
Abstract
Resistance to bacteriophage infections protects bacteria in phage-replete environments, enabling them to survive and multiply in the presence of their viral predators. However, such resistance may confer costs for strains, reducing their ecological fitness as expressed as competitiveness for resources or virulence or both. There is limited knowledge about such costs paid by phage-resistant plant pathogenic bacteria in their natural habitats. This study analyzed the costs of phage resistance paid by the phytopathogenic pectinolytic bacterium Dickeya solani both in vitro and in potato (Solanum tuberosum L.) plants. Thirteen Tn5 mutants of D. solani IPO 2222 were identified that exhibited resistance to infection by lytic bacteriophage vB_Dsol_D5 (ΦD5). The genes disrupted in these mutants encoded proteins involved in the synthesis of bacterial envelope components (viz. LPS, EPS and capsule). Although phage resistance did not affect most of the phenotypes of ΦD5-resistant D. solani such as growth rate, production of effectors, swimming and swarming motility, use of various carbon and nitrogen sources and biofilm formation evaluated in vitro, all phage resistant mutants were significantly compromised in their ability to survive on leaf surfaces as well as to grow within and cause disease symptoms in potato plants.
Similar content being viewed by others


Introduction
Bacteriophages (bacterial viruses) are the most abundant biological entities in the biosphere. With their total estimated number of ca. 1031 phage particles, they are the main driving force of bacterial adaptation and evolution1,2,3,4. Likewise, bacterial viruses play a critical role in maintaining bacterial diversity in the environment5,6,7. It is estimated that up to 20% of bacterial cells are killed daily due to phage infections among all habitats8. However, surprisingly, this enormous mortality of bacterial cells does not lead to the global disappearance of sensitive host populations or a dominance of resistant ones in a given ecological niche9,10,11. On the contrary, phage-susceptible and phage-resistant bacterial populations are frequently reported to coexist both in natural (e.g. ocean, phyllosphere)12,13 as well as human-controlled and engineered environments (e.g. wastewater treatment facilities, agricultural fields)14,15,16.
It is hypothesized that the stable coexistence of phage-resistant and phage-susceptible bacterial populations occurs because bacteria pay a cost for phage resistance, irrespective of whether the viruses are present in the environment or not17,18,19,20. The altered fitness of resistant bacteria is usually manifested by reduced competitiveness for resources and/or reduced virulence or both21,22. However, it should also be highlighted that not all strains that have evolved phage resistance suffer such costs23,24. Such costs may also depend on the environmental context of the bacterium as well as the mechanisms by which phage resistance is conferred25,26.
Even though the costs of resistance to phage infection have been reported in various phage-host systems20,27,28, relatively little is known about these costs paid specifically by phage-resistant phytopathogenic bacteria residing in their natural habitats, in agricultural fields. This is an important issue given the diversity of conditions such bacteria encounter in these settings29. Indeed, no such studies have addressed the costs of phage resistance in Soft Rot Pectobacteriaceae (SRP) species which, due to their life-style, have particularly complex interactions with varied habitats.
SRP bacteria including Pectobacterium spp. and Dickeya spp. are an excellent model for studying phage-host interaction and co-adaptation in the environment. These bacteria are considered among the ten most important agricultural phytopathogens worldwide30. SRP cause significant losses in crop production (up to 40%), with disease severity dependent on weather conditions, plant susceptibility, pathogen inoculum, and other factors29,31. Pectobacterium spp. and Dickeya spp. are widespread in various ecological niches, including natural and agricultural soils, water, sewage, the surface of host and non-host plants, and the surface and interior of insects32,33,34. Because of the diverse environments in which SRP bacteria are found, these pathogens apparently experience ecological and life-style tradeoffs because of their frequent transfer between these diverse environments: for example, from host plant to soil, plant to plant, host plant to non-host plant, surface/irrigation water to plant and vice versa29. In all of these settings, SRP bacteria can encounter lytic bacteriophages and, as a result, may become repeatedly infected35. Under conditions of high infection, resulting in large populations of the pathogen, the emergence of phage and subsequently, phage-resistant SRP variants, is inevitable36,37. However, it remains unclear what the ecological fitness costs are for SRP bacteria to become resistant to such viral infections and thus whether phage infection can reduce disease severity both directly by, at least temporarily, reducing pathogen populations or subsequently by reducing pathogen virulence or fitness.
The purpose of this study was to assess the fitness costs paid by phage-resistant variants of D. solani both in vitro and in planta. D. solani is an emerging plant pathogen that causes soft rot disease symptoms in a variety of crops and nonfood plants worldwide38,39. This species was first reported to infect potato in the early 2000s40 and has since become a serious worldwide problem in agriculture. At present, D. solani is an important pathogen in a majority of European countries38,39 as well as in Israel41, Georgia42, Turkey43 and Brazil44 causing large losses in crop production. Surprisingly, most of the D. solani strains analyzed so far belong to the same haplophyte and express only a small number of genetic differences45.
Our study investigated the interaction of D. solani strain IPO 222238, the type strain of this species, that has received considerable attention, with bacteriophage vB_Dsol_D5 (ΦD5)46,47. ΦD5 is a broad host range lytic bacteriophage able to infect D. solani strains as well as those of 3 other Dickeya species (D. zeae, D. dianthicola and D. dadantii)46. Phage ΦD5 is in the genus Limestonevirus and family Ackermannviridae48,49,50. Of all bacteriophages infecting D. solani, those within the Limestonevirus genus are the most abundant in the environment50. Limestoneviruses, including ΦD5, have been isolated in various European countries, including Belgium, Poland, Russia and the United Kingdom and exhibit a high level of genetic homogeneity50. Furthermore, ΦD5 has been extensively studied as a biological control agent useful in the control of disease caused by D. solani in the potato ecosystem47.
Using random Tn5-based mutagenesis we identified D. solani genes and operons that encoded structures required for ΦD5 attachment and susceptibility to infection to better understand the molecular determinants of phage-resistant bacterial variants as well as the fitness of mutants. We thus have addressed the hypothesis that the level of fitness tradeoff conferred by phage resistance is dependent on the environmental context in which fitness is assessed.
Results
Identification of disrupted genes in ΦD5-resistant D. solani mutants
A total of 1000 Tn5 mutants of D. solani was screened for resistance to infection by bacteriophage ΦD5. Thirteen mutants (ca. 1.3%) were found to be resistant to this phage (Table 1). The genomes of these mutants were sequenced to identify the Tn5 insertion sites. A single insertion of Tn5 was found in each of the ΦD5-resistant mutants analyzed. Interestingly, in multiple cases distinct Tn5 insertions were found in the same locus (Fig. 1, Table 1); a gene encoding glycosyltransferase family 1 was disrupted in both mutants M22 and M25; mannose-1-phosphate guanylyl-transferase/ mannose-6-phosphate isomerase was disrupted in mutants M61 and M1026; a sugar ABC transporter ATP-binding protein was disrupted in mutants M73, M144, M626 and M720; a hypothetical protein homologous to protein WbeA involved in O-antigen transport system was disrupted in mutants M83, M399 and M534 and a GDP-fucose synthase was disrupted in mutants M177 and M1004 (Table 1, Fig. 1). In total, five distinct bacterial loci were required for infection of D. solani with ΦD5. All loci disrupted by Tn5 encoded proteins associated with synthesis, metabolism, storage and/or modification of bacterial surface features, including LPS, EPS and capsular polysaccharide (Table 1, Fig. 1). The sequences (ca. 1000–5000 bp.) bordering these five loci were analyzed using BlastP to obtain additional insights into their genomic context and transcriptional organization. All five loci were associated with a total of only 2 operons (Fig. 1, Table 1). One operon is a component of the rfa gene cluster involved in the biosynthesis of the core region of LPS (mutants M22 and M25). The second operon encodes an O-antigen LPS biosynthesis cluster51 (mutants M61, M73, M83, M144, M177, M399, M534, M626, M720, M1004 and M1026). Examination of the KEGG pathways corresponding to these 5 transcriptional units enabled their assignment to the cellular pathways involved in the biosynthesis of cell surface lipopolysaccharides and exopolysaccharides. Likewise, the putative interacting partners of the these 5 proteins assessed using STRING revealed that the products of all 5 loci interact with proteins associated with bacterial cell surface features (involved in the synthesis and remodeling of the capsule, LPS and EPS) (Supplementary Table 2). These 5 loci are all conserved in other Dickeya species, with homologs found in D. dianthicola, D. dadantii, D. fangzhongdai, D. zeae, D. oryzae, D. unidicola and D. chrysanthemi strains (Supplementary Table 2).

Two operons of D. solani involved in the interaction of the bacterium with lytic bacteriophage ΦD5. (A) Operon associated with rfa gene cluster involved in the biosynthesis of the core region of LPS in Gram-negative bacteria and (B) Putative O-antigen LPS biosynthesis cluster51. The D. solani ORFs affected by the Tn5 insertion are marked in blue. The directions of the arrows represent the direction of the transcription.
Adsorption of ΦD5 to WT and phage resistant mutants in vitro
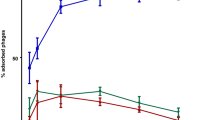
The adsorption of ΦD5 to both viable and dead (chloramphenicol-killed) cells of wild-type D. solani cells was rapid (Fig. 2). Within 5 min, nearly 90% of the phage particles had adsorbed to both living and dead cells of the WT strain and more than 99% had bound by 20 min. In contrast, the adsorption of ΦD5 to 11 phage-resistant mutants (M22, M25, M61, M73, M83, M144, M399, M534, M626, M720, M1026) was completely abolished, and only between 1 and 7% had bound to mutants M177 and M1004 within 20 min (Fig. 2). The lack of binding of ΦD5 to most mutants was confirmed using transmission electron microscopy (TEM); while abundant adsorption of the phage WT cells was observed, no adsorption of ΦD5 to phage-resistant Tn5 mutants occurred (Fig. 3).

