactions in the range from typically 1.6–6.0 nm, or even up to 15.0 nm in optimized samples27,28. The resulting distance distributions yield information about structural changes, flexibility, and intermolecular alignments27,29. Thus, DEER constitutes a valuable alternative method to study rhodopsin oligomerization in solution30. Here, we utilize this integrative structural biological tool to access the intermolecular functional context of TaHeR that reaches beyond our high resolution data from X-ray crystallography31. Conveniently, since TaHeR is dimeric, only a single cysteine mutation is required for spin-labelling, and thus distances between spin labels on adjacent protomers can be obtained. This DEER study revealed that TaHeR can form transient dimer-of-dimer associations, akin to those observed in high-speed atomic force microscopy (HS-AFM) experiments16. We determined that these potential dimer-of-dimer associations are pH- and light-dependent, suggesting that the transient formation of dimer-of-dimers could be relevant to its functionality, say, as a sensory rhodopsin binding a transducer protein.
Results and discussion
Structure of TaHeR crystallized at low pH
Purified TaHeR (Supplementary Fig. S1) was crystallized using the bicelle method32 to yield large diamond shaped crystals (Supplementary Fig. S2). Notably, this is the first example of a HeR being crystallized via bicelles, whereas previously lipidic cubic phase crystallization5,11,16,33 was employed. We determined the TaHeR structure at 1.97 Å resolution (Table 1) using a single crystal grown at pH 4.5, enabling comparison with the other available TaHeR structure (PDB ID 6is6), which has been determined at 2.4 Å resolution using more than 100 crystals grown at pH 8 in lipidic cubic phase. The pH 4.5 TaHeR structure contains a single protomer in the asymmetric unit (Fig. 3a). It also possesses 37 water molecules, an all-trans-retinal chromophore covalently linked to Lys-238 (Fig. 3b), and two chloride ions. The electron density is well-resolved for all seven transmembrane helices and loops (Fig. 3c).

Electron density map and model of TaHeR crystallized at pH 4.5. (a) Asymmetric unit of TaHeR with electron density of water molecules (red spheres), chloride ions (green spheres) and retinal (yellow sticks). A red box highlights a single water molecule present in the highly hydrophobic extracellular half of TaHeR. (b) Electron density map of the SBC, with black dashed lines revealing a water-mediated hydrogen bonding network. (c) Electron density map of the protein backbone with the corresponding residue numbers listed above. Conserved residues in the suggested intramolecular signal transduction pathway have been labelled. For all images, the 2Fo − Fc map is contoured at 1 σ (blue mesh), while Fo–Fc is contoured at − 3 σ (red mesh) and in green at + 3 σ (green mesh).
The structures of TaHeR at pH 4.5 and pH 8 are very similar, with an RMSD of 0.535 Å with 220 aligned Cα atoms calculated using PyMOL. Both structures possess an asymmetric unit containing only one protein chain, and pack as a symmetric dimer. The higher resolution of the pH 4.5 structure revealed a single, isolated water molecule present in the highly hydrophobic extracellular half of TaHeR (Fig. 3a), which was not resolved in the pH 8 structure. Importantly, this water molecule can be also observed in both high resolution HeR 48C12 structures at pH 4.3 and pH 8.8 (Fig. 5b). The location of this water, close to the β-ionone ring of retinal and adjacent to the fenestration, appears to be conserved and unique to HeRs.
Furthermore, by comparing the two TaHeR structures, three key structural differences were observed. First, TaHeR at pH 4.5 binds two chloride ions while TaHeR at pH 8 has none. Second, the tail end of the A–B loop undergoes a 6 Å shift in position, and the B–C loop shifts ~ 1 Å. Finally, several conserved internal residues change orientation or become disordered. These differences provide valuable insights about anion binding sites and a potential intramolecular signalling pathway which are explored in the subsequent sections.
Anions in the SBC under acidic conditions
This structural study of HeRs at different pH values gives insight into the interplay between the SBC and the counterion of the RSBH+. The previously published structure of TaHeR at pH 8 showed no negatively charged solvent ions bound to the protein16. This result is consistent with several ATR-FTIR studies showing wildtype TaHeR exhibits no specific binding of monovalent cations or anion at physiological pH (although the specific binding of Zn2+ was detected)16,34. In contrast, our pH 4.5 TaHeR structure revealed the presence of two chloride ions. One chloride resides in the SBC (Fig. 3b) and the other resides on the extracellular side adjacent to helices F and G (Fig. 3a).
Let us first explore the relevance of the chloride ion in the SBC. TaHeR possesses a large hydrophilic SBC on the intracellular half of the transmembrane region. The SBC is surrounded by numerous polar residues and bookended by two electrostatic interactions. As shown in Fig. 4a, at pH 8 the SBC of TaHeR accommodates four water molecules in a volume of 189 Å3 (calculated using Hollow35), and possesses two ionic interactions: (1) the RSBH+/Glu-108 pair, and (2) the Arg-105/Glu-227 pair. The surrounding histidine residues, His-23 and His-82, are assumed to be neutral because there are no other negatively charged residues in this space16. In the pH 4.5 TaHeR structure, the SBC shrinks to a volume of 156 Å3 and accommodates a chloride ion and three water molecules. The uptake of the chloride ion suggests that a positive charge has also been introduced, presumably by the RSBH+ counterion Glu-108, which becomes neutralized at the low pH. The protonation of Glu-108 is consistent with the observed 18 nm red-shift of TaHeR at low pH (Supplementary Fig. S3 and Shihoya et al.16). Acid-induced chloride ion binding is also present in solution, where TaHeR at pH 5.0 underwent a 12 nm red-shift, from 543 nm in a chloride-depleted form to 555 nm at 300 mM NaCl, with an estimated Kd of 67 mM (Supplementary Fig. S4). Coincidently, the counterion mutant, TaHeR-E108A with a neutral side chain, exhibits the same λmax in the presence of 100 mM NaCl16. Our low pH structure suggests that a chloride anion in the SBC can serve as a stabilizing counterion to the RSBH+ when the Glu-108 counterion has been neutralized by protonation at low pH or by mutation.

Schiff base cavity (SBC). SBC (grey surface) of (a) TaHeR and (b) HeR 48C12 under acidic and basic conditions. Under slightly basic conditions, the SBC of both TaHeR (PDB ID 6is6) and HeR 48C12 (PDB ID 6su3) contain only water molecules (red spheres) and are capped by two ionic interactions: (1) the RSBH+/Glu counterion pair and (2) the Arg/Glu pair. Under acidic conditions, the Glu counterion accepts a proton and is neutral. The SBC of TaHeR accommodates a negatively charged chloride ion (green sphere) while HeR 48C12 (PDB ID 6su4) accommodates a negatively charged acetate ion to maintain charge balance with the RSBH+. The negative charges are indicated by red dashes, while the positive charges are shown by blue pluses. ‡ Weak electron density adjacent to Ser-76 in the pH 4.3 HeR 48C12 structure has been modeled as a water molecule, but this molecule may also be absent, as in TaHeR.
