
Abstract
Red maple (Acer rubrum L.) is a type of colorful ornamental tree with great economic value. Because this tree is difficult to root under natural conditions and the seedling survival rate is low, vegetative propagation methods are often used. Because the formation of adventitious roots (ARs) is essential for the asexual propagation of A. rubrum, it is necessary to investigate the molecular regulatory mechanisms of AR formation in A. rubrum. To address this knowledge gap, we sequenced the transcriptome and small RNAs (sRNAs) of the A. rubrum variety ‘Autumn Fantasy’ using high-throughput sequencing and explored changes in gene and microRNA (miRNA) expression in response to exogenous auxin treatment. We identified 82,468 differentially expressed genes (DEGs) between the treated and untreated ARs, as well as 48 known and 95 novel miRNAs. We also identified 172 target genes of the known miRNAs using degradome sequencing. Two key regulatory pathways (ubiquitin mediated proteolysis and plant hormone signal transduction), Ar-miR160a and the target gene auxin response factor 10 (ArARF10) were selected based on KEGG pathway and cluster analyses. We further investigated the expression patterns and regulatory roles of ArARF10 through subcellular localization, transcriptional activation, plant transformation, qRT-PCR analysis, and GUS staining. Experiments overexpressing ArARF10 and Ar-miR160a, indicated that ArARF10 promoted AR formation, while Ar-miR160a inhibited AR formation. Transcription factors (TFs) and miRNAs related to auxin regulation that promote AR formation in A. rubrum were identified. Differential expression patterns indicated the Ar-miR160a-ArARF10 interaction might play a significant role in the regulation of AR formation in A. rubrum. Our study provided new insights into mechanisms underlying the regulation of AR formation in A. rubrum.
Similar content being viewed by others


Introduction
Acer rubrum, a large deciduous tree in family Aceraceae Juss1, is often used to beautify urban gardens due to its strong adaptability and rapid growth2. However, this tree struggle to root under natural conditions, and the survival rate of naturally rooted seedlings is low3. Therefore, some red maple varieties, such as the A. rubrum hybrid ‘Autumn Fantasy’, are primarily vegetatively propagated using softwood cuttings1,4.
AR formation is a critical aspect of cultivation for most forest species that are vegetatively propagated from elite genotypes5. AR formation is a developmental process wherein new roots are generated spontaneously (or in response to certain stimuli) from stems, leaves, or the non-pericycle tissues of older roots6. In particular, AR formation provides the basis for clonal multiplication, a technique that is utilized for the breeding and propagation of many crop and forestry plants7. Auxin, which is formed in roots and stems, and which is involved in stem cell maintenance and differentiation, effectively induces AR formation8,9. Auxin dosage, gradient, and response are all important for plant root growth10. Synthetic auxins, such as indole-3-butyric acid (IBA), have been used to induce AR for almost 80 years11,12. However, the molecular mechanisms regulating hormone-induced AR development in forest tree species remain incompletely understood.
Although molecular studies have identified several genes associated with AR in model plant species, such as Arabidopsis thaliana and Populus6,13, only a few genes that regulate AR formation have been identified in horticultural plants14, and most of these encode TFs14. In total, 35 genes encoding TFs were shown to exhibit significant changes in expression level during AR growth and development in Populus15. However, the functions of most of these genes remain unknown.
By contrast, several genes and proteins associated with AR have been characterized in A. thaliana16. In particular, ARFs are the TFs that regulate the expression of auxin response genes10. Studies of A. thaliana and Populus ARF mutants have shown that ARFs play important roles in plant growth and development. For example, in A. thaliana, single mutations in ARF7 or ARF19 reduced the numbers of both lateral and adventitious roots, and the numbers of lateral and adventitious roots were even more drastically reduced in ARF7/ARF19 double mutants17. Compared with wildtype (WT) poplars, transgenic poplars overexpressing ARF17.1 (Pro35s::PeARF17.1) had numerous stems, no obvious trunk, and an increased number of ARs18. ARFs were strongly inhibited by members of the equally large Aux/Indole-3-Acetic Acid (Aux/IAA) protein family (29 members in A. thaliana) and its core inhibitor TOPLESS19. Key to the regulation of auxin signaling is the ubiquitin-26S proteasome system20-dependent disassembly of Aux/IAA proteins by a four-member family of SCF E3 ligases assembled with transport inhibitory response 1 (TIR1) F-box proteins or its relatives AuxinBinding F-Box 1 (AFB1), AFB2, and AFB320. Gretchen Hagen 3 (GH3) acyl acid amido synthase has also been reported to help regulate levels of plant hormones, including auxin21.
AR development is also regulated by miRNAs, including miR160, miR167, and miR39616. miR160, in conjunction with auxin, was shown to regulate the gene expression of ARF10, ARF16, and ARF17; plants overexpressing miR160c produced shorter roots22. miR167-IAA-Ala Resistant3 (IAR3) also influences lateral root development23,24. In cherry radish, the upregulation of miR160-ARF16, in conjunction with lengthier photoperiods, was shown to drive rapid root formation25. Using high-throughput sequencing, it was shown that the target genes of miR160 and miR390 were related to auxin signaling and involved in the formation of apple rootstock26. Finally, miR396 regulated the transition of root stem cells into transit-amplifying cells by interacting with growth-regulating factor (GRF) in A. thaliana27. miRNAs may also influence AR growth by targeting ARFs10. For example, in A. thaliana, miR160a, miR160b, and miR160c, which have the same mature sequence, target ARF10, ARF16, and ARF17, respectively22. By targeting ARFs, the miR160 family affects the auxin signaling pathway, and thus plays an important regulatory role in root growth and development16.
Despite these previous studies in model plans such as A. thaliana and poplar, the AR-associated functions of many miRNAs remain unknown in most horticultural tree species. To address this knowledge gap, we aimed to systematically identify candidate miRNAs that may be involved in AR development in A. rubrum. In addition, to investigate the AR-associated regulatory miRNA network, we aimed to characterize the expression profiles of miRNAs and their targets during A. rubrum root development. To this end, we constructed RNA-seq, sRNA-seq, and degradome libraries from the ARs of A. rubrum ‘Autumn Fantasy’. We then used bioinformatics approaches to identify AR-associated regulatory pathways, to identify TFs and miRNAs associated with those regulatory pathways, and to characterize the expression profiles of the identified miRNAs and their targets. Finally, to further explore the regulation of ArARF10 by Ar-miR160, we overexpressed Ar-miR160a in A. thaliana and assessed produced root numbers and lengths. Our study showed that the Ar-miR160-ArARF regulatory network plays crucial roles in the growth and development of ARs. These results are of great scientific significance, as they clarify the molecular mechanism of AR formation in A. rubrum, as well as provide a basis for the improvement of A. rubrum rooting ability through molecular breeding to increase its utility in landscape applications.
Results
Transcriptome, sRNA, and degradome sequencing
After 30 days of growth, all IBA-treated groups (100–500 mg/L IBA) had developed ARs, while only a small amount of AR development was observed in the control group (Fig. 1). Importantly, substantially more ARs were developed by the cuttings treated with 300 mg/L IBA; the ARs of these cuttings were also of more consistent length and more densely packed than those formed on the cuttings treated with other concentrations of IBA (Fig. 1). Cuttings treated with 300 mg/L IBA had significantly more roots than any other cuttings. Therefore, the ARs from the cuttings treated with 300 mg/L IBA (henceforth referred to as IBA300), as well as those formed by the control cuttings, were selected for transcriptome profiling.

AR formation in A. rubrum cuttings treated with 100–500 mg/L IBA and deionized water (CK). As IBA concentration increased, the number of ARs showed a trend of increasing and then decreasing and reached a peak at IBA 300 mg/L. Scale bar, 5 mm.