Adsorption of ΦD5 to viable and chloramphenicol-killed cells of WT and phage-resistant D. solani . A MOI of 0.01 of ΦD5 was used for adsorption assay and the total assay time was 20 min. Phage adsorption was calculated as follows: the percentage adsorption = (the average titer of unabsorbed phages per sample/average titer of phages in negative control) × 100. The averages and standard deviations of three independent repetitions per strain (WT or mutants) are shown.

Visualization of adsorption of ΦD5 particles to WT and phage-resistant D. solani mutants by transmission electron microscopy (TEM). Bacterial cells and phage particles were mixed at MOI of 10 and incubated for 20 min at room temperature (ca. 20–22 °C) to allow the phages to attach to host bacterial cells. At least 10 individual images were gathered for each analyzed strain, and the experiment was repeated once (two biological replicates of the assay). Representative photos are shown. Scale bar—200 nm.
Biochemical phenotypes of phage-resistant mutants
The phage-resistant mutants were tested for changes in phenotypes that may be important for the ecological fitness and virulence of D. solani under natural and agricultural settings. No differences were found between the mutants and the WT strain in most metabolic phenotypes examined using BIOLOG GEN III and Ecoplate phenotypic microarrays. The ΦD5-resistant mutants differed from the WT strain in a total of only 3 features out of 125 tested using these systems. Mutants M73, M144, M626 and M720, having a disrupted ATP-binding protein in the O-antigen export system, lost the ability to utilize D-cellobiose as a sole carbon source, and mutants M22 and M25, having a disrupted glycosyltransferase family protein, became susceptible to troleandomycin. In addition, mutants M73, M144, M626 and M720 with a disrupted wzt, M83, M399 and M534 with a disrupted wbeA, and M177 and M1004 with a disrupted fcl, gained the ability to utilize inosine, a feature absent both in the WT and 4 other phage-resistant mutants (M22, M25, M61 and M1026) tested. All mutants shared with the WT strain the ability to produce cavities on CVP, produced proteases and degraded carboxymethylcellulose and polygalacturonic acid, while lacking the ability to produce siderophores or grow on TSA medium supplemented with 5% NaCl. Furthermore, while all mutants retained the ability to form some biofilm, all mutants except M22 and M25 produced more abundant biofilm than the WT strain. The LPS purified from the phage-resistant mutants was not distinguishable from that of the WT strain (Supplementary Fig. 1). No differences in cell morphology or size diameter were noted in the mutants compared to the WT strain by examination using transmission electron microscopy (Fig. 3). Likewise, all mutants exhibited similar colony morphology and colony diameter to that of the WT strain. None of the mutants differed significantly in their average generation times in rich (TSB) medium although M22 and M25 grew more slowly in a minimal medium (M9 + 0.4% glucose) compared to that of the WT strain (Fig. 4). The growth rate of the mutants over a range of temperatures (8, 15, 28, 37 °C) did not differ from that of the WT strain and none of the strains grew at either 4 or 42 °C. The growth rate of the mutants at pH 5.0 and pH 10.0 was similar to that of the WT strain (data not shown). While the WT was capable of both swimming and swarming motility, mutants M83, M177, M399, M534 and M1004 were nonmotile for both phenotypes and the other mutants exhibited reduced swimming motility compared to the WT strain. None of the mutants were capable of swarming motility. All mutants expressed similar susceptibility/resistance to all of the antibiotics tested as the WT strain, except for their resistance to kanamycin conferred by Tn5. All phage-resistant D. solani mutants were also significantly affected in their production of extracellular polymeric substances (EPS); M22, M25, M83, M399 and M534 produced less EPS (ca. 8 to 23% as much) than the WT strain. In contrast, mutants M61, M73, M144, M177, M626, M720, 1004 and M1026 produced more EPS (ca. 175–210% as much) than the WT strain.

Growth of D. solani WT and phage-resistant mutants M22 and M25 in minimal growth medium (M9 + 0.4% glucose). The experiment was performed in two biological replicates containing two technical replicates each (n = 4). The results were averaged for presentation. The bars show standard deviation (SD).
Differential permeability of outer membrane of phage mutants
Given that all ΦD5-resistant mutants had putative alterations in their envelope, their permeability was compared with that of the WT strain. The rate of death was much higher for the phage resistant mutants than the WT strain. While 70% of the cells of the WT strain survived exposure to a SDS solution for 1 min and ca. 15% survived for 10 min, only 30 to 60% of the mutants survived for even 1 min.
Surface features of phage-resistant mutants
Since cell surface features were apparently altered in phage-resistant mutants, we assessed the self-association properties of the mutants as such a change would be expected to influence this process. All mutants except M22 and M25 exhibited more rapid sedimentation, associated with self-aggregation, than the WT strain (Fig. 5). The highest rate of sedimentation was observed in mutants M83, M177, M399, M534 and M1004 (Fig. 5).

Self-aggregation of cells of WT and phage-resistant D. solani mutants as measured by turbidity of bacterial suspensions. The percentage of aggregation was quantified from the change in optical density (OD600) over 24 h. Percentage aggregation (sedimentation) was measured as follow: %A = 1 (OD60024h/OD600 0 h), where: %A—percentage of aggregation (sedimentation), OD6000h—OD of bacterial culture at time 0 h, OD60024h—OD of bacterial culture at time 24 h. Results were considered to be significant at p = 0.05 and pair-wise differences were obtained using the t-test. Error bars represent standard deviation (SD).
As the differences in apparent self-aggregation suggested differences in surface features of at least some phage-resistant mutants we examined the surfaces directly with both SEM and Atomic Force Microscopy (AFM). While WT cells examined by SEM were characterized by a gently folded cell wall and rounded cell poles, all phage-resistant mutants had a rougher surface than the WT strain (Fig. 6). The degree of the cell wall folding in the mutants also differed from each other. Mutants M22 and M25 had a clearly spongy surface whereas mutants M61, M144, M534 and M1004 had longitudinal furrows on their surface (Fig. 6). On the other hand, mutants M73, M83, M177 and M626 were characterized by a lobular cell surface shape. Pointed cell poles and a longitudinal surface folding was specific to mutant M399 mutant. M720 cells, apart from having longitudinal furrows in the wall, were characterized by the presence of cell membrane forms bent even at an angle of 90°. Cells of mutant M1026 cells were clearly shorter than the other mutants and formed compact aggregates (Fig. 6).

SEM imaging of D. solani WT and phage-resistant mutants. Scale bar corresponds to 1 µm.
Given the obvious differences in surface features seen in phage-resistant mutants by SEM we compared mutants M399 and M1004 with the WT strain by Atomic Force Microscopy (AFM) to provide more details of their distinctive surfaces. These mutants both possessed a rougher surface than the other strains. Mutant M399 had a less regular surface than the WT strain while mutant M1004, had a more folded surface than either the WT or mutant M399. AFM imaging of the WT strain showed a regular and symmetrical cell profile (Fig. 7). Slight folds characterized its cell surface both at three-dimensional image and height profiles, as shown in (Fig. 7, panels A1-A3). The topological features of mutants M399 and M1004 was quite different from that of the WT strain, having distinct folding, and an irregular surface with furrows visible in the height profiles (Fig. 7, panels B1-B3, C1-C3). Moreover, the surface roughness of the WT was lower (average roughness, Ra = 3.05 nm) than both mutant M399 (Ra = 4.41 nm) and M1004 (Ra = 4.89).

Atomic force microscopy (AFM) imaging of D. solani WT and selected phage-resistant mutants M399 and M1004. (panel A1–A3)—cell-surface D. solani IPO2222 WT; (panel B1–B3)—cell surface of phage-resistant mutant M399; (panel C1–C3)—cell surface of phage-resistant mutant M1004. (panel A1, B1, C1) –three-dimensional images of D. solani WT and mutant cells; (panel A2, B2, C2) –height mode images of cells; (panel A3, B3, C3)—height profiles of cells, the profiles were made along the lines shown in the height images.
To compare the profile of membrane proteins present on the surface of phage-resistant mutants and the WT strain an intact cell MALDI technique was used. While all mutants exhibited similar proteins patterns as the WT strain, subtle differences in proteins content, presumably due to compensatory alterations in membrane structure were seen in some phage mutants (Supplementary Results, Supplementary Fig. 2).
Survival of phage-resistant mutants on potato leaves
Since Dickeya spp. may exist as an epiphyte on plant surfaces before invading the plant to cause disease52, any effect of phage resistance on epiphytic survival would be an important factor in their fitness as a pathogen under field conditions. The tolerance of stresses of the mutants on the adaxial surface of detached potato (S. tuberosum) leaf surfaces over a period of 14 days was thus investigated. (Fig. 8). As expected of such assays, the numbers of bacteria differed widely between leaves but relative differences between strains did not vary between replicate experiments, enabling the experiments to be analyzed together. Immediately after inoculation viable population sizes of 105 to 106 CFU per leaf for all strains were observed. No significant changes in the population size of the WT strain were observed over a 14-day incubation period (Fig. 8). In contrast, the viable population size of all phage-resistant mutants decreased significantly over this time to only ca. 102–103 CFU per leaf (Fig. 8). No pectinolytic, cavity-forming bacteria similar to D. solani were found on the leaves of non-inoculated leaves.