A similar result was obtained from HeR 48C12 structures (PDB ID 6su3, 6su4), which possess the same polar residues surrounding the SBC as TaHeR. At pH 8.8, HeR 48C12 has a SBC of 227 Å3 (calculated with chain A of the structure) that accommodates seven water molecules (Fig. 4b). At pH 4.3, its SBC shrinks to 216 Å3 and accommodates a negatively charged acetate ion and three water molecules induced by the protonation of its Schiff base counterion, Glu-1071. Singh et al.15 showed that HeR 48C12 wildtype does not bind specific anions, whereas the counterion neutralizing mutants, E107A and E107Q, both bind anions at pH 7.0, which are needed to keep the RSBH+ protonated. The E107A mutant of HeR 48C12 in the presence of 500 mM chloride shows the same λmax as its wildtype15, similar to TaHeR. The authors proposed that since E107A absorbance exhibited a large ion dependence (12 nm redshift from chloride to iodide), the anion may be directly hydrogen bonded to the RSBH+. In contrast, the E107Q mutation shows small ion dependence (2 nm redshift from chloride to iodide), suggesting that the anion may be hydrogen bonded to the N–H group of Glu-107. This hypothesis can now be specified by our pH 4.5 TaHeR structure, where the analogous protonated O–H group of Glu-108 is located 4.6 Å from the chloride anion and linked by two hydrogen bonded water molecules (Supplementary Fig. S3).
Given the same behavior for TaHeR and HeR 48C12 and the well conserved amino acids enveloping the SBC among the HeR family, we conclude that upon replacement of the primary counterion with a neutral residue or under acidic conditions, most HeRs will likely accommodate exogenous anions as surrogate counterions to stabilize RSBH+. This behavior has also been observed in type I rhodopsins, like the outward-proton pump bacteriorhodopsin (BR). Wildtype BR does not bind chloride. However, BR was found to bind anions from solution to stabilize the RSBH+ in the D85S and D85T single counterion mutants36,37, the D85N/D212N double counterion mutant38, and at very low pH in the acid-purple form39.
A putative intramolecular signal transduction pathway
If HeRs function as sensors, they must have a mechanism to propagate conformational changes to a transducer protein. It was previously shown for TaHeR prepared with a “locked” all-trans-retinal (where isomerization is prevented by a 5-membered ring) that light-induced protein conformational changes occur even in the absence of retinal isomerization, which are instead triggered by the retinal excited state40. In a similar way, pH changes have also been shown to induce conformational changes due to protonation or deprotonation of key residues, which may reflect possible rearrangements in protein structure during its photocycle5,41. By comparing the available HeR structures at different pH values, we observe a conserved propagation of conformational changes originating from protonation of the RSBH+ counterion, which we later demonstrate computationally (see below).
Previously, Kovalev et al. compared the pH 4.3 and pH 8.8 structures of HeR 48C12 and observed two main changes, which are depicted in Fig. 5b. Adjacent to the RSB, residues Ser-76 and Ser-237 underwent rearrangement. Further, in the basic form, there exists a hydrogen-bonded network on the extracellular half of the protein comprising a water molecule, Ser-242, Gln-26, and Trp-246 linking helices A and G. In the acidic form, the adjacent His-23 rotates and displaces the water to induce a large conformational change in the network, flipping the orientation of Trp-246 and breaking the Glu-26–Trp-246 hydrogen bond. It was proposed that if HeRs are light sensors, Trp-246 reorientation may trigger interaction with a signal transducer protein5.

A putatively conserved intramolecular signaling pathway for TaHeR and HeR 48C12. (a) TaHeR dimer structure at pH 8 (cyan, PDB ID 6is6) and pH 4.5 (yellow, PDB ID 7u55). (b) HeR 48C12 dimer structure at pH 8.8 (green, PDB ID 6su3) and pH 4.3 (pink, PDB ID 6su4). The conserved intramolecular signaling mechanism involves a negatively charged anion (chloride or acetate) occupying the SBC (grey surface), switching the orientation of His-23, displacing water molecules (red arrows), disordering Gln-26 and disordering Trp-243 (in TaHeR) or Trp-246 (in HeR 48C12). The overlay reveals there is a 1 Å movement of the intracellular B–C loop. TaHeR has an additional 6 Å shift of the A − B tail from the electrostatic attraction of Tyr-48 and Asn-53 to an external chloride ion. Waters are shown as small spheres colored to match protein, and chloride ions as large green spheres (belonging to pH 4.5 TaHeR only). The 2Fo − Fc maps (blue mesh) are contoured at 1 σ, and Fo − Fc is contoured at + 3 σ for positive (green mesh) and − 3 σ for negative (red mesh) electron density, respectively. ‡ In HeR 48C12 at pH 4.3, there is no electron density to account for this water in the chain A protomer.
By comparing the pH 8 and pH 4.5 TaHeR structures (Fig. 5a), we see very similar behavior to HeR 48C12. His-23 shows a well-defined reorientation, a water molecule adjacent to Ser-78 is displaced, and Trp-243 and Gln-26 become disordered by occupying two conformations rotated ~ 180° apart. A difference to HeR 48C12 is that the backbone of TaHeR at both pH are well aligned, except at the apex of the large A–B loop encompassing residues 46–56. At its largest, there is a ~ 6 Å shift between the Cα atoms of Ile-51. This A–B loop movement observed between the two TaHeR structures may originate from the different crystallization methodology or different crystal packing. In the low pH structure, the side chain nitrogen of Asn-53 is electrostatically attracted by the extracellular chloride ion in the other protomer and the sidechain oxygen of Asn-53 is hydrogen bonded to Tyr-48. These polar interactions pull the tip of the A–B loop 6 Å closer to the other protomer at residue Ile-51. Out of the four compared low/high pH structures of TaHeR and HeR 48C12, the pH 8 TaHeR structure is different. It has the lowest resolution and shows three-times larger B-factors for the tip of the A–B loop than for the overall structure. The observed A–B loop movement may be explained by a reduction in loop dynamics as a consequence of crystallization, which motivated us to apply computational methods and EPR/DEER spectroscopy.