Illumina HiSeq sequencing generated200.4 million raw reads from the control samples and 213.29 million raw reads from IBA300. After quality filtration, 55.48 G of clean data (Q30 ≥ 93.69%) were remained. The percent GC content was greater than 38.16%, the AT and GC ratios were within a reasonable range, and there was no separation. We identified 82,468 DEGs (fold-change ≥ 2, FDR < 0.01) between the control and IBA300. Of these, 69,777 were upregulated and 12,691 were downregulated. Cross all DEG sequences, 31,385 CDS were detected, and 1417 Unigene-encoding transcription factors were predicted. These sequencing data were submitted to the SRA database with the accession numbers SRR13808891, SRR13808890, SRR13808889, and SRR13808888.
In total, approximately 51.22–52.42 million high-quality clean reads from the control and IBA300 groups were obtained using sRNA-seq. Most of the sRNAs were 21–24 nt long, with 24 nt sequences being the most common across all samples (Fig. S1). The reads remaining after the removal of tRNAs, rRNAs, snRNAs, snoRNAs, and degraded mRNAs were considered endogenous sRNAs and used in subsequent analyses. These sequencing data were submitted to the SRA database with the accession numbers SRR13808887, SRR13808886, SRR13808885 and SRR13808884.
Analysis of the sRNA-seq data showed that identified 48 known (Table S1) and 95 novel miRNAs. The length of the mature miRNAs varied from 20 to 30 nt. Due to the recognition and cleavage of DCL1 enzyme, the first base at the 5′ end of each miRNA was biased to U and resistant to G. Across all datasets, the first nucleotide of the novel miRNA was primarily biased towards U, followed by A (Fig. S2). In total, 1744 target genes cleaved by known miRNAs and 1956 miRNA-mRNA pairs were predicted using Targetfinder.
To generate a miRNA-cleaved target library (degradome) from A. rubrum, we first identified the mRNA transcripts targeted by miRNAs in the total RNA samples using high-throughput sequencing. We obtained 34,373,253 short reads from the control RNA samples and 28,061,482 short reads from the IBA300 RNA samples, representing the 5′ ends of uncapped, poly-adenylated RNAs. After initial processing, equal numbers of 20 and 21 nt sequence reads remained. In total, 47.66% of the unique reads were successfully mapped to the A. rubrum transcriptome. We identified 172 mRNAs targeted by the known miRNAs and 243 miRNA-mRNA pairs using CleaveLand (Table S2). These sequencing data were submitted to the SRA database with the accession numbers SRR13808883 and SRR13808882. Across the 172 mRNAs targeted by the known miRNAs, most were associated with plant hormone signal transduction. Of these, 80 mRNAs were either transcription factors or related to TFs, including GRFs, ARFs, SQUAMOSA promoter-binding protein-like (SPL) proteins, MYBs, scarecrow-like3 (SCL), and ethylene response factors (AP2/ERF). Consistent with the RNA-seq results, many genes were associated with the plant hormone signal transduction pathway. However, only 8.8% of the target genes were identified by degradome sequencing. This suggested that most of the predicting miRNA target genes were false positives.
The T-Plot of five conserved miRNAs and one predicted novel miRNA is shown in Fig. 2. Ar-miR172a-2 targeted AP2 (CL321.Contig7_All), at position 2063 nt; Ar-miR160a-5P targeted ARF (CL3897.Contig1_All), at position 1793 nt; Ar-miR156f targeted SPL (CL2023.Contig7_All), at position 1497 nt; Ar-miR171 targeted SCL (CL2444.Contig4_All), at position 1501 nt. All peaks were category 0. Three of the validated differentially expressed miRNAs (Ar-miR160a-5, Ar-miR171d-1, and Ar-miR156f) had previously been shown to target transcription factors.

Identification of candidate miRNA targets using degradome sequencing. Six targets of five conserved and one novel miRNAs are shown in the panels as examples. The positions of the target genes are shown on the x-axis, while numbers of sequenced reads are shown on the y-axis. Each line corresponds to a degradome fragment successfully mapped to the corresponding target gene and the red lines indicate the expected miRNA positions. Category 0 indicates that, where the original data fragment was located, the abundance was equal to the maximum abundance of the mRNA, and that there was only one maximum.
Pathways and functions associated with the DEGs in A. rubrum
Across all DEGs, GO analysis indicated that 14,451 were significantly enriched in 30 GO terms between the control and IBA300 groups (27% cellular component terms, 23% molecular function terms, and 50% biological process terms; Fig. S3). In particular, the biological process terms “regulation of biological process”, “biological regulation”, “cellular process”, “metabolic process”, “signaling”, and “response to stimulus”; the cellular component terms “macromolecular complex”, “cell junction”, “nucleoid”, “organelle”, “cell part”, and “membrane part”; and the molecular function terms “binding”, “catalytic activity”, “transcription regulator activity”, and “signal transducer activity” were overrepresented in the DEGs (Fig. S3).
Of the 14,451 DEGs, 9507 were successfully mapped to the KEGG database (P-value < 0.05). KEGG mapping indicated that 59 terms and 12 metabolic pathways were significantly enriched in these DEGs, with the largest numbers of DEGs associated with the MAPK signaling pathway and the plant hormone signal transduction pathways (Fig. S4). The log-normalized FPKM values of the 60 significantly enriched metabolic pathways were used for hierarchical clustering analysis (Fig. 3). Pathways related to basal TFs, hormone transduction, and exogenous IBAs play a role in promoting gene expression. The significantly upregulated pathways were ubiquitin mediated proteolysis and hormone signal transduction (Table S4).

Hierarchical clustering of the differentially expressed genes associated with the major regulatory pathways. Bars represented scales of pathway expression levels (log 2), from upregulated (red) to downregulated (blue).
Many of the DEGs associated with the plant hormone signaling pathway were involved in auxin signaling transduction. For example, numerous genes in the families associated with auxin signaling (e.g., AUX, IAA, GH3, ARF, and SAUR) were significantly differentially expressed between the control group and the IBA300 group, including Aux/IAA5 (Unigene3732_All), SAUR11 (Unigene73966_All), Aux/IAA4 (Unigene5767_All), GH3 (CL4559.Contig2_All), and ARF18-1 (Unigene5190_All). The expression levels of 24 genes (i.e., the IAAs, GH3s, ARFs, and SAURs) were increased in IBA300 as compared to the control (Fig. 4a). Our results indicated that 42 DEGs were involved in the ubiquitin-mediated proteolysis pathway (Fig. 4b). Some of these DEGs, which were associated with the mediation of Aux/IAA protein ubiquitination, were significantly upregulated, including S phase kinase-associated protein 1 (SKP1) (Unigene7049_All), cullin 1 (CUL1) (Unigene20934_All), and F-box (SCF) E3 (Unigene17129_All). The patterns of upregulation we observed suggested that the plant hormone signaling pathway and the ubiquitin mediated proteolysis pathway jointly regulate A. rubrum root development.

Hierarchical clustering of the differentially expressed genes in the two key pathways. (A) Plant hormone signaling pathway. (B) Ubiquitin mediated proteolysis pathway.
We also performed GO and KEGG analyses of the miRNA target genes. We found that the GO term “DNA binding pathway (GO: 0003677)” was significantly enriched in the target genes (p = 0.003258). KEGG pathway analysis also showed that 10 of the targets of the 220 differentially expressed miRNAs that played a significant regulatory role in the plant hormone signaling pathway belonged to three gene families (the ARF, SCL and TGA families) (Fig. S5).
qRT‑PCR validation of the miRNAs and their targets
Nine miRNAs were selected for qRT-PCR validation. All nine miRNAs were significantly downregulated in the IBA300-treated samples as compared to the control (Fig. S6), which was consistent with our sequencing data. Thus, these nine miRNAs might play important regulatory roles in A. rubrum growth, especially during rooting.