Population size of viable D. solani WT and phage-resistant mutants on the adaxial surface of detached leaves of S. tuberosum measured 14 days after inoculation. In replicated experiments, 20 leaves (10 leaves per experiment per mutant) were samples at two time points (0 [control] and 14 days post inoculation). Both at 0 and 14 dpi, five leaves (third to six from the shoot terminal) spray-inoculated with 2 ml of 106 CFU mL−1 of bacterial suspension (WT or individual Tn5 mutant) in 1/4 Ringer’s buffer and incubated adaxial side up on 0.5% water agar in square plastic Petri dishes (100 × 100 mm) were sampled. At 0 and 14 dpi, five randomly chosen leaves from five randomly chosen Petri dishes were collected and individually shaken in 10 ml Ringer’s buffer in 50-mL Falcon tubes at 50 rpm at room temperature for 30 min to wash bacterial cells off the leaf surface. The serial diluted leaf washings were plated in duplicates on CVP supplemented with 200 µg mL-1 cycloheximide (for isolation of IPO 2222 WT) or on CVP containing 50 µg mL-1 neomycin ad 200 µg mL-1 cycloheximide (for isolation of phage-resistant Tn5 mutants). Inoculated plates were incubated at 28 °C for 24–48 h, and the resulting colonies were counted. The results were averaged for presentation. Results were considered to be significant at p = 0.05 and pair-wise differences were obtained using the t-test. Error bars represent standard deviation (SD).
Virulence of ΦD5-resistant mutants to potato
Although all 13 phage-resistant mutants expressed some level of virulence, as measured by the degree of tissue maceration when introduced into the interior of potato tubers by stab-inoculation, they all were significantly affected in their ability to macerate potato tuber tissue compared with the WT strain (Fig. 9). Likewise, all mutants were significantly less virulent when tested for their ability to incite blackleg symptoms on potato plants grown in potting compost infested with bacteria (Fig. 10), a setting that reproduces the process by which the pathogen naturally invades the roots of growing plants from inoculum in the soil. Most potato plants inoculated with phage-resistant mutants in phytochamber experiments did not exhibit any blackleg symptoms. In contrast, between 80 and 100% of the plants inoculated with the WT strain developed severe and typical blackleg symptoms, which usually led to the death of some of the infected plants. As expected, no symptoms were observed at any time in plants inoculated with sterile buffer.

Maceration of potato tuber tissue by D. solani WT and phage-resistant mutants. Five individual potato tubers were inoculated with a given phage-resistant bacterial mutant using a whole tuber injection method (stab inoculation) 103,106. Bacterial strains were grown in TSB (WT strain), or TSB supplemented with 50 µg mL-1 of neomycin (Tn5 mutants) for 24 h at 28 °C. After incubation, bacterial cultures were separately collected, washed two times with 1/4 Ringer’s buffer and resuspended in the initial volume of Ringer’s buffer. Optical density (OD600 = 0.1) was used to normalize the number of bacterial cells in all treatments (ca. 108 CFU mL-1). Surface-sterilized potato tubers were stab-inoculated with sterile yellow pipette tip filled with 50 µl of bacterial suspension (treatments) or sterile Ringer’s buffer (negative control). IPO 2222 WT was used as a positive control. Results were considered to be significant at p = 0.05 and pair-wise differences were obtained using the t-test. Error bars represent standard deviation (SD).

Population size of D. solani WT and phage-resistant mutants within stems of potato plants after introduction of the pathogen into the soil. Results were considered significant at p = 0.05, and the pair-wise differences were obtained using the t-test. The means that do not share the same letters above each bar differ. Results were considered to be significant at p = 0.05 and pair-wise differences were obtained using the t-test. Error bars represent standard deviation (SD).
In addition to the large reduction in disease incidence incited by the mutants compared to the WT strain, the severity of infection, as measured by the number of viable bacterial cells within the tissue was also greatly reduced. Although the population size of the WT differed significantly between infected plants when measured 14 days after inoculation, it was recovered from all inoculated plants at densities ranging from 103 to 15 × 103 CFU g−1 of the stem tissue (Fig. 10). Phage-resistant mutants were recovered with both: (i) a much lower incidence and (ii) lower population size from stems after soil infestation. Among all phage mutants tested (10 plants per mutant), viable cells were detected in only 4 plants expressing typical blackleg symptoms; M73, M144, M626 and M720 were each recovered only once, and each with a very low population size (ca. 1000 CFU g−1 of stem tissue). None of the other mutants was detected inside stems of inoculated plants in both experiments (Fig. 10). As expected, D. solani was never detected in the stems of non-inoculated control plants.
Competition of D. solani strains tagged with GFP or DsRed
The degree of competition of mutant and wild type strains, a sensitive method for comparing fitness differences, was assessed in different setting by quantifying changes in the proportion of the different strains in a mixture that differed in their color of fluorescence. The WT and phage-resistant mutants were transformed with plasmids pPROBE-AT-gfp and/or pRZ-T3-dsred conferring green and red fluorescence respectively. Both plasmids (Supplementary Table 1) were stably maintained in these strains as evidenced by maintenance on media without supplementation with ampicillin or tetracycline. Fluorescently-labelled bacterial variants displayed similar growth characteristics in liquid media to their parental strains (data not shown), indicating that their growth was not altered either by the presence of plasmids carrying gfp or dsred genes or by expression of fluorescent proteins in transformants.
The competition of fluorescently-labelled WT and phage-resistant mutants co-inoculated into tubers by stabbing was assessed after 72 h incubation under conditions conducive for development of disease symptoms (28 °C, ca. 80–90% relative humidity (RH)) in phytochamber experiments (Fig. 11). The numbers of bacteria recovered differed among individual tubers but similar numbers were found for a given strain among replicate experiments and consequently, the results from both experiments were analyzed together. The population size of the WT strain inoculated alone into tubers reached between 106 and 107 CFU g−1 of tuber tissue by 72 h. Furthermore, the average populations of the WT strain co-inoculated with a phage-resistant mutant were only slightly reduced (to ca. 5 × 105 CFU g−1) compared to that in controls inoculated only with the WT strain. The population size of any given mutant inoculated alone into tubers was greatly reduced (to ca. 103–104 CFU g−1) compared to that of the WT strain (Fig. 11). Importantly, the population sizes of any given mutant co-inoculated into tubers with the WT strain were greatly decreased (to ca. 10–5 × 102 CFU g−1) compared to their already low population sizes achieved when inoculated alone into tubers (Fig. 11). These results strongly indicate that mutations that confer phage-resistance in D. solani impart severe decreases in its ability to infect plants.

Population size of differentially marked cells of D. solani WT and phage-resistant mutants in potato tubers after co-inoculation or single inoculation into tubers. Potato tubers were inoculated either with fluorescently labelled D. solani IPO 2222 WT, fluorescently labelled individual phage-resistant D. solani Tn5 mutants or co-inoculated with WT strain and individual Tn5 mutant. In the first experiment, a GFP-tagged D. solani IPO 2222 WT and DsRed-tagged phage resistant D. solani mutants were used. In contrast, DsRed-tagged IPO 2222 WT and GFP-tagged phage resistant D. solani mutants were applied in the second experiment. Inoculated tubers were kept under conditions that promote rotting (28º and 80–90% relative humidity). Three days (72 h) post inoculation tubers were sampled and analyzed for the presence of fluorescently tagged bacteria using pour plating. The GFP and DsRed positive colonies were counted. The experiment was replicated one time with the same setup, and the results from both repetitions were averaged for analyses. The numbers inside bars represent the average log (CFU + 1/g of tuber tissue) for the respective treatment. Results were considered to be significant at p = 0.05 and pair-wise differences were obtained using the t-test. Error bars represent standard deviation (SD).
Discussion
The results of this study reveal that the cost of resistance of D. solani to lytic phage ΦD5 is strongly dependent on the environmental context. Although resistance to ΦD5 did not affect many of the phenotypes tested in vitro, all mutants were significantly impacted in their ability both to survive on the plant surface and to cause disease symptoms in either potato plants or tubers. Unfortunately, many details of the interactions of soft rot Pectobacteria such as D. solani and its host plant have not yet been revealed, so it is not possible to understand what aspects of its interaction with its host are most impacted by changes associated with phage resistance. Likewise, their interactions with lytic bacteriophages, especially the ecological context of such associations, remain insufficiently explored53,54. This study used a random Tn5-based transposon mutagenesis method to identify D. solani mutants resistant to infection caused by lytic phage ΦD5. The power of this approach was to unambiguously associate disruption of a given gene with changes in the behavior and features of the pathogen. By focusing on phenotypes likely to be involved in the ecological success of the pathogen in the plant environment and interrogating the mutant directly in these plant settings, this approach allowed us to investigate the tradeoff hypothesis in which the ΦD5 resistance confers fitness costs for the phage-resistant D. solani variants during infection.
Screening a collection of 1000 random Tn5 mutants allowed us to rather completely interrogate those genes and traits required for susceptibility to this phage. It was interesting to find that the 13 Tn5 insertional mutations conferring resistance to ΦD5 were located in only a small pool of 5 genes with multiple insertions in a given gene of interest. Furthermore, the disrupted genes are components of only two operons (Fig. 1, Table 1), both previously described as involved in the envelope status of Gram-negative bacteria, including bacteria belonging to Enterobacteriaceae and Pectobacteriaceae families55. Specifically, these operons participate in the biosynthesis of the core region of LPS together with production of the O-antigen of LPS in Gram-negative bacteria (Fig. 1)51,56. As no phage resistant-mutants analyzed in this study had insertions in other genes and operons, we believe that bacterial envelope components (viz. LPS, EPS and extracellular capsule (CPS)) encoded by the operons mentioned above are particularly essential for interaction of ΦD5 with D. solani IPO 2222. To gain more insight about the role of LPS in the context of phage resistance, we compared the lipopolysaccharides from phage-resistant mutants with that of the WT strain. Surprisingly, the characterization of LPSs by SDS-PAGE did not reveal apparent structural differences between the LPS of the WT strain and Tn5 mutants that could immediately explain the ΦD5-resistant phenotype suggesting that small structural changes not revealed in such an analysis were sufficient to disrupt interaction with the phage.