The A–B loop movement is not observed in HeR 48C12, perhaps since its pH 4.3 and pH 8.8 structures were both crystallized using LCP and possessed similar crystal packing. Additionally, HeR 48C12 has different residues comprising the bend in the A–B loop. HeR 48C12 has a non-polar Gly-Pro-Pro-Gly (GPPG) motif, while TaHeR has a polar Tyr-Asp-Glu-Ile (YDEI) motif, both spanning residues 48–51 in the A–B loop. As discussed above, the first residue in the YDEI motif, Tyr-48 in TaHeR, may contribute to the A–B loop movement. The GPPG motif is conserved among many HeRs including those in Ornithinimicrobium pekingense (OpHeR), Nocardioides dokdonensis (NdHeR) and Demequina lutea (DlHeR)17. The polar motif is more variable with FDEA, FDTT, FDEI, and other variants in HeRs from Thermococcus sp. 2319 × 1 (TcHeR), Halorhabdus tiamatea SARL4B (HtHeR), and Dehalogenimonas alkenigignens (DaHeR)17, respectively. It was previously shown that mutating GPPG to FDEA in OpHeR yielded a + 1 nm redshift of the maximum retinal absorbance, while the reverse FDEA to GPPG mutation in TcHeR yielded a + 3 nm redshift17, suggesting that disrupting the polar motif causes conformational changes which reach to the retinal binding pocket/RSB to affect the HeR's absorption properties.
Allostery and dynamics in HeRs
We investigated how a charge perturbation in the SBC base cavity of HeRs could facilitate signal transmission using rigidity transmission allostery (RTA) algorithms42,43. RTA utilizes concepts in mathematical rigidity theory42,43,44,45,46 to analyze allosteric networks within protein structures. RTA is based on the idea that a binding event at one site introduces constraints which perturb local conformational degrees of freedom and changes in rigidity that could propagate and cause a change in degrees of freedom in distant parts of a structure. The allosteric communication is characterized by significant changes in degrees of freedom. Upon rigidifying the anion in the SBC, chloride for TaHeR and acetate for HeR 48C12, an allosteric pathway across the membrane emerges (Fig. 6a,c). An allosteric pathway can be defined between the perturbed SBC and the A–B and B–C loops in the extracellular and intracellular space, respectively, traversing a central four-helix bundle comprising transmembrane helices B, C, F, and G for both HeRs. Interestingly, applying RTA to the SBC of one protomer leads to allosteric changes propagating to the other protomer, suggesting that allostery is enhanced by the dimer (data not shown). Ultimately, if HeRs behave as light sensors, the SBC may be critical in facilitating allosteric transmission for signaling.

Allosteric transmission and conformational dynamics modelling in TaHeR and HeR 48C12 dimers. (a, c) The allosteric network within HeR dimers is revealed through rigidity theory allostery analysis. Allosteric transmission is measured by changes in conformational degrees of freedom (red/blue gradient bar) experienced upon rigidification of the negatively charged ion (chloride or acetate, green) occupying the SBC (grey surface) of the acidic pH structure. Amino acids within 3 Å of the charged ion (grey portions of the helix) were omitted from the coloring scheme to prevent skewed coloring biases, as neighbouring residues would naturally have high allosteric effects. (b, d) Constrained geometric Monte Carlo-based dynamics analysis predicts individual residue dynamics measured as the root mean squared fluctuation (RMSF). The extracellular β-sheet (A–B loop) that was further on investigated by DEER is highlighted in pink.
Constrained geometric Monte Carlo simulations reveal that the A–B loop is the most dynamic portion of TaHeR and HeR 48C12 (Fig. 6b,d). TaHeR exhibits a greater root mean squared fluctuation (RMSF) in the A–B loop than HeR 48C12. Since the A–B loop in HeR 48C12 retains more rigidity and possesses more optimal interaction with the rest of the protein, allosteric transmission can propagate further into the β-sheet (Fig. 6c, red β-sheet) compared to TaHeR (Fig. 6a, light blue β-sheet). The large amplitude dynamics are somewhat unexpected since the A–B loop in HeR is essential in dimer formation. A higher amplitude of motion may be required to facilitate unlatching of the A–B loop for a light-activated function. In a previously published HS-AFM video of HeR 48C1216, two dimers can be observed interacting as a pseudo tetramer, where they exchange protomer partners and then dissociate back into dimers (Supplementary Fig. S5). This oligomeric exchange would require that the A–B loop possesses sufficiently high dynamics to switch binding partners by detaching from one protomer and binding to another.
Electron paramagnetic resonance spectroscopy
The pH 4.5 and pH 8.0 TaHeR structures suggested that the A–B loop conformation may play a role in the TaHeR photocycle. Thus, the dynamics of the A–B loop subject to different pH and illumination conditions were explored using CW-EPR and DEER.
For EPR experiments, Ile-51 was mutated to cysteine (TaHeR-I51C) and then combined with a cysteine-reactive spin label, MTSL (1-oxyl-2,2,5,5-tetramethyl-∆3-pyrroline-3-methyl methanethiosulfonate) or IAP (3-(2-iodoacetamido) proxyl)47. Native cysteines, Cys-168 and Cys-205, were not accessible for the spin labels as shown by two control experiments (Fig. 7a and Supplementary Fig. S7). This can be accounted for by topological shielding of the detergent micelles. Since TaHeR-I51C initially formed unwanted covalent oligomers via Cys-51 linked disulfide bonds, we designed a purification protocol that successfully reduced covalent oligomers and selected for spin-labelled dimers (Supplementary Fig. S6).

DEER of TaHeR-I51P1 suggests HeR can adopt dimer-of-dimer assemblies that are pH- and light-dependent. (a) The DEER dipolar evolution function of TaHeR-I51P1 with indicated modulation depth (Δ) at pH 8 and pH 4.5 in dark and light conditions. The inset (red) shows data from wildtype TaHeR incubated with IAP spin label. Here, the lack of dipolar interactions indicated that native cysteines could not be sufficiently spin-labelled. (b) The DEER distance distribution, P(r), revealed two spatially separated peaks at 4.7 nm and 6.6 nm. The experimental parameters allow for accurate distance distributions up to 5.9 nm and mean distances up to 7.4 nm24. Relative to the pH 8 dark sample, light or acidification increases the population of the 6.6 nm peak (up arrow). Relative to the pH 4.5 dark sample, illumination slightly decreases the population of the 4.7 nm peak (down arrow). DEER experiments were performed in duplicate. (c) MMM distance distributions of dimer and dimer-of-dimer assemblies compared to the pH 8 dark sample. Dimer-of-dimer assemblies were generated from AlphaFold (square, L-shaped) or PyMOL (staggered, V-shaped), with corresponding models shown in panel e. (d) Model of the TaHeR-I51P1 dimer with the P1 rotamer cloud (orange surface) from the pH 4.5 structure. The two native cysteines, Cys-168 and Cys-205, are inaccessible to spin-labelling. (e) Dimer-of-dimer models with P1 rotamer clouds (orange surface). (f) HS-AFM images of TaHeR and HeR 48C12 from the supplementary videos of ref.16 showing similar dimer-of-dimer assemblies. Reprinted by permission from CCC: Springer Nature Shihoya et al. (2019)16.