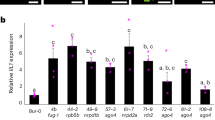
Next, to reveal the expression patterns of miRNA-mRNA pairs associated with plant hormone signal transduction in A. rubrum roots, we quantified the expression levels of six miRNAs and six target genes that were verified to interact by degradome sequencing, and where the target gene was also differentially expressed in the hormone transduction pathway. After IBA treatment, these six miRNAs showed inverse expression patterns to the corresponding six target genes (Fig. 5). This was consistent with our RNA and degradome sequencing results. Therefore, we speculated that the Ar-miR160-ArARF10 interaction might be critical for the regulation of AR development in A. rubrum. The primers used in all qRT-PCR experiments are listed in Table S3.

Relative expression levels of six miRNAs and target genes. Actin6 was used as a reference for the mRNA qRT-PCR while U6 snRNA was used as a reference for the miRNA qRT-PCR. The expression in the control was set to 1.0. Mean values of three replicates are shown with standard error bars.
Comparative phylogenetic analysis of the ArARF protein family
Putative ARF genes in A. rubrum were identified in the RNA-seq data using BLAST. After the removal of redundant sequences and alternative splice forms, 18 ARF proteins were identified as potentially encoded by the ArARF genes (Table S5). In this study, we designated these proteins ArARF1–ArARF10 and ArARF16–ArARF19. We named the encoding genes as follows: ArARF1–ArARF10, ArARF16–ArARF19, ArARF19-2, and ArARF19-3 (Table S5). The ORFs of the ArARF genes varied from 1836 bp (ArARF10) to 3510 bp (ArARF19-3). These genes encoded polypeptides of 612–1170 amino acids, with predicted molecular masses of 67.17–131.19 kDa and theoretical pIs of 5.30–8.39 (Table S5).
Our neighbor-joining (NJ) phylogenetic tree, which included 18 ArARFs, 22 AtARFs, 16 DiARFs, and 18 CsARFs, suggested that ArARF homologs were more common in Dimocarpus longan and Citrus reticulate than in A. thaliana (Fig. S7A). The ARF genes were clustered into three major, well-supported clades (I–III; BS > 74; Fig. S7A). Seven ArARF genes fell into clade Ia, four into clade Ib, one into clade IIa, three into clade IIb, and two into class III (Fig. S7A, highlighted in red). Alignment of the ARF proteins indicated that most of the ArARF proteins harbored three characteristic regions (Fig. S7C). Unsurprisingly, most of the close homologs in our phylogenetic tree shared common motifs. The three domains of ArARF proteins were comprised of a total of eight different motifs: motifs 1, 2, and 3 constituted the DNA-binding domains; motifs 6, 8, and 9 constituted the ARF domain; and motifs 5 and 10 constituted the C-terminal Aux/IAA domains. Motifs 1, 2, 3, 6, 8, and 9 were found in all 18 ArARF proteins (Fig. S7B).
Construction of three miRNA–mRNA regulatory networks
The minimum free energy (MFE) values for the precursors of our three focal miRNAs (Ar-miR160a-5p, Ar-miR171d-1, and Ar-miR156f) were − 87.10 kcal/mol, − 72.90 kcal/mol, and − 34.70 kcal/mol, respectively. The precursor sequences had typical stem-loop structures (Fig. S8). High-throughput degradome sequencing indicated that Ar-miR160a targeted ArARF10 (CL3897.Contig1_All) and ArARF18-1 (Unigene5190_All); Ar-miR171d-1 targeted ArSCL6-1 (Unigene23148_All) and ArSCL6-2 (CL2444.Contig4_All); and Ar-miR156f targeted ArSPL-2 (CL2023.Contig1_All) (Fig. 6).

The miRNA-mRNA regulation networks of Ar-miR160a-5p, Ar-miR171d-1 and Ar-miR156f. Upregulated genes/miRNAs are shown in red; downregulated genes/miRNAs are shown in green.
Identification of ArARF10
Expression of fluorescent-tagged ArARF10 in N. benthamiana protoplasts showed that, while the green fluorescent protein (GFP) control was dispersed throughout the cell (Fig. S9), GFP-tagged ArARF10 proteins were located in the nucleus (Fig. S9), consistent with their putative function as transcription factors. In addition, ArARF10 fused to the GAL4 DNA-binding domain activated the expression of the His-3 reporter gene in yeast (Fig. S10), indicating that this gene was a transcriptional activator.
Overexpression of Ar-miR160a and ArARF10 in A. thaliana
GUS staining indicated that the transgenic A. thaliana seedlings successfully overexpressed Ar-miR160a or ArARF10 (Fig. S11). Transgenic plants overexpressing ArARF10 had many more ARs than WT plants (Fig. 7A,B,E,F), while transgenic plants overexpressing Ar-miR160a had fewer roots than WT plants (Fig. 7A,C–F). Consistent with this, Ar-miR160a and ArARF10 were significantly more upregulated in the transgenic lines than in the WT lines (A. thaliana Columbia-0) (Fig. 7G). However, none of these genes were significantly differentially expressed in the transgenic plants except AtGH3.6, which was significantly upregulated in the transgenic plant overexpressing ArARF10 as compared to the WT (Fig. 7H). Overexpression of ArARF10 significantly increased the number of ARs compared with WT plants, whereas overexpression of Ar-miR160a had the opposite effect, indicating that this pair of target-regulated genes participates in the control of AR number.

AR formation in transgenic A. thaliana overexpressing ArARF10 and Ar-miR160a. (A) Comparison of transgenic plants (OE-ArARF10 and OE-Ar-miR160a) to Col-0 plants. Scale bar, 5 mm. (B) ARs of WT phenotypes incubated under long-daylight conditions for 8 days. (C) Overexpression of ArARF10 promoted AR formation. (D) Overexpression of Ar-miR160a inhibited AR formation. Scale bar, 1 mm. (E) Left panel shows changes in the number of ARs in Col-0, OE-ArARF10 and OE-Ar-miR160a lines over 8 days. Right panel showed the number recorded on day 8, showing a significant increase in OE-ArARF10 ARs and a significant decrease in OE-Ar-miR160a ARs compared to the WT. Error bars show SEM (**p < 0.01, n = 3). (F) Left panel shows the changes in AR length in Col-0, OE-ArARF10 and OE-Ar-miR160a lines over eight days. Right panel shows the AR length recorded on day 8, with few changes in OE-ArARF10 and OE-Ar-miR160a AR lengths compared to the WT. (G) The relative expression levels of ArARF10 in OE-ArARF10 (left panel) and Ar-miR160a in OE-Ar-miR160a (right panel). (H) Expression patterns of endogenous genes in transgenic and Col-0 plants. Error bars show SEM (**p < 0.01, n = 3).
Discussion
The plant hormone auxin is commonly used to promote vegetative propagation in woody plants that are difficult to root6,28. Our results suggested that 300 mg/L IBA most effectively induced rooting in A. rubrum cuttings. It was found that the efficiency of AR formation was a Gaussian distribution that correlated with the concentration of IBA, indicating that, as in many other woody plants, auxin might play an important role in AR formation in A. rubrum.
To further investigate the molecular mechanisms regulating AR formation in A. rubrum in response to IBA treatment, we first identified 9507 genes that were differentially expressed in IBA-treated A. rubrum ARs compared to untreated A. rubrum ARs. KEGG pathway analysis suggested that these genes were associated with a total of 134 metabolic pathways. In particular, genes in the tryptophan metabolism, anthocyanin biosynthesis, and nitrogen metabolism pathways were generally downregulated in response to IBA treatment.