Modification of bacterial envelope is one of the most common strategies of phage evasion in Gram-negative bacteria57,58. To date, however, only limited data exist on how frequently such a mechanism is employed by plant pathogenic bacteria such as D. solani to prevent phage infection. In this study, phage adsorption was completely blocked in 11 of 13 phage-resistant mutants expressed a complete inhibition and was substantially reduced in 2 others. This observation cleanly connects phage adsorption with the nature of the D. solani envelope. In earlier studies in which we analyzed the interaction of Pectobacterium parmentieri with its lytic bacteriophage ΦA38, we found out that the phage requires intact LPS to infect its host and that alterations in LPS synthesis protected P. parmentieri from viral infection59. Similar observations linking phage-resistance and cell surface features have appeared for other SRP bacteria, including P. atrosepticum strain SCRI 1043, P. carotovorum strain Pcc27 and P. brasiliense strain F152 (PB29)60,61,62 but not for D. solani strain IPO 2222.
Most knowledge of the ecological functions of bacterial envelope components such as LPS, EPS and CPS polysaccharides, have come from studies of human and animal pathogenic bacteria63,64, microorganisms that encounter quite different environments in their life cycle than plant pathogenic bacteria. The highly variable and often stressful habitat in which plant pathogens commonly exist may make them particularly dependent on a suitable cell envelop. Unfortunately, the role of bacterial surface status in the interaction of plant pathogens and their plant hosts has received much less attention65. The gene encoding a glycosyltransferase that is linked to extracellular polysaccharide synthesis was disrupted in mutants M22 and M25. Glycosyltransferases are involved in LPS and EPS synthesis in plant pathogenic and plant beneficial bacteria66,67. Glycosyltransferase was reported to be required for EPS and LPS production in the plant pathogenic bacterium Xanthomonas citri subsp. citri (Xac). Furthermore, glycosyltransferase defective Xac mutants were impaired in biofilm and EPS production, exhibited delayed and reduced growth and were more susceptible to environmental stresses than the wild type strain66. In the plant-beneficial bacterium Rhizobium sp. YAS34, a polysaccharide-associated glycosyltransferase was involved in the synthesis of EPS, and was critical for colonization of roots as this function was required for cell adherence to roots during plant colonization67. Mutants M22 and M25 exhibited similar phenotypic defects in this study, having reduced growth in minimal medium in vitro, reduced synthesis of EPS, and decreased swimming and complete lack of swarming motility. The cpsB (manC) gene disrupted in mutants M61 and M1026 encodes a mannose-1-phosphate guanylyltransferase/mannose-6-phosphate isomerase. cpsB was reported to be required for virulence of Streptococcus penumoniae and Klebsiella pneumoniae in mice68,69. A homolog of cpsB is a part of the GDP-fucose synthesis pathway involved in the synthesis of a capsular polysaccharide colanic acid in the plant pathogenic bacterium Erwinia amylovora. Consequently, E. amylovora mutants lacking a functional cpsB were avirulent to its plant host70. Likewise, colanic acid was required for full virulence of a soft rot Pectobacterium spp. closely related to Dickeya spp.71. In Xanthomonas campestris pv. campestris (Xcc), a homolog of cpsB, gene xanA is required for the synthesis of extracellular polysaccharide xanthan, involved in stress tolerance and required by Xcc both for attachment to plant surfaces and biofilm formation72. Here, D. solani phage-resistant mutants M61 and M1026 with a disrupted locus encoding mannose-1-phosphate guanylyltransferase/mannose-6-phosphate isomerase exhibited elevated biofilm formation, reduced swimming motility and increased EPS production. The wzt (rbfB) gene was disrupted in D. solani mutants M73, M144, M626 and M720. This gene encodes the O-antigen export system (LPS export system) ATP binding protein involved in the translocation of O-antigen across the inner cell membrane to the periplasm, attaching it to a lipid A core73. In Rhizobium tropici CIAT899, a wzt deletion mutant had a lower growth rate, reduced motility, and lower colonization of roots than the wild type strain74. In D. dadantii A1828, wzt mutants had reduced survival of osmotic stress imposed by NaCl75. In the gliding bacterium Myxococcus xanthus blockage of its O-antigen export system ATP binding protein led to defects in social motility and colony formation under laboratory conditions76. In our study, all 4 wzt mutants shared similar phenotypes that were divergent from that of the WT strain including elevated EPS production, higher biofilm formation and reduced motility. wbeA encoding a hypothetical protein most closely related to the glycosyltransferase WbeA was disrupted in mutants M83, M399 and M534. The role of WbeA has not been assessed in plant pathogenic bacteria and the knowledge of its function in the ecology and pathogenicity of SRP bacteria is limited51. wbeA gene is a conserved member of the putative O-antigen LPS gene cluster present in various Dickeya species51. Its glycosyltransferase activity and genomic location suggest its involvement in LPS synthesis, together with another glycosyltransferase, WbeB, present in the same operon51. In our study, mutants M83, M399 and M534 had elevated biofilm formation, reduced EPS production and were incapable of swarming. Mutants M177 and M1004 had a mutation in fcl encoding GDP-L-fucose synthase. This enzyme is required for the production of the extracellular polysaccharide colanic acid in Escherichia coli strain K-12 and other members of Enterobacteriaceae77. Fcl was also reported to be involved in the synthesis of EPS in Pectobacterium carotovorum78. A fcl knockout mutant of P. carotovorum produced elevated biofilm but was less virulent to plants78. These phenotypes were similar to that seen here in phage-resistant mutants M177 and M1004.
To further support a role of surface features of D. solani for infection by ΦD5 we compared the surface of the WT and phage-resistant mutants using several techniques. While phage-resistant mutants were generally indistinguishable from the WT strain in overall the colony and cell morphology, more detailed analyses with SEM and AFM revealed that all had rougher and more irregular cell surfaces than the WT strain. In line with our observations, several other studies have showed that bacterial mutants with rough envelope phenotypes are more resistant to viral infection79,80. Furthermore, envelope regularity and smoothness resulting from LPS, EPS and capsule cooperation are also among the key factors determining the ability of bacterial to form biofilms on various surfaces and to survive in harsh environments81,82. Alterations of envelope properties may therefore increase antibiotic susceptibility, influence biofilm formation and reduce resistance to environmental stresses83. In our study, while all phage-resistant mutants did not differ from the WT in resistance to various antibiotics they did have elevated cell aggregation and biofilm formation phenotypes as well as higher membrane permeability than the WT strain.
All phage-resistant D. solani IPO 2222 mutants were compromised in their ability to cause disease symptoms in planta. Their decreased survival on the leaf surface and their low virulence in both potato tubers and growing potato plants were not due to a reduced growth rate of the mutants. The mutants and WT had similar generation times in rich media and only two mutants M22 and M25 had a slightly reduced growth in a minimal medium compared to the WT strain. The lower relative fitness of the phage-resistant mutants in planta was most clearly revealed in competition assays in which potato tubers were inoculated with mixtures of the WT strain and an individual phage-resistant mutant. Not only did all phage-resistant mutants achieve lower population sizes in potato than the WT strain when applied individually to potato tubers, but were particularly impaired in growing in planta when co-inoculated with the WT strain. Therefore, it is clear that the modifications of the cell envelope strongly impacts both the survival of D. solani in planta and its virulence. Interestingly, wbeA and gmd, the two members of the putative O-antigen LPS biosynthesis operon involved in interaction of D. solani with phage ΦD5, were also found to be induced in the presence of potato tuber tissue in our prior studies of this pathosystem84. As gmd and wbeA, together with cpsB, cpsG, wzm, wzt, flc, and wbeB constitute one operon (Fig. 1), we can assume that this operon is required both during infection of potato tubers as well as for infection of D. solani by ΦD5. It is thus clear that the bacterial envelope plays a central role both in the communication of D. solani with both their external world and its interaction with lytic bacteriophages. For this reason, alternations of the envelope to avoid viruses will likely impact their ecological fitness. The practical relevance of this observation is that by using phage therapy in agricultural applications (e.g. under field conditions or in greenhouses) we may be able not only to kill pathogens in situ but also to select for less virulent bacterial variants that under high and constant phage pressure may spread in the given niche and cause less problems in these environments in the future85.
Indeed, SRP bacteria, including D. solani, encounter harsh conditions inside the host during infection86. These include oxidative and osmotic stresses and exposure to antimicrobial compounds produced by plant injured tissues87. To cope with such stresses during infection, the bacterial envelope undergoes an extensive remodeling involving changes in LPS, EPS and capsule87. Mutations in the genes encoding envelope components in D. solani have been shown to lead to decreased survival and performance during infection88. Similar observations were made for other plant pathogenic bacteria. For example, envelope-defective mutants of Ralstonia solanacearum exhibit decreased virulence in tobacco plants89 and mutations altering envelope polysaccharides of Erwinia amylovora resulted in a reduced ability of the mutant to survive in planta and cause disease in pear90.