CW-EPR revealed that TaHeR-I51C was successfully spin-labelled with MTSL or IAP (Supplementary Fig. S7), forming TaHeR-I51R1 or TaHeR-I51P1, respectively. Moreover, CW-EPR spectra of TaHeR-I51R1 and TaHeR-I51P1 show relatively narrow linewidths, which are characteristic for a spin label attached to a flexible loop region25, as expected. The spectral shape was largely unaffected by changes in pH or light exposure, suggesting no major changes in secondary structure, polarity, or solvent exposure around this label. Because the R1-labelled samples precipitated under acidic conditions, we focused on the P1-labelled samples, which gave consistent and reproducible data for all tested conditions.
Thereafter, DEER was employed to yield distance distributions between spin labels on adjacent A–B loops in the TaHeR dimer. The DEER experiment on TaHeR-I51P1 at pH 8.0 showed a rather broad distance population centered at ~ 4.7 nm and a peak at 6.6 nm that could be only clearly separated and identified after sample perdeuteration for better long-distance resolution (Fig. 7b). We assume that the broad feature in P(r) highlights highly flexible loop dynamics that are blurring out further conformational details. Based on the computed DEER distributions using MMM software48, employing our TaHeR structures at pH 8 and 4.5, we expected to observe distances only between 2 and 5 nm for both pH values, with a shift towards larger distances under acidic conditions (Fig. 7c). Initially, an unexpected long distance beyond this predicted distance range was also measured for TaHeR-I51R1 (Supplementary Fig. S8). The long-distance peak at 6.6 nm is already visible in the dipolar evolution function, appearing as a low frequency dipolar oscillation that persists even after background correction. The presence of this oscillation cannot be explained by the sample concentration of 220 µM, which would give rise to an estimated mean distance of ca. 25 nm in between individual TaHeR dimers (see Supplementary methods for calculation).
For TaHeR-I51P1, beyond the effect of pH (4.5, 8.0), also the effect of illumination (dark, light) on the A–B loop was investigated. The distance distributions for all samples exhibited similar shapes with slightly varying peak populations (Fig. 7b), which we further validated (Supplementary Fig. S9). The DEER data did not show a clear pH-induced shift to larger distances as predicted from MMM, which reflects the wide ensemble of conformations available to the flexible A–B loop in solution. This dynamic feature may be inaccessible in the crystal due to packing constraints and constitutes a major advantage of applying DEER spectroscopy in general31. MMM also did not account for the 6.6 nm population. For a standalone dimer (Fig. 7d) to achieve this long distance, the A–B loops would need to undergo extremely large conformational changes.
As an explanation for this long distance, we propose that TaHeR can form transient dimer-of-dimers associations. These pseudo-tetramers could account for the broad long-distance peak at 6.6 nm. This dimer-of-dimers hypothesis is further supported by slight changes in the modulation depth (Δ, Fig. 7a), which can serve as an indirect measure for the number of coupled spins per cluster24,29,49,50. Relative to the pH 8.0 dark sample (Δ = 0.094), there was an increase in the modulation depth under illumination (Δ = 0.116), acidification (Δ = 0.126), or both (Δ = 0.103), denoting an increase in the number of spins per cluster and therefore a slight change in oligomerization can be anticipated. According to our data, an increase in modulation depth is generally also accompanied by an increase in the 6.6 nm peak population. Thus, light irradiation, or lowering pH could slightly increase the number of dimer-of-dimer assemblies. Unusually, illumination of the acidic dark sample decreases its modulation depth, while still increasing the 6.6 nm distance peak. We can conclude that light exposure and protein net charge can be both used to induce slight variations in the intermolecular dimer-of-dimers assembly of TaHeR.
The idea of ‘dimer-of-dimers’ is also strongly supported by high-speed atomic force microscopy (HS-AFM) as performed on TaHeR and HeR 48C12 reconstituted into lipid bilayers16. HS-AFM of TaHeR at pH 8 shows prolonged dimer-of-dimers interactions oriented in a V-shape or square shape16 (Fig. 7f). Similarly, HS-AFM of HeR 48C12 showed transient dimer-of-dimers associations that existed in an L-shaped, staggered shaped, or V-shaped alignment16 (Fig. 7f). Both phenomena can be observed throughout the full HS-AFM recording, although not explicitly discussed in the publication.
Dimer-of-dimer models were generated from either the X-ray crystal packing, Alphafold51,52, or structural modelling using PyMOL53. MMM was used to simulate distance distributions from these pseudo tetramers, all of which are all presented in Supplementary Fig. S10, and the best fit assemblies in Fig. 7c. Briefly, the crystallography-derived pseudo tetramers, including vertically stacked and inverted-L shaped, did not fit the DEER distribution and are also considered as biologically irrelevant arrangements in the cell membrane. AlphaFold predicted more biologically plausible L-shaped and square-shaped tetrameric assemblies (Fig. 7e), with all protomers oriented in the same plane and membrane orientation. The L-shape conformation fits the DEER data exhibiting appropriate short and long distances, while the square-shaped alignment only populates the short distances (Fig. 7c). Finally, PyMOL was employed to generate staggered and V-shaped tetramer orientations of TaHeR (Fig. 7e) based on the conformations observed in the aforementioned HS-AFM videos16. Both V-shaped and staggered orientations fit the experimental DEER data, resembling the short and long-distance features quite well (Fig. 7c).
Ultimately, the combination of DEER, structural modelling, and MMM computations revealed that TaHeR-I51P1 exhibits long distance dipolar interactions from potential dimer-of-dimer assemblies, and short distance dipolar interactions from a convolution of dimer and pseudo tetramer contributions. These potential dimer-of-dimer assemblies occur even in detergent micelles and are pH- and light-dependent. Specifically, light and acidification promote dimer-of-dimer assemblies increasing the population of the long distance peak at 6.6 nm. We propose that these dimer-of-dimer assemblies may be necessary for a putative light sensory function, such that two TaHeR dimers adopt a specific relative orientation to bind a transducer protein.
In summary, the investigation of TaHeR using X-ray crystallography, computational methods, and DEER enabled us to identify an anion binding site that compensates for the neutralization of the Schiff base counterion at low pH, propose a conserved intramolecular signaling pathway, illustrate a mechanism for allosteric signal transmission, and identify higher order assemblies of heliorhodopsin in detergent. This work provides insights into conformational changes of heliorhodopsin that might be used by the microbial rhodopsin to serve as a possible light sensor.