Because auxin has such a profound effect on plant growth and development, it is likely that certain checks must be in place to prevent excessive responses. Consistent with this, it has been shown that the PIN auxin efflux carrier genes transport excess auxin out of the cell via a feedback regulation loop29. Here, genes in the pathway responsible for synthesizing the auxin precursor tryptophan were generally downregulated after exogenous IBA treatment. Thus, we speculated that exogenous auxin might inhibit the synthesis of internal auxin in A. rubrum. However, the changes in expression profiles of genes in the plant hormone signal transduction pathway suggested that appropriate exogenous auxin might prolong AR development, improving rooting.
AR development is characterized by high energy requirements30,31. A previous transcriptomic study in carnation indicated that sucrolytic enzymatic activity was regulated at the transcript level during AR induction32. Consistent with this, we found that 384 of the DEGs were enriched in the “starch and sucrose metabolism” (ko00500), while several genes associated with the sucrose metabolism, including α-glucosidase 3 (Unigene42466_All) and glucoamylase 1 (Unigene10485_All), were significantly upregulated at the transcriptional level. This implied that the energy provided by the sucrose metabolism is crucial for the development of ARs in A. rubrum.
The multisubunit ubiquitin ligases of the ubiquitin–26S proteasome system (UPS), which comprise 6% of the A. thaliana proteome20, are a large and complex system of degraded proteins that play a prominent role in hormone regulation. Among the UPS, the SCF group, consisting of the Skp1, CUL1, and SCF E3 subunits33,34, was the most numerous and well-characterized35,36. Auxin mediates the binding of Aux/IAA to TIR1/AFBs, and then brings the complex to the SCF for ubiquitination and subsequent degradation37. Several studies have shown that auxin promotes the SCFTIR1/AFB degradation of Aux/IAA proteins38,39. Here, most of the ubiquitination-associated DEGs were implicated in the ubiquitination and degradation of Aux/IAA by the SKP1-CUL1-SCF complex, mediated by auxin. In particular, genes that mediate the ubiquitination of Aux/IAA proteins, including SKP1 (Unigene7049_All), CUL1 (Unigene20934_All), and SCF E3 F-box (Unigene17129_All), were significantly upregulated in response to exogenous auxin treatment. This result suggested that auxin-mediation ubiquitination and degradation of Aux/IAA might facilitate AR formation in A. rubrum.
Members of the Aux/IAA family are short-lived nuclear proteins that play a critical role in suppressing the expression of ARF-activated genes19,40. Aux/IAA proteins have been known to bind to ARFs and prevent the activation of auxin-responsive genes in the absence of auxin41. At high levels of auxin, Aux/IAA proteins are ubiquitinated by interaction with the TRANSPORT INHIBITOR RESPONSE 1/AUXIN SIGNALING F-BOX (TIR1/AFB) (Unigene20637_All) receptor and subsequently degraded by the 26S proteasome42,43. The SCFTIR1/AFB-Aux/IAA-ARF signaling system thus allows for a diversity of auxin responses. Interestingly, our transcriptome sequencing results indicated that this signaling regulatory system in A. rubrum ARs was significantly affected by treatment with exogenous auxin. Altering the transcription of genes in the auxin signaling pathway might be a genetic strategy used by A. rubrum in response to auxin alterations. Analysis of these genes might help to identify those that play a key role in AR development.
We identified 14 putative ArARF genes in the AR transcriptome of A. rubrum, most of which contained an amino-terminal DNA binding domain, a middle region that functioned as the activation or repression domain, and a carboxy-terminal dimerization domain. Four of these genes were significantly upregulated in response to IBA treatment: ArARF5 (CL11296.Contig1_All), ArARF17 (CL5608.Contig3_All), ArARF18-1 (Unigene5190_All), and ArARF19-1 (CL1642.Contig1_All). The nuclear targeting of the ArARF10 protein was consistent with transcriptional regulation and transcriptional activation functions. Structural domain function analysis implied that ArARF10 promoted AR development in A. rubrum by activating downstream auxin-responsive genes. These results indicated that ArARFs might play different roles during AR development in A. rubrum cuttings.
Phylogenetic analysis of the ARF proteins from A. rubrum, Longan, Citrus sp., and A. thaliana suggested that ARF proteins fell into three major clades, consistent with previous evolutionary analyses44,45,46,47. Consistent with this, previous studies have shown that ARF proteins include a highly conserved region of about 100 amino acid residues in the N-terminal region, corresponding to the DNA-binding domains48. Motif analysis identified 10 unique motifs across the 18 ArARF proteins49. Notably, ArARF8, ArARF10, and ArARF6-2 exhibited a high degree of homology with DiARF8, DiARF1, CsARF6, CsARF8, and CsARF10. In addition to the high degree of domain conservation and homology in ARF proteins among species, numbers of ARF copies are similar between A. rubrum and other species in the Sapindales, including Longan, and Citrus sp.44,50. This indicated that species in this order might undergo similar whole-genome duplications, and it was speculated that ARF diversification might represent an adaptation to environmental changes51.
Although ARF TFs have been shown to play a critical role in the regulation of root development and auxin correspondence52, miRNA interactions with TFs and other targets might also influence the regulatory networks that control the root transcriptome and might play a key role in the eventual translation of mRNAs53. Using sRNA sequencing, we identified six differentially expressed miRNAs associated with plant hormone signal transduction. miR171, SCL6-II, SCL6-III, and SCL6-IV are expressed ubiquitously in plants54,55. Transgenic plants overexpressing MIR171c (35Spro–MIR171c) and scl6-II scl6-III scl6-IV triple mutant plants exhibited pleiotropic phenotypes56, including alterations in shoot branching, plant height, chlorophyll accumulation, root development, flower architecture, and leaf shape and pattern. We found that Ar-miR171 cleaved ArSCL in A. rubrum. Ar-miR171 was significantly downregulated after exogenous IBA treatment, suggesting that the expression of miR171/SCL might be related to auxin activity in A.rubrum. miR171/SCL may play an important role in AR development. In Arabidopsis, SPL was targeted by miR156, while SPL3, SPL9 and SPL10 were involved in suppressing lateral root growth57. In addition, both miR156 and SPLs were sensitive to auxin signaling58. In our work, we found that Ar-miR156 was sensitive to exogenous IBA and targeted ArSPL, suggesting that miR156/SPL might be involved in AR development in A. rubrum.
Ar-miR160a-5p and Ar-miR160b-1 targeted and cleaved ArARF10 and ArARF18-1. Ar-miR160 and its target gene ArARF10 showed opposite expression patterns during AR formation in the presence of exogenous auxin. Most miRNAs in the miR160 family regulate plant development through the targeted cleavage of ARFs. Transgenic A. thaliana and Medicago truncatula overexpressing miR160 had shorter roots than their WT counterparts22,59, and a study in Longan showed that Dlo-miR160 negatively regulated DiARF10, DiARF16, and DiARF17 to affect hormone signaling and somatic embryogenesis60. In developing ARs of poplars, peu-miR160a was downregulated, while its target (peARF17.1) was upregulated61; AR formation in transgenic poplar overexpressing peu-miR160a was inhibited, whereas AR formation in polar overexpressing PeARF17.1 or PeARF17.2 was promoted18. Finally, A. thaliana overexpressing miR160 exhibited reduced AtARF10 and AtARF16 expression, shortened roots, and abnormal root cap development, as well as uncontrolled cell division and root apical meristem (RAM) differentiation22. Considering the highly conserved nature of the miR160 family62, we speculated that Ar-miR160 plays an important role in AR development through the targeted cleavage of ARF genes.
Despite this evidence supporting the important role of miR160 in AR development, the specific effects of miR160 expression tend to vary among species. For example, miR160 overexpression in poplar suppressed AR development and elongation, but overexpression of miR160a in A. thaliana promoted AR development16. Here, A. rubrum seedlings overexpressing Ar-miR160a generally had fewer ARs than the WT. Similar results were observed in poplar seedlings overexpressing Peu-miR160a18.