In conclusion, this study is one of few to investigate the tradeoff between phage resistance and fitness of bacterial plant pathogens in their natural environment. Likewise, this study is an initial step toward better understanding of how lytic bacteriophages interact with Dickeya spp., including D. solani. Using several complementary approaches, we showed that phage-resistant D. solani variants pay a substantial fitness penalty during their interaction with potato plants, suggesting that the resistance to phage ΦD5 strongly reduces its virulence. Furthermore, although these tradeoffs are linked with several modifications of the bacterial envelope, these costs are paid only in planta and not under in vitro conditions. This, in turn, suggests that fitness costs due to phage resistance in plant pathogenic bacteria is more frequently in nature than has been projected from in vitro studies.
Methods
Bacteriophages, bacterial strains, and growth media
The lytic bacteriophage vB_Dsol_D5 (ΦD5) was previously described46,47,91. For this work, ΦD5 was propagated on its wild-type host, D. solani strain IPO 222238, and quantified as described earlier46. A stock of ΦD5 phage particles (ca. 108—109 plaque-forming units (PFU) mL−1 in tryptone soya broth (TSB, Oxoid) or quarter-strength (1/4) Ringer’s buffer (Merck) was used in all experiments unless otherwise stated. Bacterial strains used in this study are listed in Supplementary Table 1. The pool of 1000 mutants from a collection of 10,000 D. solani Tn5 mutants previously generated84,92 was interrogated as a source of ΦD5-resistant mutants. The D. solani wild type (WT) strain was cultivated for 24–48 h at 28 °C on tryptic soy agar (TSA, Oxoid), in tryptone soy broth (TSB, Oxoid) or M9 minimal medium (MP Biomedicals) supplemented with glucose (0.4%) (Sigma-Aldrich). Bacteriological agar (Oxoid) (15 g L−1) was added to solidify the media. As appropriate, media were supplemented with neomycin (Sigma-Aldrich) (50 µg mL−1), ampicillin (Sigma-Aldrich) (150 µg mL−1) or tetracycline (Sigma-Aldrich) (40 µg mL−1). Bacterial cultures were agitated during incubation (120 rpm). To prevent fungal growth, cycloheximide (Sigma-Aldrich) was added to the growth medium at a final concentration of 200 µg mL−1.
Selection of ΦD5-resistant D. solani mutants
Phage-resistant D. solani Tn5 mutants were preselected as previously described93. Briefly, the overnight cultures of individual Tn5 mutants, grown in TSB supplemented with neomycin to the final concentration of 50 µg mL−1 and containing ca. 108 colony-forming units (CFU) mL−1, were diluted 100-times in the same fresh medium (final bacterial density ca. 106 CFU mL−1). Per Tn5 mutant to be analyzed, two wells of a 96-well plate were inoculated with 50 µl of individual IPO 2222 mutant culture (final bacterial density per well: ca. 5 × 104 CFU). Subsequently, they were filled with 200 µl of ΦD5 solution (final phage concentration per well: ca. 2 × 107 PFU). To each well of the microtiter plate, 5 µl of sterile 0.7% resazurin (Sigma) solution in demineralized water was added. After inoculation, 96-well plates were sealed with an optically transparent sealing type (Sarstedt) to prevent contamination and evaporation of bacterial cultures during incubation. The plates were incubated for 16 h with shaking (150 rpm) at 28 °C. After this time, the development of the pink/yellowish color (positive reaction) in the inoculated wells was determined by eye. The positive reaction indicated the growth of the respective Tn5 mutant in the presence of viable bacteriophages (= phage resistance). ΦD5-susceptible D. solani strain IPO 2222 and ΦD5-resistant Escherichia coli strain DH5α (ThermoFisher Scientific), grown under the same conditions and treated similarly as described above in the case of IPO 2222 Tn5 mutants, were used as controls93. Each Tn5 mutant was tested in four replicates for its resistance against ΦD5 phage. To confirm the resistance of the candidate Tn5 mutant to bacteriophage infection, each putative mutant obtained in the resazurin assay screen was additionally exposed to repeated phage challenge (at least 2 independent assays per mutant) and plaque formation assays (at least two independent assays per mutant) as previously described46.
Identification of the transposon insertion sites in phage-resistant D. solani mutants
To identify the Tn5 insertion sites in the genomes of phage-resistant mutants, the genome of each mutant was sequenced. Genomic DNA of each mutant was isolated, sequenced, and assembled into a draft genome at the Laboratory of DNA Sequencing and Oligonucleotide Synthesis (Institute of Biochemistry and Biophysics of the Polish Academy of Science, Warsaw, Poland) using Illumina technology. Structural and functional annotations of the draft Tn5 genomes were determined using RAST (Rapid Annotation using Subsystem Technology (http://rast.nmpdr.org/).94The position of the Tn5 transposons in the draft genomes was established using BlastN and BlastX alignments accessed via the NCBI website (http://blast.ncbi.nlm.nih.gov/Blast.cgi)95. Using the available complete genome sequence of D. solani WT strain IPO 2222 (Genbank accession: CP015137)45 and the draft genomes of the Tn5 mutants the insertion sites was determined as described before84. For each mutant, sequences (ca. 1,000- to 5,000-bp) adjacent to the Tn5 transposon site were examined to determine the genomic context of each Tn5-disrupted gene84,92. The function of the disrupted genes was inferred using BlastN and BlastX alignments accessed via the NCBI website (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Similarly, the functions of any unannotated open reading frames (ORFs) encoding hypothetical proteins or proteins without apparent sequence homology to proteins deposited in protein databases were inferred using GeneSilico Protein Structure Prediction meta-server96, together with PSI-BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) 97. The gene functions with the highest scores obtained were judged to be the most probable.
Characterization of D. solani transcriptional units disrupted by the presence of Tn5 transposon
The putative transcriptional organization of D. solani genes interrupted by Tn5 was established using Operon-mapper (https://biocomputo.ibt.unam.mx/operon_mapper/). Inference of the biochemical pathways in which the genes of interest might participate was made using KEGG98. The results were visualized using iPath99. Likewise, proteins were evaluated for their predicted biological, functional, and metabolic roles in cellular networks using STRING (Search Tool for Retrieval of Interacting Genes/Proteins) v11.5 accessed via the website (https://string-db.org/) (parameters: network type: full network, network edges: high confidence, interaction sources: text mining, experiments, databases, co-expression, co-occurrence, gene fusion), providing information regarding interactions of proteins of interest100 using the proteome of the WT D. solani strain as a reference.
Kinetics of ΦD5 adsorption to WT D. solani and Tn5 mutants
The rate of ΦD5 adsorption to wild-type D. solani and phage-resistant mutants was determined as before59,101. Briefly, log-phase bacterial cultures were inoculated with a phage suspension (Multiplicity of Infection (MOI) of 0.01) and incubated at 28 °C for up to 20 min. Two individual samples of each bacterial strain were collected after 0 (control), 1, 2, 5, 10, 15, and 20 min after inoculation with phage, and the number of unadsorbed phages was quantified. As a negative control, bacteriophages were suspended in sterile TSB and recovered at different times as above. The experiment was repeated three times, and the results were averaged. Phage adsorption efficiency was calculated as the average titer of unabsorbed phages per sample/average titer of phages in the negative control) x 10059.
Kinetics of ΦD5 adsorption to killed D. solani cells
To test whether bacteriophage ΦD5 can adsorb to non‐viable cells of D. solani, a dead cell adsorption assay was employed59,102. Briefly, the WT strain was grown for 16 h in TSB at 28 °C with shaking and chloramphenicol (Sigma‐Aldrich, Darmstadt, Germany) was then added to a final concentration of 5 mg mL−1. The complete killing of the cells after 1 h was confirmed by plating 100 μL aliquots of treated bacterial culture on TSA plates. The chloramphenicol‐killed D. solani cells were mixed with a phage suspension (MOI = 0.01) and analyzed for phage adsorption as above. Three repetitions of the experiment were performed, and the results were averaged for analysis. Phage adsorption efficiency was calculated as described above.
Determination of growth dynamics of phage-resistant Tn5 mutants of D. solani in rich and minimal media
To determine whether the Tn5 insertions affect the growth rate of the mutants, the growth of the selected D. solani Tn5 phage-resistant mutants was assessed both in TSB (rich medium) and in M9 containing 0.4% glucose (minimal medium) at 28 °C for 16 h as previously described103. The experiment was replicated once, and the results were averaged. The average generation time of each Tn5 mutant was analyzed using the Doubling Time calculator (parameters: C0 = 3 h, Ct = 7 h, t = 4 h) (http://www.doubling-time.com/compute.php) 104. The ability of ΦD5-resistant Tn5 mutants and the WT strain to grow was also tested qualitatively on solid rich and minimal media at temperatures: 4, 8, 15, 28, 37, and 42 °C as described before105. For this, 5-µL aliquots of 50-fold diluted in TSB or M9 + glucose overnight bacterial cultures grown in either TSA and M9 + glucose, respectively, were placed on the surface of either TSA or M9 + glucose and incubated for 120 h at 5 and 8 °C, or for 48 h at 15, 28, 37 and 42 °C. Growth was assessed visually daily. The experiment was replicated one time using the same setup.
To assess whether the transposon-mediated ΦD5-resistance differentially affected the growth rate of the D. solani mutants at different pHs, the growth of selected D. solani ΦD5-resistant Tn5 mutants was compared in TSB at pH 5 and pH 10, similarly to other studies106. Briefly, overnight cultures in TSB (ca. 109 CFU ml−1) were diluted 50-fold in fresh growth broth with pH 5 or pH 10. Aliquots (100 µL of the diluted bacterial cultures) were aseptically transferred to the wells of 96-well microtiter plates and sealed with optically clear sealing tape (Sarstedt) to prevent evaporation. Bacterial growth rate was determined by measuring the optical density (λ = 600 nm) every 0.5 h for 12 h in an Epoch2 Microplate Spectrophotometer (BioTek). The experiment was repeated once, the generation time was calculated as above, and the results were averaged.