Methods and materials
HeR expression and purification
For expression of TaHeR wildtype, a gene encoding Thermoplasmatales archaeon SG8-52–1 HeR (GenBank ID: KYK26602.1) with an N-terminal 6 × His tag was obtained from GenScript and cloned into the pET21a(+) vector using NdeI-XhoI restriction sites. For expression of mutant TaHeR-I51C, codon exchange from ATC to TGC was done using the QuikChange Lightning (Agilent) toolkit. The mutation was verified by DNA sequencing (ACGT Corporation, Toronto). TaHeR was expressed in E. coli OverExpress™ C43(DE3) chemically competent cells using a similar protocol described previously16,33. Transformed E. coli cells were grown (37 °C, 200 rpm) in LB media supplemented with 100 µg/mL ampicillin until an OD600 of 0.6 was obtained. Protein expression was induced by the addition of 0.5 mM isopropyl-β-D-thiogalactopyranose and 5 µM of all-trans retinal, and incubated overnight at a reduced temperature (25°, 200 rpm, 20 h). The collected cells were lysed using an Emulsiflex C3 (Avestin) in buffer containing 50 mM MES, pH 6.5, 300 mM NaCl, and a SIGMAFAST™ protease inhibitor cocktail tablet (Sigma). The crude membranes were pelleted via ultracentrifugation (4 °C, 1 h, 130 000× g, Beckman, 45Ti rotor), and then solubilized in 50 mM MES, pH 6.5, 300 mM NaCl, 2% w/v of n-octyl β-D-glucopyranoside (OG, Glycon) overnight at 4 °C. Solubilized TaHeR was recovered through ultracentrifugation (4 °C, 1 h, 130 000 × g) and the pellet was discarded. By approximating ε550 = 42,100 M−1 cm−1 at pH 6.5, the yield of TaHeR was ~ 10 mg per litre of expression medium.
A two-step purification protocol of nickel immobilized metal affinity chromatography (Ni2+ IMAC) and size exclusion chromatography was employed to achieve TaHeR at > 95% purity as seen by gel electrophoresis and western blotting (Supplementary Fig. S1). For IMAC purification, the solubilized solution was loaded onto a HisTrap™ 1 mL Ni–NTA column (GE Healthcare) equilibrated with 20 mM imidazole, 50 mM MES, pH 6.5, 300 mM NaCl, 1% OG. TaHeR was eluted using a 30-column volume linear imidazole gradient from 20 to 500 mM imidazole. Fractions with sufficient purity (A550nm/A280nm > 0.65) were combined and concentrated using a 30 kDa concentrator tube (Amicon). Size exclusion chromatography (SEC) was run on 1 mL of concentrated protein loaded on a Superdex™ 200 10/300 GL column (Cytiva) using 20 mM MES, pH 6.5, 300 mM NaCl, 1% OG. Consecutive fractions with A550nm/A280nm > 0.7 were combined and prepared for crystallization.
Crystallization and harvesting
24% w/v bicelles composed of 2.8:1 DMPC:CHAPSO (molar ratio) were prepared in advance32, and stored at –20 °C. Prior to crystallization, the bicelles were thawed at room temperature and then kept on ice. Purified TaHeR was concentrated to 7.5 mg/mL and combined with bicelles in a 2:1 ratio (resulting in 5 mg/mL protein: 8% bicelle), then kept on ice for two hours to encourage mixing. The crystallization buffer was prepared containing 26% polyethylene glycol 3350, 0.1 M sodium phosphate monobasic monohydrate at pH 4.5 (Hampton Research), 0.28 M ammonium sulfate, and 0.18 M 1,6-hexanediol. Hanging-drop vapour-diffusion crystallization experiments were set up on standard pre-greased 24-well crystallization trays (Hampton). 4 µL of TaHeR-bicelle mixture was gently added to 1.5 µL of crystallization buffer without mixing on a thick siliconized cover slide, which was held over 0.5 mL of crystallization buffer in the reservoir. Crystal trays were stored at 34 °C and left undisturbed for several months. Large 50–100 µm diamond-shaped crystals formed (Supplementary Fig. S2) and were harvested after four months using various MicroLoop LD™ 50–200 μm (MiTeGen), and then flash frozen in liquid nitrogen without addition of cryoprotectant.
Data collection and analysis
X-ray diffraction experiments were carried out on beamline 23-ID-B of the Advanced Photon Source at Argonne National Laboratory (Lemont, Illinois). Diffraction data were indexed using xia2/DIALS54 and scaled using AIMLESS55 from the CCP4i program package56. The acidic HeR structure was solved by molecular replacement with phaser57 using basic TaHeR16 as a search model (PDB ID 6is6). The model was manually built in Coot58 and iterative refinement was done with the phenix.refine routine of Phenix59. Refinement statistics are summarized in Table 1. Figures with 3D structures were prepared using PyMOL53.
Data availability
The TaHeR structure at pH 4.5 was deposited to the Protein Data Bank under PDB ID 7u55.
Phylogenetic tree
A multiple amino acid alignment was carried out using ClustalW60. The evolutionary distances were estimated by employing the Maximum Likelihood method and JTT matrix-based model61 using 100 bootstrap replicates. Evolutionary analyses were conducted in MEGA X62. The phylogenetic tree was rendered using iTOL63. The NCBI searchable sequence codes are provided in Supplementary Table S1.
Site-directed spin-labelling of TaHeR-I51C
TaHeR-I51C protein expression was identical to the wildtype. However, the purification protocol was modified because the cysteine mutation introduced intermolecular disulfide bonds that formed covalent oligomers as shown by the two peaks in the size exclusion chromatogram (Supplementary Fig. S6). Thus, following Ni2+ IMAC purification, tenfold molar excess of the reducing agent 1,4-dithiothreitol (DTT) was added to TaHeR-I51C for 1 h at 4 °C in the dark, which successfully reduced disulfide bonds between Cys-51 residues. SEC was run on the reduced protein using a Superdex™ 200 10/300 GL column to remove the DTT and change the buffer to 20 mM MES, pH 7.2, 300 mM NaCl, 1% OG for optimal spin-labelling. Immediately after protein elution from the SEC, a tenfold molar excess of the spin label, either MTSL (1-oxyl-2,2,5,5-tetramethyl-∆3-pyrroline-3-methyl methanethiosulfonate) or IAP (3-(2-iodoacetamido) proxyl) was added and incubated for 2 h at 4 °C with gentle rotation to form TaHeR-I51R1 or TaHeR-I51P1. A second SEC was performed to remove excess label and unwanted residual covalent oligomers. The SEC fractions containing only the labelled protein dimer were then combined. See Supplementary Fig. S6 for a detailed overlay of the SEC spectra.