Determination of target gene function may help to clarify miRNA function. Transgenic A. thaliana overexpressing ArARF10 had more ARs than the WT; GH3.6 expression was also significantly upregulated in the transgenic plants. GH3 genes are highly responsive to exogenous auxin63; auxin stimulates AR development by inducing the expression of the GH3.3, GH3.5, and GH3.6 genes via the positive regulators ARF6 and ARF8. Consistent with this, the three-null mutant gh3.3gh3.5gh3.6 had fewer ARs than its wild-type counterpart64. These results suggested that, in A. thaliana, high levels of auxin led to the overexpression of ArARF10, stimulating AR formation. Interestingly, although ArARF10 was functionally similar to AtARF8 and AtARF6, ArARF10 overexpression did not upregulate GH3.3 and GH3.5, implying that auxin regulation pathways might differ between A. rubrum and A. thaliana. Our examination of Ar-miR160-ArARF10 function in AR development in A. rubrum revealed differences with its role in AR development in A. thaliana. AtARF17 suppresses AR development in A. thaliana16, whereas ArARF10 played a positive role in AR development in the model. Interestingly, this difference was also present between Populus and A. thaliana. PeARF17.1 plays a similar role to ArARF1018.In this study, we identified two pathways involved in the auxin response and analyzed the ArARF genes in those pathways. We characterized the TF ArARF10, and separately verified the role of the Ar-miR160a-ArARF10 interaction in AR development. We found that, during AR development, Ar-miR160 acted as a negative regulator to cleave the positive regulator ArARF10; this process was also integrated into the plant hormone signaling pathway and the ubiquitin mediated proteolysis pathway. Based on our results, we propose a model clarifying the mechanisms underlying the promotion of AR growth by exogenous auxin (Fig. 8): under low auxin conditions, Aux/IAA co-inhibits ARF by recruiting the OPLESS (TPL) family and preventing the ARF activation of downstream auxin-inducible genes, regulating AR formation at the protein level65,66. Simultaneously, Ar-miR160 suppresses ArARF10 expression by specifically cleaving ArARF10 mRNA, thus regulating AR formation at the transcriptional level (Fig. 8A). When exogenous auxin levels are elevated, Aux/IAA is ubiquitinated and degraded by the SCF-type E3 ubiquitin protein ligase complex, and ARF binds to AREs in the promoters of the auxin-inducible genes to promote transcription (Fig. 8B)20. The downregulation of Ar-miR160 also implies that more ArARF10 mRNAs are translated into proteins and perform downstream functions (Fig. 8C). In this way, A. rubrum responds sensitively and effectively to exogenous auxin during AR formation, leading to increased AR production. Thus, our work provides new insights into the key regulatory roles played by this module in the AR development of A. rubrum. These results are of great scientific significance, as they clarify the molecular mechanism of AR formation in A. rubrum, as well as provide a basis for the improvement of A. rubrum rooting ability through molecular breeding to increase its utility in landscape applications.

Schematic presentation of the regulation of AR growth in A. rubrum. (A) Under low auxin conditions, Ar-miR160 specifically cleaves mRNA-ArARF10 to repress ArARF10 expression, while Aux/IAA prevents auxin-inducible gene transcription by recruiting TPL to co-inhibit the binding of ArARF10 AREs in the auxin-inducible gene promoter. (B) Under increased levels of exogenous auxin, Aux/IAA is ubiquitinated and degraded by the SCF-type E3 ubiquitin protein ligase complex, and ArARF10 binds to AREs in the promoters of auxin-inducible genes to increase transcription. (C) The downregulation of Ar-miR160 also caused the translation of additional mRNA-ArARF10 into protein, as well as additional downstream functions that promoted AR formation.
Conclusion
In this study, AR development in A. rubrum was significantly increased by treatment with 300 mg/L IBA. Changes in gene expression after exogenous auxin treatment were explored using high-throughput sequencing. We found that two key regulatory pathways (ubiquitin mediated proteolysis and plant hormone signal transduction), the miRNA Ar-miR160a, and its target gene ArARF10, were involved in the auxin response. Experiments overexpressing ArARF10 or Ar-miR160a showed that ArARF10 increased AR development, while Ar-miR160a inhibited AR development. Our results may help to clarify the key regulatory pathways and genes associated with AR formation in woody plants, and provide a basis for the eventual improvement of A. rubrum rooting success.
Methods
The use of plant materials and ethical approval statement
This study including sample collection was conducted following China’s Biodiversity Conservation Strategy and Action Plan (2011–2030) (Index number: 000014672/2010-00714) and the Seed Law of the People’s Republic of China (2015 Revised Version), which together permit use of biological resources by Chinese researchers for scientific purposes.
Plant materials
We collected full and semi-woody annual branches (60 cm in length) from A. rubrum hybrid ‘Autumn Fantasy’ trees. The branches were divided into 8–10 cm cuttings, each containing two nodes. Cuttings were disinfected with 0.5% potassium permanganate solution, and then soaked in 100 mg/L, 200 mg/L, 300 mg/L, 400 mg/L, or 500 mg/L IBA for 1 h to induce AR formation (n = 50 branches per group). The depth of each cutting was 3 cm. Control cuttings were soaked in clean water for one hour (n = 50). Cuttings were then grown in a greenhouse under a 16 h light (25 °C)/8 h dark (18 °C) photoperiod. After 30 days of growth, roots were washed and the numbers of ARs were recorded. The ARs from branches soaked in 300 mg/L IBA and in clear water were stored at − 80 °C for RNA sequencing (RNA-seq), small RNA sequencing (sRNA-seq), and degradome sequencing.
RNA isolation, library construction, and sequencing
Total RNA was isolated from the CK and IBA300 groups using Trizol Reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. After verification of RNA sample quality using a Nanodrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA), mRNA was reverse transcribed using PrimeScriptTM (Takara, China). Finally, the double-stranded cDNA was purified using a DNA purification kit (QIAGEN, Germany) to generate high quality cDNA. Libraries were prepared from 300 to 500 bp size-selected fractions following adapter ligation and agarose gel separation. The libraries were sequenced using a paired-end read protocol with 100 bp of data collected per run on the Illumina HiSeq 2000.
The sRNA libraries were constructed using a NEBNext Multiplex Small RNA Library Prep Set for Illumina and sequenced on a BGISEQ-500 system. mRNA was captured using magnetic beads. Mixed biotinylated random primers and mRNA were used for reverse transcription. After library preparation was completed, the constructed library was sequenced using an Illumina HiSeq2500, with a single-ended read length of 1 × 50 bp.
Bioinformatics analysis of the sequencing data
The raw transcriptome sequencing data were filtered by removing adapters and low quality sequences. The clean data were assembled using Trinity version 2.0.667. The transcripts were clustered and redundant transcripts were eliminated using Tgicl to obtain Unigenes68. The assembled transcripts were quality-checked by comparison with conserved genes using the single-copy direct homology database BUSCO69. Potential duplicate molecules were removed from the aligned BAM format records. Fragments per kilobase of exon per million fragments mapped (FPKM) values were used determined relative gene expression levels in Cufflinks70. Novel genes were identified based the transcripts newly discovered using Cufflinks. Genes encoding peptides with less than 50 amino acid residues were filtered. DEGs were detected using DEGseq; gens were considered differentially expressed when thefold change in relative expression was ≥ 2 and the Q-value was ≤ 0.00171.
The raw sRNA sequencing data were filtered by removing adapters and low-quality data to generate clean reads. Unique sRNAs were aligned to the miRNA precursors of corresponding species in miRBase to obtain a miRNA count. We used miRA, which identifies miRNAs based on the ability of the miRNA precursors to form hairpin secondary structures, with default plant parametersto predict novel miRNAs72. Fold-change in miRNA expression levels were calculated using Transcripts Per Kilobase Millions (TPMs)73. DEGseq was used to identify differentially expressed miRNAs between IBA300 and CK71. The P-values calculated for each gene were adjusted to Q-values to correct for multiple testing using two alternative strategies74. miRNAs were considered differentially when fold change was ≥ 2 and Q-value was ≤ 0.001.