Phenotypes of phage-resistant D. solani Tn5 mutants analyzed using BIOLOG Phenotypic Microarrays
The metabolism of phage-resistant D. solani mutants was analyzed in GEN III and EcoPlate microplates in a BIOLOG phenotypic microarray system (Biolog Inc.) as described before59. Inoculum was grown on TSA plates for 24 h at 28 °C, and then suspended in inoculation fluid (IF-A) (GENIII) or in 10 mM phosphate buffer pH 7.4 (EcoPlate) using a sterile cotton swab (Bionovo). The turbidity of the bacterial suspensions was adjusted to ca. 90% T with a spectrophotometer [A = log(%T)]. Bacterial suspensions (100 µl) were inoculated into duplicate wells of a 96-well microplate. The wells were then examined for a color change as evidence of growth and resource consumption. Color development was quantified using an Epoch2 microplate spectrophotometer (BioTek) equipped with a λ = 595-nm wavelength filter. Plates inoculated with the wild-type D. solani strain were used as controls.
Phenotypes of phage-resistant D. solani Tn5 mutants
ΦD5-resistant D. solani mutants were screened for phenotypic features, putatively important for their interaction with plant tissues and/or environmental fitness, including swimming and swarming motility106, biofilm formation107, the ability to grow on TSA medium supplemented with 5% NaCl108, production of enzymes: pectinolytic enzymes109, cellulases110, proteases111 and siderophores112.
Phage‐resistant mutants were also interrogated by whole‐cell MALDI‐TOF MS spectral analysis as previously described59. Briefly, WT and mutants were grown on TSA at 28 °C for 24 h and then sampled with a loop and placed onto a MALDI plate. A mixture 1:1 (v/v) of ferulic acid (FA, 10 mg/ml in 33% acetonitrile, 13% formic acid, water) and dihydroxybenzoic acid (DHB, 10 mg/ml in 50% acetonitrile in water and 0.1% trifluoroacetic acid) was used as a matrix. 1 μL of matrix solution was used to overlay a bacterial spot, and the plate was then left to crystallize at room temperature for 15 min. Protein mass fingerprints were then obtained using a 5800 MALDI-TOF/TOF mass spectrometer (AB Sciex, Framingham, MA, USA), with detection in the linear middle mass in the range from 5000 to 20 000 Da, positive ion mode for a total of 1000 laser shots with a 1 kHz OptiBeam laser (YAG, 349 nm)59. The MS spectra obtained were examined with Data Explorer Software (AB Sciex). All MALDI‐TOF MS spectra reported were averages of six replicated measurements (2 independent measurements of each of 3 technical repetitions) for each strain analyzed as described before59.
Measurement of morphological features of cells and colonies
The colony morphology of phage-resistant mutants was analyzed using a Leica MZ10 F stereomicroscope with 10 × and 40 × magnifications coupled to a Leica DFC450C camera (Leica) as previously described59,92.
The cell morphology of phage resistant mutants was evaluated using transmission electron microscopy (TEM) as previously described103. TEM analyses were performed at the Laboratory of Electron Microscopy (Faculty of Biology, University of Gdansk, Poland). Bacteria were adsorbed onto carbon-coated grids (GF Microsystems), directly stained with 1.5% uranyl acetate (Sigma-Aldrich), and visualized with an electron microscope (Tecnai Spirit BioTWIN, FEI) as described previously103. At least ten images of each mutant and the wild-type strain were taken to estimate cell diameter.
WT and phage-resistant mutant cells were fixed for Scanning Electron Microscopic analyses using a standard procedure113. Briefly, bacterial cells were fixed with 4% glutaraldehyde in 0.1 M phosphate buffer, pH 7.0, treated with OsO4, dehydrated in a graded series of acetone concentrations (15%, 30%, 50%, 70% and twice 100%), followed by drying the specimens using silica gel for 24 h, The samples were then gold-sputtered using a K550X sputter coater (Quorum Technologies) and visualized with a Vega 3 Scanning Electron Microscope (Tescan)113.
Detailed imaging of the surface of selected strain cells was done using atomic force microscopy (AFM) as previously described113. The measurements were taken in Peak-Force Quantitative Nanomechanical Mapping mode using a NanoScope V AFM (Bruker, Vecco Instruments Inc., Billerica, MA, USA) equipped with NanoScope 8.15 software. The force constant of the NSG01 probe (NT-MDT Spectrum Instruments, Russia) was in the range 1.45–15.1 N/m. Three fields of 3 µm × 3 µm were scanned for each sample. The roughness was read from 10 areas of 100 nm × 100 nm from each image, and the arithmetic mean was calculated from the obtained results. Images were analyzed with Nanoscope Analysis version 1.4.
Autoaggregation of Tn5 mutants
The autoaggregation of strains was assessed as the degree of accelerated sedimentation and phase separation of aqueous bacterial suspensions114. Phage-resistant mutants were assessed for the speed of autoaggregation (sedimentation) as described in115,116. Briefly, WT (control) and phage-resistant mutants were grown in 10 ml TSB at 28 °C for 24 h with shaking and then aliquoted (1 ml) transferred to sterile cuvettes (Eppendorf), and the optical density (OD600) was measured initially (time = 0) and again after cuvettes (covered with parafilm) were incubated for 24 h at 28 °C without shaking. Two replicates were done per each bacterial strain and the experiment was repeated once. The results from both repetitions were averaged for analysis. Percentage aggregation (sedimentation) was measured as follow: %A = 1-(OD60024h/OD600 0 h), where: %A—percentage of aggregation (sedimentation), OD6000h—OD of bacterial culture at time 0 h, OD60024h—OD of bacterial culture at time 24 h115.
Permeability of cell outer membrane
Permeability of cellular outer membranes was assessed by the degree of cell lysis observed following exposure of bacteria to sodium dodecyl sulphate (SDS)117. Briefly, cells were grown in TSB (WT strain) or in TSB supplemented with 50 μg mL−1 neomycin (Tn5 mutants) for 16 h at 28 °C with shaking. After incubation 2 ml of culture was washed two times with PBS pH 7.2 (Sigma-Aldrich) and resuspended in 2 ml of PBS to an OD600 = 0.1(ca. 108 CFU mL−1). Aliquots (180 µL) were then transferred to wells of a 96-well plate (Greiner Bio-One) and either 20 µL of sterile water (control) or 10% SDS (Sigma-Aldrich) was then added. Cell lysis was monitored at room temperature via a decrease of optical density (OD) of the bacterial culture measured every minute for 10 min using an Epoch2 Microplate Spectrophotometer (BioTek). Two replicates were done for each bacterial strain and the entire experiment was repeated once. The results were averaged for analysis.
Antibiotic susceptibility of Tn5 mutants
The antibiotic susceptibility of D. solani mutants was determined with a disc diffusion method as previously described59,118. Antibiotic discs (BD BBL—Sensi-Disc antimicrobial test discs) containing chloramphenicol (30 µg), gentamicin (10 µg), tigecycline (15 µg), doxycycline (30 µg), sulfametoxazol/trimetropin (23,75/1.25 µg), ciprofloxacin (5 µg), ceftaroline (5 µg), imipenem (10 µg), piperacillin/tazobactam (30/6 µg), cefuroksym/ceftaroline (30/5 µg), cefuroxime (30 µg), aztreonam (30 µg), ampicillin (10 µg), ampicillin/sulbactam (10/10 µg), colistin (10 µg), fosfomycin (200 µg) were applied to bacterial seeded plates of Mueller–Hinton (MH medium, BD) supplemented with 1.5% agar (Oxoid). Inoculum of phage-resistant mutants grown for 16 h in TSB supplemented with neomycin (50 μg mL−1) or WT grown in TSB at 28 °C with shaking (120 rpm). The plates were inoculated using a sterile cotton swab soaked in a bacterial suspension and the antibiotic discs were placed on the agar surface in such a way to ensure a minimum distance between each disc of ca. 1.5—2 cm. Plates were incubated at 28 °C for 24 h and examined for the presence of a clear halo surrounding a given disc. The experiment was repeated once.
Isolation and visualization of lipopolysaccharide (LPS) from wild-type D. solani strain IPO 2222 and phage-resistant D. solani Tn5 mutants
Lipopolysaccharides (LPS) of bacterial strains were isolated using a Lipopolysaccharide Extraction Kit (Abcam, Symbios, Gdansk, Poland) with a modified protocol59. Lipopolysaccharides were separated using 4–20% sodium dodecyl sulfate‐polyacrylamide gradient gel Mini‐PROTEAN® TGX™ Precast Protein Gel, BioRad, Hercules, USA) electrophoresis (SDS‐PAGE) according to119 and visualized with silver staining as described before120.