Sample preparation for EPR
To prepare TaHeR-I51R1 or TaHeR-I51P1 for EPR, a PD MidiTrap G-25 column was employed to exchange the solvent to a basic buffer (pH 8, 20 mM Tris, 300 mM NaCl, 1% OG, D2O) or acidic buffer (pH 4.5, 20 mM sodium phosphate, 300 mM NaCl, 1% OG, D2O). D2O was used to increase the phase memory time and therefore the accessible time trace length for DEER experiments up to 7.0 μs27,28,29. Samples were concentrated to ~ 220 µM for TaHeR-I51P1, or ~ 110 µM for TaHeR-I51R1. 20% v/v D8-glycerol was added to the samples as a cryo-protectant for all subsequent CW-EPR and DEER experiments.
EPR control experiments and CW-EPR
See supplementary methods, Fig. 7a, and Supplementary Fig. S7 for further details.
DEER spectroscopy
High sensitivity Q-band (33.6–33.8 GHz) 4-pulse DEER experiments were conducted on a Bruker Elexsys E580 spectrometer equipped with a Super Q-FTu microwave bridge, a 10 W AmpQ and an EN 5107D2 Q-band flexline resonator. Spin-labelled samples (TaHeR-I51R1 or TaHeR-I51P1) were pipetted into quartz capillaries (1.5 mm inner diameter, 1.8 mm outer diameter, VitroCom) under dark or illuminated conditions. The dark samples were kept under light exclusion for 1 h before conducting measurements under dim red light (red LED lamps, λmax ~ 625 nm). The light samples were illuminated with yellow light using a Fiber-Lite MI-150 lamp (Dolan-Jenner) equipped with a yellow filter (< 500 nm) for 60 s prior to flash freezing. The samples containing D8-glycerol as a cryoprotectant, were vitrified using a dry-ice/ethanol bath and stored at − 80 °C prior to data collection. The sample temperature was adjusted to 80 K with an Oxford Instruments continuous-flow cooling system and data collection was performed for 16–24 h to achieve optimum signal-to-noise ratios. A 32-ns π-pump pulse was applied to the low field peak of the nitroxide field swept spectrum, and the observer π/2 (16 ns) and π (32 ns) pulses were positioned 50–56 MHz (18–20 G) upfield. In order to cancel out receiver offsets and unwanted echoes, a 2-step phase cycle was employed. Distance distributions were obtained from the raw time traces using the Matlab®-based open-source program DeerAnalysis 201964. To obtain the distance distributions P(r), a 3-D background correction was used exclusively for all data sets. After Tikhonov regularization, the GCV algorithm65 was chosen to determine an optimum regularization parameter (794 ≤ α ≤ 1000) from the resulting L curves. The Tikhonov validation error estimates of all P(r) are given in Supplementary Fig. S9. Theoretical DEER distance distributions of the TaHeR crystal structures and dimer-of-dimer models were calculated using the Matlab®-based, open-source program, Multiscale Modeling of Macromolecules (MMM)66. DeerAnalysis and MMM are both available at https://epr.ethz.ch/software.html. For all routines, Matlab® version R2020b was used.
Dimer-of-dimer TaHeR models
Dimer-of-dimer HeR arrangements were acquired from all available HeRs crystal structures, Alphafold51,52, and using manipulation in PyMOL. Details are presented in the supplementary methods. All models are presented in Supplementary Fig. S10 and the best fit models are given in Fig. 7c.
Allostery predictions
Allostery was analyzed using the rigidity-transmission allostery (RTA) algorithms42,43,67. The RTA method is based on mathematical rigidity theory42,68. Starting with TaHeR (PDB ID 7u55) and HeR 48C12 (PDB ID 6su4) dimers, a constrained network representation of protein structure was generated with the method FIRST44, which consists of nodes (atoms) and edges representing covalent and non-covalent interactions. The RTA was used to quantify the available conformational degrees of freedom at individual residues before and after perturbation of rigidity of the negatively charged ion (chloride or acetate) pocket occupying the SBC. The extent of the “degree of freedom transmission” was then extracted and visualized on the structure based on the strength of allosteric communication.
Constrained geometric Monte Carlo simulations
To probe the dynamical features of TaHeR and HeR 48C12, we have applied a constrained geometric Monte Carlo simulation, based on methodology Framework Rigidity Optimized Dynamics Algorithm New (FRODAN)68,69,70. This approach, which can be regarded as a low computational complexity alternative to MD simulations, utilizes a coarse-grained molecular mechanics potential and rigidity theory to explore the wide regions of conformational space, probing the conformational ensemble well outside the starting structure. Starting with TaHeR and HeR 48C12 crystal structures, we first added hydrogen atoms using the MolProbity server, and ran FRODAN in the non-targeted mode, generating 30,000 candidate structures for each case. Simulations were carried out at different hydrogen bond energy cut-offs, from − 1.0 to − 2.0 kcal/mol; during each individual run, the cut-off value was kept constant. To evaluate the dynamics, backbone Root Mean Square Fluctuations (RMSF) were calculated.
References
Pushkarev, A. et al. A distinct abundant group of microbial rhodopsins discovered using functional metagenomics. Nature 558, 595–599. https://doi.org/10.1038/s41586-018-0225-9 (2018).
Rozenberg, A., Inoue, K., Kandori, H. & Beja, O. Microbial rhodopsins: The last two decades. Annu. Rev. Microbiol. 75, 427–447. https://doi.org/10.1146/annurev-micro-031721-020452 (2021).
Shibukawa, A. et al. Photochemical characterization of a new heliorhodopsin from the gram-negative eubacterium Bellilinea caldifistulae (BcHeR) and comparison with heliorhodopsin-48C12. Biochemistry 58, 2934–2943. https://doi.org/10.1021/acs.biochem.9b00257 (2019).
Chazan, A. et al. Diverse heliorhodopsins detected via functional metagenomics in freshwater Actinobacteria, Chloroflexi and Archaea. Environ. Microbiol. 24, 110–121. https://doi.org/10.1111/1462-2920.15890 (2022).
Kovalev, K. et al. High-resolution structural insights into the heliorhodopsin family. Proc. Natl. Acad. Sci. USA 117, 4131–4141. https://doi.org/10.1073/pnas.1915888117 (2020).
Bulzu, P. A. et al. Heliorhodopsin evolution is driven by photosensory promiscuity in monoderms. mSphere https://doi.org/10.1128/mSphere.00661-21 (2021).
Sakmar, T. P. & Huber, T. Ancient family of retinal proteins brought to light “sight-unseen”. Biochemistry 57, 6735–6737. https://doi.org/10.1021/acs.biochem.8b01188 (2018).
Flores-Uribe, J. et al. Heliorhodopsins are absent in diderm (Gram-negative) bacteria: Some thoughts and possible implications for activity. Environ. Microbiol. Rep. 11, 419–424. https://doi.org/10.1111/1758-2229.12730 (2019).
Hososhima, S. et al. Proton-transporting heliorhodopsins from marine giant viruses. BioRxiv https://doi.org/10.1101/2022.03.24.485645 (2022).