Standard sequences were compared to the degradome sequences using reads per million (RPM) to remove redundancy. The Needle program in the EMBOSS package was applied to derive all sequences that matched the sequences in the previous sRNA library, and then the columns were scored according to the plant miRNA/target pairing criteria75,76. The mRNA sequences of target genes paired with the sRNA sequences of A. rubrum were predicted by Targetfinder, with a prediction score cutoff of four. The target genes corresponding to the predicted miRNAs and the mRNAs in the degradome were combined and analyzed to identify shared mRNAs targeted by the miRNAs. Degradome peak classifications and scores, as well as the prediction results, were plotted77. The MFE values for these precursors were predicted using the classic algorithm of Zuker and Stiegler78.
GO and KEGG enrichment of the differentially expressed genes (DEGs)
DEGs were annotated by searching the sequences against the NR, Swiss-Prot, GO, KEGG databases using BLAST79,80,81. We investigated the GO enrichment of DEGs using the Gene Ontology Consortium (http://geneontology.org/); p-values were FDR corrected using DEGseq71, and terms with Q-value ≤ 0.05 were considered significantly enriched in the DEGs. We identified KEGG pathways significantly associated with the DEGs based on relative expression levels. Hierarchical clustering of expression data was performed using TBtools82.
Identification and classification of ARF genes
We searched 23 protein sequences from A. thaliana (https://www.arabidopsis.org/) against the transcript of A. rubrum using Tblastn, with the following settings: matrix, BLOSUM62; expect value, < 1e − 005; gap-existence, 11; gap-existension extension, 1; and filter, low-complexity. Previously published ARFs from A. thaliana, C. sinensis, and D. longan were identified using a BLASTP search with the score value set to ≥ 100 and e-value set to ≤ e−10 45,83. Next, the Pfam database (http://pfam.xfam.org/) was used to determine whether each candidate ARF sequence belonged to the ARF gene family84. To exclude overlapping genes, all candidate ARF genes were aligned with ClustalW and checked manually. The SMART (http://smart.embl-heidelberg.de/) and InterProScan (http://www.ebi.ac.uk/Tools/pfa/iprscan/) web servers were used to examine the conserved domains of the identified A. rubrum genes. Obtained ArARF genes were named based on their A. thaliana homologs. Protein characteristics, including molecular weight (MW), isoelectric point (pI), and length, were predicted using the online ProtParam tool.
Phylogenetic and conserved motif analysis of the ArARFs
To explore inter- and intra-specific phylogenetic relationships among ARF proteins, we constructed a dataset of ARF proteins including the ArARFs identified in this study, as well as 22 A. thaliana ARFs, 16 D. longan ARFs, and 18 citrus ARFs (referred to herein as AtARF, DiARF, and CsARF respectively). Neighbor-joining (NJ) phylogenetic analysis of the 74 ARF protein sequences was performed using MEGA785, with 1000 bootstrap (BS) replicates. The best-fit model of nucleotide substitution (i.e., that with the lowest BIC score) was JTT + G + I. All positions with less than 80% site coverage were eliminated; that is, fewer than 20% alignment gaps, missing data, and ambiguous bases were allowed at any position86. To investigate the structural differences among ArARF genes, conserved motifs in the predicted ARF proteins were investigated using online MEME analysis (http://meme.nbcr.net/meme/), with an optimum motif width of 6–50. The maximum number of motifs was set to 1087. Results were visualized using TBtools88.
Quantitative reverse-transcription PCR (qRT-PCR) assay
qRT-PCRs were performed using a CFX Connect TM Real-Time System. Each total RNA sample (2 μg) was digested with RNase-free DNase I (Invitrogen) to remove genomic DNA contamination and subsequently reverse transcribed into cDNA using an Oligo (dT)18 primer. For the qRT-PCR of the miRNAs, U6 snRNA was used as the internal control. For the qRT-PCR analyses of the miRNA target genes89, Actin6 was used as the internal control90. PCR volumes (20 μl) contained 1 μl of 20× diluted cDNA, 5× SYBR buffer, and 0.25 μM forward and reverse primers; the gene-specific primers used for qRT-PCR are given in Table S3. The thermal cycling conditions were as follows: 95 °C for 30 s, followed by 40 cycles of 5 s at 95 °C and 30 s at 60 °C. All reactions were performed in triplicate in three independent experiments. The relative fold-changes in miRNA and gene expression levels were calculated using the 2−ΔΔCt method while the relative fold-change in ArARF10 expression was calculated using the 2−ΔCt method91.
Subcellular localization and transcriptional activation
The full-length cDNA sequences of the target gene ArARF10 were cloned from the cDNA of ARs from A. rubrum. The subcellular location of ArARF10 was determined by transfecting GFP-tagged ArARF10 into 3–4-week-old Nicotiana benthamiana protoplasts. The full-length cDNA of ArARF10 was fused in-frame with GFP cDNA and ligated between the CaMV 35S promoter and the nopaline synthase terminator. The fluorescence signals in the tobacco protoplasts were examined under a confocal laser scanning microscope (Axio Lab A1)92. The transcriptional activation of ArARF10 was investigated by transforming the pGBKT7 construct (in which ArAFR10 was fused with the GAL4 DNA-binding domain) into yeast strain AH109. Yeast strain AH109 contains the His-3 reporter genes. The transformants were streaked on the SD/Trp- and SD/Trp-/His-media. After incubation at 28 °C for 3 days, the growth of the transformants was evaluated. The transformed yeast cells were grown on synthetic defined (SD) media (with or without His); yeast with active His-3 reporter genes were able to grow on synthetic defective plates.
Constructs and plant transformation
We next tested whether the overexpression of Ar-miR160a and ArARF10 affected the growth and development of A. thaliana. The Ar-miR160a-5p precursor was cloned from A. rubrum genomic DNA. The precursor sequence of Ar-miR160a-5p and the ORFs of ArARF10 were cloned into the entry vector pTOPO-T. After verification by sequencing, the fragments of the Ar-miR160a-5p precursor and ArARF10 inserted into the entry vector were transferred to the destination vector pCAMBIA1301 via an LR reaction. The constructed vectors (Pro35S::Ar-miR160a and Pro35S::ArARF10) were driven by the CaMV 35S promoter. The pCAMBIA1301 vector contained a GUS reporter gene initiated by the 35S promoter.
The constructs (Pro35S::Ar-miR160a and Pro35S::ArARF10) were introduced into WT A. thaliana (Columbia-0) using electroporation and Agrobacterium-mediated transformation. Seeds of the plants exhibiting transgenic phenotypes were sterilized and sown on 1/2 Murashige and Skoog (1/2 MS) medium, as described previously93. Plates were incubated at 4 °C for 48 h for stratification and then exposed to light for several hours to induce germination. Plates were next wrapped in three layers of aluminum foil and placed in the dark until the seedlings had an average hypocotyl length of 6 mm (48 h). The A. thaliana primordium was excised using a sterile blade, preserving about 6 mm of the hypocotyl, and then the seedlings were exposed to a natural photoperiod (16 h light/8 h dark) to induce AR formation. AR lengths and numbers were measured using ImageJ94 8 days after transfer to lighted conditions. For each biological replicate, at least ten seedlings were analyzed, and each experiment was repeated at least three times. One-way ANOVAs, combined with Tukey's multiple comparison tests, were performed using GraphPad Prism 8 to analyze differences in means and variances among genotypes.
GUS staining
After eight days of cultivation, transgenic and WT A. thaliana seedlings were washed and completely immersed in GUS staining solution (Coolaber, China). After overnight staining at 37 °C in the dark, seedlings were decolorized in 70% ethanol, examined, and photographed.