Population dynamics of D. solani phage-resistant mutants on adaxial leaf surfaces of S. tuberosum
Potato plants (S. tuberosum) cultivar Kondor, cultivated as described earlier47, were grown in a growth chamber at 22 °C and 16/8 h (day/night) photoperiod (white cool fluorescent light, Philips, TLD 58 W/84o, 30–35 μmol m−2 s−1) and 80% relative humidity (RH). Leaves (third to sixth from the shoot tip) were detached from the plants and inoculated with bacteria as described earlier121. Briefly, bacterial suspensions (2 ml) adjusted to 106 CFU mL−1 in sterile Ringer’s buffer (Merck) were sprayed onto the adaxial surface of each detached leaf using a manual sprayer. The moisture on sprayed leaves were then allowed to dry in a laminar flow hood and placed, adaxial side up, on 0.5% water agar (Oxoid) supplemented with 200 µg mL−1 cycloheximide (Sigma-Aldrich), to prevent fungal growth. Sterile Ringer’s buffer was used instead of bacterial suspensions as a control. The detached, inoculated leaves were incubated on the agar plates in the growth chamber under the same conditions used to grow the plants. Samples were collected immediately after placement in the growth chamber and after 14 days and assayed for the presence of viable D. solani. At each sample time 5 randomly chosen leaves were collected for a given strain and shaken in 10 ml of Ringer’s buffer in 50 ml Falcon tubes at 50 rpm at room temperature for 30 min to remove bacterial cells from the surfaces of the leaves. The leaf washings were then serially diluted in Ringer’s buffer and 100 μl of appropriate dilutions plated in duplicate on CVP supplemented with 200 μg ml−1 cycloheximide (Sigma) or on CVP containing 50 μg ml−1 neomycin and 200 μg ml−1 cycloheximide as appropriate. The inoculated plates were incubated at 28 °C and cavity-forming D. solani colonies enumerated. The experiment was replicated once, and the results were averaged.
The ability of selected phage-resistant mutants to cause maceration of potato tubers
Potato tubers (5 replicate tubers per bacterial strain) of cv. Bryza purchased locally in Gdansk, Poland and selected for their uniform size (diameter of ca. 5–6 cm and weight of ca. 50 -70 g) were inoculated with a given strain and assessed for subsequent disease symptoms, using a whole tuber injection method (stab inoculation), as described before103,106. Bacterial suspension (ca. 108 CFU mL−1) (100 µl per strain/site) was delivered to potato tuber by stab inoculation into tuber pith using a 200 µl pipette tip. Wild‐type D. solani was used as a positive control, and negative control plants were inoculated with sterile demineralized water. The experiment was repeated once.
Virulence of D. solani Tn5 mutants in potato plants
Replicated experiments of plants grown in a growth chamber were performed in November and December 2021 using the previously developed protocol47. Certified potato tubers (cv. Kondor) were acquired from the Plant Breeding and Acclimatization Institute—National Research Institute, Bonin, Poland. S. tuberosum potato plants were cultivated in a growth chamber at 22 °C and 16/8 h (day/night) photoperiod (white cool fluorescent light, Philips, TLD 58 W/84o, 30–35 μmol m−2 s−1) and 80% relative humidity (RH) as described earlier47. After two weeks of cultivation, rooted plants with a height of ca. 10–15 cm were transferred to 1 L pots and cultivated in potting soil for 2 additional weeks under similar conditions. Potato plants (5 plants per replication) were inoculated with bacterial strains by the application of 50 ml of bacterial suspensions (108 CFU mL−1) in sterile 1/4 Ringer’s buffer) directly to the soil surrounding stems 1 h after plants had been well watered to ensure uniformly moist soil. As a negative control, soil was treated with only sterile Ringer’s buffer (50 ml per plant). Pots were then randomized within a growth chamber with 5 blocks of 15 pots per treatment. Plants were visually inspected daily for the development of disease symptoms consisting of chlorosis, black rotting of the stem, haulm wilting, and the death of the plants. Plants were sampled 14 days post-inoculation by excising ca. 2 cm long stem segments, located ca. 5 cm above ground level, and pooled per analyzed plant59. The stem fragments were surface-sterilized as described before47. The presence of D. solani cells inside potato stems was determined by plating stem macerates on CVP supplemented with 200 μg mL−1 cycloheximide (WT strain) or CVP supplemented with 50 μg mL−1 neomycin and 200 μg mL−1 cycloheximide (Tn5 mutants) and counting the resulting bacterial colonies. The experiment was replicated once, and the results were averaged for analysis.
Competition of bacterial strains on potato tubers
Generation of fluorescently-tagged strains
Plasmids pPROBE-AT-gfp122 and pRZ-T3-dsred123, used previously for stable tagging of D. solani cells with fluorescent proteins124,125, were used to generate GFP- and dsRed-tagged variants of phage-resistant D. solani mutants and/or WT strain, respectively. Plasmids pPROBE-AT-gfp and pRZ-T3-dsred were introduced into bacterial cells by electroporation as described in125. After electroporation, bacterial cultures (100 µL) were plated on TSA plates containing 100 µg mL−1 ampicillin or 40 µg mL−1 tetracycline as appropriate and incubated for 24–48 h at 28 °C. The GFP and dsRed-tagged mutant strains were grown on TSA supplemented with neomycin (50 µg mL−1) and additionally supplemented with ampicillin (100 µg mL−1) for selection of GFP-tagged strains or tetracycline (40 µg mL−1) for selection of DsRed-tagged D. solani strains unless stated otherwise. About 20 to 50 transformants were recovered per strain obtained per transformation event. One bacterial colony expressing a high fluorescent signal was collected and used for further analysis in each case. In addition, the growth rate of fluorescent protein-tagged strains was evaluated as described above.
Co-inoculation of potato tubers with fluorescently tagged WT and ΦD5-resistant mutants
To determine whether phage-resistant mutants were altered in their fitness during infection of potato tubers compared to the WT strain, two independent tuber co-inoculation experiments were conducted. In the first experiment, a GFP-tagged WT (IPO 2254)125 strain together with DsRed-tagged phage-resistant Tn5 D. solani mutants were used, whereas in the second experiment, DsRed-tagged WT and GFP-tagged phage-resistant mutants were employed. The tagged WT strain was grown in TSB supplemented with ampicillin (100 µg mL−1: GFP-tagged) or tetracycline (40 µg mL−1: DsRed-tagged), respectively for 16 h at 28 °C with shaking (200 rpm). Phage-resistant Tn5 mutants tagged either with GFP or DsRed were grown under the same conditions, but the medium was additionally supplemented with neomycin (Sigma-Aldrich) to a final concentration of 50 µg mL−1. Bacterial cultures were collected by centrifugation (6000 × RCF, 5 min.), washed twice with sterile 1/4 Ringer’s buffer and resuspended in sterile Ringer’s buffer to a density of ca. 108 CFU mL−1. Potato tubers of cultivar Bryza (5 replicate tubers/strain), selected for their uniform size (diameter of ca. 5–6 cm and weight of ca. 50 -70 g) that were obtained locally in Gdansk, Poland (and shown to be free of Pectobacterium spp. and Dickeya spp. as described earlier126) were rinsed with running tap water to remove soil particles, surface-sterilized for 20 min in 5% commercial bleach solution in water, and washed twice for 1 min with demineralized, sterile water. Surface-sterilized tubers were then dried in a laminar flow hood. Tubers were injected with bacterial strains as described earlier103,106. A volume of 100 µl of bacterial culture was delivered to each tuber by stab inoculation of 100 µl bacterial suspension with a pipette tip as above. Treatments included: (i): 106 CFU mL−1 of WT strain, (ii): 5 × 105 CFU mL−1 of WT together with 5 × 105 CFU mL−1 of a phage-resistant mutant or (iii): 106 CFU mL−1 of a phage-resistant mutant alone. Inoculated tubers were kept in humid boxes (80–90% relative humidity) at 28 °C for 72 h to allow bacteria to rot potato tissue. After incubation, ca. 2 g of rotted potato tissue was collected from each tuber and resuspended in 2 ml Ringer’s buffer/g of tissue126. To protect bacterial cells from oxidative stress, the Ringer’s buffer was supplemented with the anti-oxidant, 0.02% diethyldithiocarbamic acid (DIECA, Sigma-Aldrich). Aliquots (100 µL of appropriate dilutions of tissue macerates were mixed with 300 µL of PT medium109 prewarmed to 45–50 °C and containing 200 µg mL-1 cycloheximide, 50 µg mL-1 neomycin and either 150 µg mL-1 ampicillin (for selection of the GFP-tagged strains) or 40 µg mL-1 tetracycline (for selection of the DsRed-tagged strains) in wells of a 48-well plate (Greiner Bio-One). Plates containing solidified medium were incubated for 24 to 48 h at 28 °C. Wells were inspected for the presence of GFP- and DsRed-fluorescent bacterial cells using an epifluorescence stereomicroscope (Leica MZ10 F and Leica DFC450C camera system)127 and GFP-positive and DsRed-positive colonies were counted. The experiment was replicated once, and the results were averaged for analysis.
Statistical analyses
Statistical analyzes were performed as previously described59. Briefly, bacterial colony counts were transformed as log(x + 1) to achieve normality. Each time, the treatments were analyzed matching the experimental pattern in which two replicated experiments were done per each treatment of replicated plants/tubers/leaves. The Shapiro–Wilk test (p < 0.05)128 was used for those samples where the distribution of bacterial population size was described by a normal distribution. For those samples not normally distributed (e.g. control vs. treatment or treatment vs. treatment), the Welch’s T‐test was applied129. The variance homogeneity was validated using the Fisher–Snedecor test130. Pair‐wise differences were evaluated using a two‐tailed Student’s t‐test131. The linear model was a complete block design with replicates as individual blocks132. The main effects observed were analyzed for the impact of time and treatment type and a two‐way interaction between time and treatment type.
Data availability
Data generated or analyzed during this study are included in this published article (including its Supplementary Information files). In addition, the raw genome sequences of the D. solani Tn5 phage-resistant mutants are deposited at Zenodo (www.zenodo.org) under https://doi.org/10.5281/zenodo.6587967, protein mass fingerprints (MALDI-TOF/TOF) are deposited at Zenodo under https://doi.org/10.5281/zenodo.6588037 and all TEM, SEM and AFM raw photos and accompanying data are deposited at Zenodo under https://doi.org/10.5281/zenodo.6587895.