Ernst, O. P. et al. Microbial and animal rhodopsins: Structures, functions, and molecular mechanisms. Chem. Rev. 114, 126–163. https://doi.org/10.1021/cr4003769 (2014).
Lu, Y. et al. Crystal structure of heliorhodopsin 48C12. Cell Res. 30, 88–90. https://doi.org/10.1038/s41422-019-0266-0 (2020).
Otomo, A. et al. Resonance Raman investigation of the chromophore structure of heliorhodopsins. J. Phys. Chem. Lett. 9, 6431–6436. https://doi.org/10.1021/acs.jpclett.8b02741 (2018).
Tahara, S. et al. Ultrafast dynamics of heliorhodopsins. J. Phys. Chem. B 123, 2507–2512. https://doi.org/10.1021/acs.jpcb.9b00887 (2019).
Singh, M., Inoue, K., Pushkarev, A., Béjà, O. & Kandori, H. Mutation study of heliorhodopsin 48C12. Biochemistry 57, 5041–5049. https://doi.org/10.1021/acs.biochem.8b00637 (2018).
Singh, M., Katayama, K., Béjà, O. & Kandori, H. Anion binding to mutants of the Schiff base counterion in heliorhodopsin 48C12. Phys. Chem. Chem. Phys. 21, 23663–23671. https://doi.org/10.1039/c9cp04102h (2019).
Shihoya, W. et al. Crystal structure of heliorhodopsin. Nature 574, 132–136. https://doi.org/10.1038/s41586-019-1604-6 (2019).
Kim, S. H. et al. Color-tuning of natural variants of heliorhodopsin. Sci. Rep. 11, 854. https://doi.org/10.1038/s41598-020-72125-0 (2021).
Bogomolni, R. A. & Spudich, J. L. The photochemical reactions of bacterial sensory rhodopsin-I. Flash photolysis study in the one microsecond to eight second time window. Biophys. J. 52, 1071–1075. https://doi.org/10.1016/S0006-3495(87)83301-5 (1987).
Sasaki, J. & Spudich, J. L. The transducer protein HtrII modulates the lifetimes of sensory rhodopsin II photointermediates. Biophys. J. 75, 2435–2440. https://doi.org/10.1016/S0006-3495(98)77687-8 (1998).
Chizhov, I. et al. The photophobic receptor from Natronobacterium pharaonis: Temperature and pH dependencies of the photocycle of sensory rhodopsin II. Biophys. J. 75, 999–1009. https://doi.org/10.1016/S0006-3495(98)77588-5 (1998).
Mironova, O. S. et al. Functional characterization of sensory rhodopsin II from Halobacterium salinarum expressed in Escherichia coli. FEBS Lett. 579, 3147–3151. https://doi.org/10.1016/j.febslet.2005.05.010 (2005).
Gellini, C., Luttenberg, B., Sydor, J., Engelhard, M. & Hildebrandt, P. Resonance Raman spectroscopy of sensory rhodopsin II from Natronobacterium pharaonis. FEBS Lett. 472, 263–266. https://doi.org/10.1016/s0014-5793(00)01472-1 (2000).
Tateishi, Y. et al. Spectroscopic evidence for the formation of an N intermediate during the photocycle of sensory rhodopsin II (phoborhodopsin) from Natronobacterium pharaonis. Biochemistry 50, 2135–2143. https://doi.org/10.1021/bi1019572 (2011).
Jeschke, G. DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 63, 419–446. https://doi.org/10.1146/annurev-physchem-032511-143716 (2012).
Reichenwallner, J., Liu, B., Balo, A. R., Ou, W. L. & Ernst, O. P. Electron paramagnetic resonance spectroscopy on G-protein-coupled receptors: Adopting strategies from related model systems. Curr. Opin. Struct. Biol. 69, 177–186. https://doi.org/10.1016/j.sbi.2021.06.003 (2021).
Van Eps, N. et al. Conformational equilibria of light-activated rhodopsin in nanodiscs. Proc. Natl. Acad. Sci. USA 114, E3268–E3275. https://doi.org/10.1073/pnas.1620405114 (2017).
Schmidt, T., Walti, M. A., Baber, J. L., Hustedt, E. J. & Clore, G. M. Long distance measurements up to 160 A in the GroEL tetradecamer using Q-band DEER EPR spectroscopy. Angew. Chem. Int. Ed. Engl. 55, 15905–15909. https://doi.org/10.1002/anie.201609617 (2016).
Endeward, B. et al. Long-range distance determination in fully deuterated RNA with pulsed EPR spectroscopy. Biophys. J. 121, 37–43. https://doi.org/10.1016/j.bpj.2021.12.007 (2022).
Bordignon, E. & Bleicken, S. New limits of sensitivity of site-directed spin labeling electron paramagnetic resonance for membrane proteins. Biochim. Biophys. Acta Biomembr. 841–853, 2018. https://doi.org/10.1016/j.bbamem.2017.12.009 (1860).
Morizumi, T. et al. X-ray crystallographic structure and oligomerization of gloeobacter rhodopsin. Sci. Rep. 9, 11283. https://doi.org/10.1038/s41598-019-47445-5 (2019).
Jeschke, G. The contribution of modern EPR to structural biology. Emerg. Top. Life Sci. 2, 9–18. https://doi.org/10.1042/ETLS20170143 (2018).
Agah, S. & Faham, S. Crystallization of membrane proteins in bicelles. Methods Mol. Biol. 914, 3–16. https://doi.org/10.1007/978-1-62703-023-6_1 (2012).
Tanaka, T. et al. Structural basis for unique color tuning mechanism in heliorhodopsin. Biochem. Biophys. Res. Commun. 533, 262–267. https://doi.org/10.1016/j.bbrc.2020.06.124 (2020).
Hashimoto, M., Katayama, K., Furutani, Y. & Kandori, H. Zinc binding to heliorhodopsin. J. Phys. Chem. Lett. 11, 8604–8609. https://doi.org/10.1021/acs.jpclett.0c02383 (2020).
Ho, B. K. & Gruswitz, F. HOLLOW: Generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct. Biol. 8, 49. https://doi.org/10.1186/1472-6807-8-49 (2008).
Facciotti, M. T., Cheung, V. S., Nguyen, D., Rouhani, S. & Glaeser, R. M. Crystal structure of the bromide-bound D85S mutant of bacteriorhodopsin: Principles of ion pumping. Biophys. J. 85, 451–458. https://doi.org/10.1016/S0006-3495(03)74490-7 (2003).
Tittor, J. et al. Chloride and proton transport in bacteriorhodopsin mutant D85T: Different modes of ion translocation in a retinal protein. J. Mol. Biol. 271, 405–416. https://doi.org/10.1006/jmbi.1997.1204 (1997).