Data availability
The sequencing data were submitted to the SRA database with the accession numbers SRR13808891, SRR13808890, SRR13808889, SRR13808888, SRR13808887, SRR13808886, SRR13808885 and SRR13808884.The data that support the conclusions are within this article and its additional files. All data and plant materials used in current study are available from the corresponding author on reasonable request.
References
Abrams, M. D. The red maple paradox. Bioscience 48, 355–364 (1998).
Lahr, E. C., Dunn, R. R. & Frank, S. D. Variation in photosynthesis and stomatal conductance among red maple (Acer rubrum) urban planted cultivars and wildtype trees in the southeastern United States. PLoS ONE 13, e0197866 (2018).
Pijut, P. M., Woeste, K. E. & Michler, C. H. 6 promotion of adventitious root formation of difficult-to-root hardwood tree species. Hortic. Rev. 38, 213 (2011).
Sibley, J. L., Joseph Eakes, D., Gilliam, C. H., Keever, G. J. & Dozier, W. A. Jr. Growth and fall color of red maple selections in the Southeastern United States. J. Environ. Horticult. 13, 51–53 (1995).
Druege, U., Franken, P. & Hajirezaei, M. R. Plant hormone homeostasis, signaling, and function during adventitious root formation in cuttings. Front. Plant Sci. 7, 381 (2016).
Geiss, G., Gutierrez, L. & Bellini, C. Adventitious root formation: New insights and perspectives. Annu. Plant Rev. Online. 127–156 (2018).
Druege, U. et al. Molecular and physiological control of adventitious rooting in cuttings: Phytohormone action meets resource allocation. Ann. Bot. 123, 929–949 (2019).
Pacurar, D. I., Perrone, I. & Bellini, C. Auxin is a central player in the hormone cross-talks that control adventitious rooting. Physiol. Plant. 151, 83–96 (2014).
Sablowski, R. Plant stem cell niches: From signalling to execution. Curr. Opin. Plant Biol. 14, 4–9 (2011).
Guilfoyle, T. J. & Hagen, G. Auxin response factors. Curr. Opin. Plant Biol. 10, 453–460 (2007).
Epstein, E. & Ludwig-Müller, J. Indole-3-butyric acid in plants: Occurrence, synthesis, metabolism and transport. Physiol. Plant. 88, 382–389 (1993).
Simon, S. & Petrášek, J. Why plants need more than one type of auxin. Plant Sci. 180, 454–460 (2011).
Lakehal, A. & Bellini, C. Control of adventitious root formation: Insights into synergistic and antagonistic hormonal interactions. Physiol. Plant. 165, 90–100 (2019).
Legué, V., Rigal, A. & Bhalerao, R. P. Adventitious root formation in tree species: Involvement of transcription factors. Physiol. Plant. 151, 192–198 (2014).
Rigal, A. et al. The AINTEGUMENTA LIKE1 homeotic transcription factor PtAIL1 controls the formation of adventitious root primordia in poplar. Plant Physiol. 160, 1996–2006 (2012).
Gutierrez, L. et al. Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell 21, 3119–3132 (2009).
Okushima, Y., Fukaki, H., Onoda, M., Theologis, A. & Tasaka, M. ARF7 and ARF19 regulate lateral root formation via direct activation of LBD/ASL genes in Arabidopsis. Plant Cell 19, 118–130 (2007).
Liu, S. et al. The peu-miR160a–PeARF17.1/PeARF17.2 module participates in the adventitious root development of poplar. Plant Biotechnol. J. 18, 457–469 (2020).
Luo, J., Zhou, J.-J. & Zhang, J.-Z. Aux/IAA gene family in plants: Molecular structure, regulation, and function. Int. J. Mol. Sci. 19, 259 (2018).
Vierstra, R. D. The ubiquitin-26S proteasome system at the nexus of plant biology. Nat. Rev. Mol. Cell Biol. 10, 385–397 (2009).
Westfall, C.S., Sherp, A.M., Zubieta, C., Alvarez, S. & Jez, J.M. Arabidopsis thaliana GH3.5 acyl acid amido synthetase mediates metabolic crosstalk in auxin and salicylic acid homeostasis. Proc. Natl. Acad. Sci. USA. 113 (2017).
Wang, J.-W. et al. Control of root cap formation by microRNA-targeted auxin response factors in Arabidopsis. Plant Cell 17, 2204–2216 (2005).
Kinoshita, N. et al. IAA-Ala Resistant3, an evolutionarily conserved target of miR167, mediates Arabidopsis root architecture changes during high osmotic stress. Plant Cell 24, 3590–3602 (2012).
Olatunji, D., Geelen, D. & Verstraeten, I. Control of endogenous auxin levels in plant root development. Int. J. Mol. Sci. 18, 2587 (2017).
Guo, R. et al. Effect of photoperiod on the formation of cherry radish root. Sci. Hortic. 244, 193–199 (2019).
Li, K. et al. miRNAs associated with auxin signaling, stress response, and cellular activities mediate adventitious root formation in apple rootstocks. Plant Physiol. Biochem. 139, 66–81 (2019).
Rodriguez, R. E. et al. MicroRNA miR396 regulates the switch between stem cells and transit-amplifying cells in Arabidopsis roots. Plant Cell 27, 3354–3366 (2015).
Yu, J., Liu, W., Liu, J., Qin, P. & Xu, L. Auxin control of root organogenesis from callus in tissue culture. Front. Plant Sci. 8, 1385 (2017).
Weijers, D. & Wagner, D. Transcriptional responses to the auxin hormone. Annu. Rev. Plant Biol. 67, 539–574 (2016).
Agulló-Antón, M. Á., Sánchez-Bravo, J., Acosta, M. & Druege, U. Auxins or sugars: What makes the difference in the adventitious rooting of stored carnation cuttings?. J. Plant Growth Regul. 30, 100–113 (2011).
Agulló-Antón, M. Á. et al. Early steps of adventitious rooting: Morphology, hormonal profiling and carbohydrate turnover in carnation stem cuttings. Physiol. Plant. 150, 446–462 (2014).
Villacorta-Martín, C. et al. Gene expression profiling during adventitious root formation in carnation stem cuttings. BMC Genom. 16, 1–18 (2015).
Kelley, D. R. & Estelle, M. Ubiquitin-mediated control of plant hormone signaling. Plant Physiol. 160, 47–55 (2012).
Calderon-Villalobos, L. I., Tan, X., Zheng, N. & Estelle, M. Auxin perception—Structural insights. Cold Spring Harb. Perspect. Biol. 2, a005546 (2010).
Vierstra, R. D. The ubiquitin/26S proteasome pathway, the complex last chapter in the life of many plant proteins. Trends Plant Sci. 8, 135–142 (2003).
Salehin, M., Bagchi, R. & Estelle, M. SCFTIR1/AFB-based auxin perception: Mechanism and role in plant growth and development. Plant Cell 27, 9–19 (2015).
Villalobos, L. I. A. C. et al. A combinatorial TIR1/AFB–Aux/IAA co-receptor system for differential sensing of auxin. Nat. Chem. Biol. 8, 477–485 (2012).
Tan, X. et al. Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 446, 640–645 (2007).
Walsh, T. A. et al. Mutations in an auxin receptor homolog AFB5 and in SGT1b confer resistance to synthetic picolinate auxins and not to 2, 4-dichlorophenoxyacetic acid or indole-3-acetic acid in Arabidopsis. Plant Physiol. 142, 542–552 (2006).
Dreher, K. A., Brown, J., Saw, R. E. & Callis, J. The Arabidopsis Aux/IAA protein family has diversified in degradation and auxin responsiveness. Plant Cell 18, 699–714 (2006).
Tiwari, S. B., Hagen, G. & Guilfoyle, T. J. Aux/IAA proteins contain a potent transcriptional repression domain. Plant Cell 16, 533–543 (2004).
Weijers, D. et al. Developmental specificity of auxin response by pairs of ARF and Aux/IAA transcriptional regulators. EMBO J. 24, 1874–1885 (2005).
Parry, G. et al. Complex regulation of the TIR1/AFB family of auxin receptors. Proc. Natl. Acad. Sci. 106, 22540–22545 (2009).
Peng, Y., Fang, T., Zhang, Y., Zhang, M. & Zeng, L. Genome-wide identification and expression analysis of auxin response factor (ARF) gene family in Longan (Dimocarpus longan L.). Plants 9, 221 (2020).
Li, S.-B. et al. Genome-wide identification, isolation and expression analysis of auxin response factor (ARF) gene family in sweet orange (Citrus sinensis). Front. Plant Sci. 6, 119 (2015).
Piya, S., Shrestha, S. K., Binder, B., Stewart, C. N. Jr. & Hewezi, T. Protein–protein interaction and gene co-expression maps of ARFs and Aux/IAAs in Arabidopsis. Front. Plant Sci. 5, 744 (2014).
Finet, C., Berne-Dedieu, A., Scutt, C. P. & Marlétaz, F. Evolution of the ARF gene family in land plants: Old domains, new tricks. Mol. Biol. Evol. 30, 45–56 (2013).
Li, S.-B., Xie, Z.-Z., Hu, C.-G. & Zhang, J.-Z. A review of auxin response factors (ARFs) in plants. Front. Plant Sci. 7, 47 (2016).
Bailey, T. L. et al. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009).
Xie, R. et al. The ARF, AUX/IAA and GH3 gene families in citrus: Genome-wide identification and expression analysis during fruitlet drop from abscission zone A. Mol. Genet. Genom. 290, 2089–2105 (2015).
Jiao, Y. et al. Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100 (2011).
Overvoorde, P., Fukaki, H. & Beeckman, T. Auxin control of root development. Cold Spring Harb. Perspect. Biol. 2, a001537 (2010).
Ha, M. & Kim, V. N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 15, 509–524 (2014).
Llave, C., Xie, Z., Kasschau, K. D. & Carrington, J. C. Cleavage of scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 297, 2053–2056 (2002).
Song, L., Axtell, M. J. & Fedoroff, N. V. RNA secondary structural determinants of miRNA precursor processing in Arabidopsis. Curr. Biol. 20, 37–41 (2010).
Wang, L., Mai, Y.-X., Zhang, Y.-C., Luo, Q. & Yang, H.-Q. MicroRNA171c-targeted SCL6-II, SCL6-III, and SCL6-IV genes regulate shoot branching in Arabidopsis. Mol. Plant 3, 794–806 (2010).
Yu, N., Niu, Q. W., Ng, K. H. & Chua, N. H. The role of miR156/SPL s modules in Arabidopsis lateral root development. Plant J. 83, 673–685 (2015).
Xu, X. et al. High miR156 expression is required for auxin-induced adventitious root formation via MxSPL26 independent of PINs and ARFs in Malus xiaojinensis. Front. Plant Sci. 8, 1059 (2017).
Bustos-Sanmamed, P. et al. Overexpression of miR160 affects root growth and nitrogen-fixing nodule number in Medicago truncatula. Funct. Plant Biol. 40, 1208–1220 (2013).
Lin, Y. et al. Endogenous target mimics down-regulate miR160 mediation of ARF10,-16, and-17 cleavage during somatic embryogenesis in Dimocarpus longan Lour. Front. Plant Sci. 6, 956 (2015).
Liu, S., Wu, L., Qi, H. & Xu, M. LncRNA/circRNA–miRNA–mRNA networks regulate the development of root and shoot meristems of Populus. Ind. Crops Prod. 133, 333–347 (2019).
Zhang, B., Pan, X., Cobb, G. P. & Anderson, T. A. Plant microRNA: A small regulatory molecule with big impact. Dev. Biol. 289, 3–16 (2006).
Park, J.-E. et al. GH3-mediated auxin homeostasis links growth regulation with stress adaptation response in Arabidopsis. J. Biol. Chem. 282, 10036–10046 (2007).
Gutierrez, L. et al. Auxin controls Arabidopsis adventitious root initiation by regulating jasmonic acid homeostasis. Plant Cell 24, 2515–2527 (2012).
Leyser, O. Auxin signaling. Plant Physiol. 176, 465–479 (2018).
Iwakawa, H.-O. & Tomari, Y. The functions of microRNAs: mRNA decay and translational repression. Trends Cell Biol. 25, 651–665 (2015).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Pertea, G. et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 19, 651–652 (2003).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Wang, L., Feng, Z., Wang, X., Wang, X. & Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010).
Evers, M., Huttner, M., Dueck, A., Meister, G. & Engelmann, J. C. miRA: Adaptable novel miRNA identification in plants using small RNA sequencing data. BMC Bioinform. 16, 1–10 (2015).
Hoen, P. A. et al. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 36, e141 (2008).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Nat. Prec. 1–1 (2010).
Bo, X. & Wang, S. TargetFinder: A software for antisense oligonucleotide target site selection based on MAST and secondary structures of target mRNA. Bioinformatics 21, 1401–1402 (2005).
Addo-Quaye, C., Miller, W. & Axtell, M. J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 25, 130–131 (2009).
Meyers, B. C. et al. Criteria for annotation of plant MicroRNAs. Plant Cell 20, 3186–3190 (2008).
Zuker, M. & Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 9, 133–148 (1981).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M. et al. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551 (2021).
Chen, C. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Peng, Y., Fang, T., Zhang, Y., Zhang, M. & Zeng, L. Genome-wide identification and expression analysis of auxin response factor (ARF) gene family in Longan (Dimocarpus longan L.). Plants (Basel) 9 (2020).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evolut. 33, 1870–1874 (2016).
Nei, M. & Kumar, S. Molecular Evolution and Phylogenetics (Oxford University Press, 2000).
Bailey, T. L. & Elkan, C. The value of prior knowledge in discovering motifs with MEME. ISMB. 3, 21–29 (1995).
Chen, C. et al. Tbtools—An integrative toolkit developed for interactive analyses of big biological data. bioRxiv. 289660 (2020).
Lin, Y. L. & Lai, Z. X. Evaluation of suitable reference genes for normalization of microRNA expression by real-time reverse transcription PCR analysis during longan somatic embryogenesis. Plant Physiol. Biochem. 66, 20–25 (2013).
Zhu, L. et al. Reference gene selection for quantitative real-time PCR analyses of Acer palmatum under abiotic stress. Phyton 89, 385 (2020).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Lichocka, M. & Schmelzer, E. Subcellular localization experiments and FRET-FLIM measurements in plants. Bio-Protoc. 4, 1–12 (2014).
Sorin, C. et al. Auxin and light control of adventitious rooting in Arabidopsis require ARGONAUTE1. Plant Cell 17, 1343–1359 (2005).
Collins, T. J. ImageJ for microscopy. Biotechniques 43, S25–S30 (2007).
Acknowledgements
The authors hope to express their appreciation to the reviewers for this manuscript.
Funding
This project was supported by the Beijing Excellent Training Project to WG (Grant no. 68), Beijing Natural Science Fund-Municipal Education Commission Jointly Funded Projects (Grant no. KZ201810020029) and Scientific Research Project of Beijing Municipal Education Commission (Grant no. KM201910020005).
Author information
Authors and Affiliations
Contributions
W.Z. conducted the experiments, data analysis and wrote the manuscript. M.Z., J.L. and H.Z. analyzed data. W.G. and K.Z. designed the experiments and revised the manuscript. All authors read and approved the final draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, W., Zhang, M., Li, J. et al. Key regulatory pathways, microRNAs, and target genes participate in adventitious root formation of Acer rubrum L. Sci Rep 12, 12057 (2022). https://doi.org/10.1038/s41598-022-16255-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-16255-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