Marti, T., Rosselet, S. J., Otto, H., Heyn, M. P. & Khorana, H. G. The retinylidene Schiff base counterion in bacteriorhodopsin. J. Biol. Chem. 266, 18674–18683 (1991).
Dunach, M., Padros, E., Seigneuret, M. & Rigaud, J. L. On the molecular mechanism of the blue to purple transition of bacteriorhodopsin. UV-difference spectroscopy and electron spin resonance studies. J. Biol. Chem. 263, 7555–7559 (1988).
Das, I., Pushkarev, A. & Sheves, M. Light-induced conformational alterations in heliorhodopsin triggered by the retinal excited state. J. Phys. Chem. B 125, 8797–8804. https://doi.org/10.1021/acs.jpcb.1c04551 (2021).
Okumura, H., Murakami, M. & Kouyama, T. Crystal structures of acid blue and alkaline purple forms of bacteriorhodopsin. J. Mol. Biol. 351, 481–495. https://doi.org/10.1016/j.jmb.2005.06.026 (2005).
Sljoka, A. Probing allosteric mechanism with long-range rigidity transmission across protein networks. Methods Mol. Biol. 2253, 61–75. https://doi.org/10.1007/978-1-0716-1154-8_5 (2021).
Sljoka, A. Algorithms in Rigidity Theory with Applications to Protein Flexibility and Mechanical Linkages (York University Toronto, 2012).
Jacobs, D. J., Rader, A. J., Kuhn, L. A. & Thorpe, M. F. Protein flexibility predictions using graph theory. Proteins Struct. Funct. Bioinform. 44, 150–165 (2001).
Ye, L. et al. Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 9, 1372. https://doi.org/10.1038/s41467-018-03314-9 (2018).
Whiteley, W. Counting out to the flexibility of molecules. Phys. Biol. 2, S116-126. https://doi.org/10.1088/1478-3975/2/4/S06 (2005).
Fielding, A. J., Concilio, M. G., Heaven, G. & Hollas, M. A. New developments in spin labels for pulsed dipolar EPR. Molecules 19, 16998–17025. https://doi.org/10.3390/molecules191016998 (2014).
Jeschke, G. MMM: A toolbox for integrative structure modeling. Protein Sci. 27, 76–85 (2018).
Milov, A., Ponomarev, A. & Tsvetkov, Y. D. Electron-electron double resonance in electron spin echo: Model biradical systems and the sensitized photolysis of decalin. Chem. Phys. Lett. 110, 67–72 (1984).
Jeschke, G., Bender, A., Paulsen, H., Zimmermann, H. & Godt, A. Sensitivity enhancement in pulse EPR distance measurements. J. Magn. Reson. 169, 1–12. https://doi.org/10.1016/j.jmr.2004.03.024 (2004).
Varadi, M. et al. AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444. https://doi.org/10.1093/nar/gkab1061 (2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. https://doi.org/10.1038/s41586-021-03819-2 (2021).
DeLano, W. L. & Lam, J. W. PyMOL: A communications tool for computational models. Abstr. Pap. Am. Chem. S 230, U1371–U1372 (2005).
Winter, G. xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190. https://doi.org/10.1107/S0021889809045701 (2010).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution?. Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214. https://doi.org/10.1107/S0907444913000061 (2013).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242. https://doi.org/10.1107/S0907444910045749 (2011).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. https://doi.org/10.1107/S0021889807021206 (2007).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. https://doi.org/10.1107/S0907444910007493 (2010).
Adams, P. D. et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221. https://doi.org/10.1107/S0907444909052925 (2010).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. https://doi.org/10.1093/bioinformatics/btm404 (2007).
Jones, D. T., Taylor, W. R. & Thornton, J. M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. https://doi.org/10.1093/bioinformatics/8.3.275 (1992).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. https://doi.org/10.1093/molbev/msy096 (2018).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. https://doi.org/10.1093/bioinformatics/btl529 (2007).
Jeschke, G. et al. DeerAnalysis2006—A comprehensive software package for analyzing pulsed ELDOR data. Appl. Magn. Reson. 30, 473–498 (2006).
Edwards, T. H. & Stoll, S. Optimal Tikhonov regularization for DEER spectroscopy. J. Magn. Reson. 288, 58–68. https://doi.org/10.1016/j.jmr.2018.01.021 (2018).
Polyhach, Y., Bordignon, E. & Jeschke, G. Rotamer libraries of spin labelled cysteines for protein studies. Phys. Chem. Chem. Phys. 13, 2356–2366. https://doi.org/10.1039/c0cp01865a (2011).
Huang, S. K. et al. Delineating the conformational landscape of the adenosine A2A receptor during G protein coupling. Cell 184, 1884–1894. https://doi.org/10.1016/j.cell.2021.02.041 (2021).
Sljoka, A. Sublinear Computation Paradigm 337–367 (Springer, 2022).
Farrell, D. W., Speranskiy, K. & Thorpe, M. F. Generating stereochemically acceptable protein pathways. Proteins 78, 2908–2921. https://doi.org/10.1002/prot.22810 (2010).
Zhu, S. et al. Hyperphosphorylation of intrinsically disordered tau protein induces an amyloidogenic shift in its conformational ensemble. PLoS ONE 10, e0120416. https://doi.org/10.1371/journal.pone.0120416 (2015).
Acknowledgements
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We specifically thank the staff at the GM/CA beamlines 23-ID. We would like to thank Professors Leonid Brown and Emil Pai for discussions and advice. This work has been supported in part by the Natural Sciences and Engineering Research Council of Canada (NSERC, Grant RGPIN-2017-06862), a Mitacs award (IT17088), and the Canadian Institute for Advanced Research (to O.P.E. and R.J.D.M.). O.P.E. was supported as Anne and Max Tanenbaum Chair in Neuroscience at the University of Toronto.
Author information
Authors and Affiliations
Contributions
O.P.E., J.E.B, and J.R, designed the research. J.E.B. and P.D.G. performed crystallography. J.E.B. and J.R. conducted EPR experiments and data analysis. A.S. designed and supervised computational analysis. A.S. performed allostery computer simulations and wrote corresponding methods. A.S., A.T and K.T. carried out computational dynamics analysis. T.M. performed the pH titration and analysis. A.K. and T.M. were involved in experimental design. R.J.D.M and O.P.E. provided supervision, resources, and funding acquisition. J.E.B. and O.P.E. wrote the paper. All authors have read, reviewed, and contributed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Besaw, J.E., Reichenwallner, J., De Guzman, P. et al. Low pH structure of heliorhodopsin reveals chloride binding site and intramolecular signaling pathway. Sci Rep 12, 13955 (2022). https://doi.org/10.1038/s41598-022-17716-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-17716-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