References
Campbell, A. The future of bacteriophage biology. Nat. Rev. Genet. 4, 471–477. https://doi.org/10.1038/nrg1089 (2003).
Cheetham, B. F. & Katz, M. E. A role for bacteriophages in the evolution and transfer of bacterial virulence determinants. Mol. Microbiol. 18, 201–208. https://doi.org/10.1111/j.1365-2958.1995.mmi_18020201.x (1995).
Mann, N. H. The third age of phage. PLoS Biol. 3, e182. https://doi.org/10.1371/journal.pbio.0030182 (2005).
Thurber, R. V. Current insights into phage biodiversity and biogeography. Curr. Opin. Microbiol. 12, 582–587. https://doi.org/10.1016/j.mib.2009.08.008 (2009).
Buckling, A. & Rainey, P. B. Antagonistic coevolution between a bacterium and a bacteriophage. Proc. Biol. Sci. 269, 931–936. https://doi.org/10.1098/rspb.2001.1945 (2002).
Dennehy, J. J. What can phages tell us about host-pathogen coevolution?. Int. J. Evol. Biol. 2012, 396165. https://doi.org/10.1155/2012/396165 (2012).
Forde, S. E., Thompson, J. N., Holt, R. D. & Bohannan, B. J. Coevolution drives temporal changes in fitness and diversity across environments in a bacteria-bacteriophage interaction. Evolution 62, 1830–1839. https://doi.org/10.1111/j.1558-5646.2008.00411.x (2008).
Suttle, C. A. The significance of viruses to mortality in aquatic microbial communities. Microb. Ecol. 28, 237–243. https://doi.org/10.1007/BF00166813 (1994).
Duffy, M. A. & Forde, S. E. Ecological feedbacks and the evolution of resistance. J. Anim. Ecol. 78, 1106–1112. https://doi.org/10.1111/j.1365-2656.2009.01568.x (2009).
Koskella, B. & Brockhurst, M. A. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 38, 916–931. https://doi.org/10.1111/1574-6976.12072 (2014).
Chibani-Chennoufi, S., Bruttin, A., Dillmann, M. L. & Brussow, H. Phage-host interaction: an ecological perspective. J. Bacteriol. 186, 3677–3686. https://doi.org/10.1128/JB.186.12.3677-3686.2004 (2004).
Waterbury, J. B. & Valois, F. W. Resistance to co-occurring phages enables marine synechococcus communities to coexist with cyanophages abundant in seawater. Appl. Environ. Microbiol. 59, 3393–3399. https://doi.org/10.1128/aem.59.10.3393-3399.1993 (1993).
Koskella, B. & Parr, N. The evolution of bacterial resistance against bacteriophages in the horse chestnut phyllosphere is general across both space and time. Phil. Trans. R. Soc. B 370, 20140297 (2015).
Hantula, J., Kurki, A., Vuoriranta, P. & Bamford, D. H. Ecology of bacteriophages infecting activated sludge bacteria. Appl. Environ. Microbiol. 57, 2147–2151. https://doi.org/10.1128/aem.57.8.2147-2151.1991 (1991).
Fernandez, L., Gutierrez, D., Rodriguez, A. & Garcia, P. Application of bacteriophages in the agro-food sector: a long way toward approval. Front. Cell Infect. Microbiol. 8, 296. https://doi.org/10.3389/fcimb.2018.00296 (2018).
Jones, J. B. et al. Bacteriophages for plant disease control. Annu Rev. Phytopathol. 45, 245–262. https://doi.org/10.1146/annurev.phyto.45.062806.094411 (2007).
Bradde, S., Vucelja, M., Tesileanu, T. & Balasubramanian, V. Dynamics of adaptive immunity against phage in bacterial populations. PLoS Comput. Biol. 13, e1005486. https://doi.org/10.1371/journal.pcbi.1005486 (2017).
Naureen, Z. et al. Bacteriophages presence in nature and their role in the natural selection of bacterial populations. Acta Biomed. 91, e2020024. https://doi.org/10.23750/abm.v91i13-S.10819 (2020).
Stern, A. & Sorek, R. The phage-host arms race: shaping the evolution of microbes. BioEssays 33, 43–51. https://doi.org/10.1002/bies.201000071 (2011).
Burmeister, A. R. & Turner, P. E. Trading-off and trading-up in the world of bacteria-phage evolution. Curr. Biol. 30, R1120–R1124. https://doi.org/10.1016/j.cub.2020.07.036 (2020).
Koderi Valappil, S. et al. Survival comes at a cost: a coevolution of phage and its host leads to phage resistance and antibiotic sensitivity of Pseudomonas aeruginosa multidrug resistant strains. Front. Microbiol. 12, 783722. https://doi.org/10.3389/fmicb.2021.783722 (2021).
Labrie, S. J., Samson, J. E. & Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317–327. https://doi.org/10.1038/nrmicro2315 (2010).
Lythgoe, K. A. & Chao, L. Mechanisms of coexistence of a bacteria and a bacteriophage in a spatially homogeneous environment. Ecol. Lett. 6, 326–334. https://doi.org/10.1046/j.1461-0248.2003.00433.x (2003).
Mizoguchi, K. et al. Coevolution of bacteriophage PP01 and Escherichia coli O157:H7 in continuous culture. Appl. Environ. Microbiol. 69, 170–176. https://doi.org/10.1128/AEM.69.1.170-176.2003 (2003).
Lennon, J. T., Khatana, S. A., Marston, M. F. & Martiny, J. B. Is there a cost of virus resistance in marine cyanobacteria?. ISME J. 1, 300–312. https://doi.org/10.1038/ismej.2007.37 (2007).
Vale, P. F. et al. Costs of CRISPR-Cas-mediated resistance in Streptococcus thermophilus. Proc. Biol. Sci. 282, 20151270. https://doi.org/10.1098/rspb.2015.1270 (2015).
Keen, E. C. Tradeoffs in bacteriophage life histories. Bacteriophage 4, e28365. https://doi.org/10.4161/bact.28365 (2014).
Abedon, S. T. Bacterial ‘immunity’ against bacteriophages. Bacteriophage 2, 50–54 (2012).
Charkowski, A. O. The changing face of bacterial soft-rot diseases. Annu. Rev. Phytopathol. 56, 269–288. https://doi.org/10.1146/annurev-phyto-080417-045906 (2018).
Mansfield, J. et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 13, 614–629. https://doi.org/10.1111/j.1364-3703.2012.00804.x (2012).
Perombelon, M. C. M. Potato diseases caused by soft rot Erwinias: an overview of pathogenesis. Plant. Pathol. 51, 1–12. https://doi.org/10.1046/j.0032-0862.2001.Shorttitle.doc.x (2002).
Perombelon, M. C. M. & Kelman, A. Ecology of the soft rot Erwinias. Annu. Rev. Phytopathol. 18, 361–387 (1980).
Rossmann, S., Dees, M. W., Perminow, J., Meadow, R. & Brurberg, M. B. Soft Rot Enterobacteriaceae are carried by a large range of insect species in potato fields. Appl. Environ. Microbiol. 84, 1. https://doi.org/10.1128/AEM.00281-18 (2018).
Fikowicz-Krosko, J., Wszalek-Rozek, K., Smolarska, A. & Czajkowski, R. First report of isolation of soft rot Pectobacterium carotovorum subsp carotovorum from symptomless bittersweet nightshade occuing in rural area of Poland. J. Plant Pathol. 99, 1 (2017).
Batinovic, S. et al. Bacteriophages in natural and artificial environments. Pathogens 8, 100. https://doi.org/10.3390/pathogens8030100 (2019).
Wright, R. C. T., Friman, V. P., Smith, M. C. M. & Brockhurst, M. A. Resistance evolution against phage combinations depends on the timing and order of exposure. mBio 10, e01652–01619, https://doi.org/10.1128/mBio.01652-19 (2019).
Borin, J. M., Avrani, S., Barrick, J. E., Petrie, K. L. & Meyer, J. R. Coevolutionary phage training leads to greater bacterial suppression and delays the evolution of phage resistance. Proc. Natl. Acad. Sci. USA 118, e2104592118. https://doi.org/10.1073/pnas.2104592118 (2021).
van der Wolf, J. M. et al. Dickeya solani sp. nov., a pectinolytic plant-pathogenic bacterium isolated from potato (Solanum tuberosum). Int J Syst Evol Microbiol 64, 768–774, https://doi.org/10.1099/ijs.0.052944-0 (2014).
Toth, I. K. et al. Dickeya species: an emerging problem for potato production in Europe. Plant. Pathol. 60, 385–399. https://doi.org/10.1111/j.1365-3059.2011.02427.x (2011).
Czajkowski, R., van Veen, J. A. & van der Wolf, J. M. New biovar 3 Dickeya spp. strain (syn. Erwinia chrysanthemi) as a causative agent of blackleg in seed potato in Europe. Phytopathology 99, S27-S27 (2009).
Tsror Lahkim, L. et al. Characterization of Dickeya strains isolated from potato grown under hot-climate conditions. Plant Pathology 62, 1097–1105, doi:https://doi.org/10.1111/ppa.12030 (2013).
Tsror, L. et al. First report of potato blackleg caused by a biovar 3 Dickeya sp. in Georgia. New Disease Reports 23, 1 (2011).
Ozturk, M. & Aksoy, H. M. First report of Dickeya solani associated with potato blackleg and soft rot in Turkey. J. Plant Pathol. 99, 298 (2017).
Cardoza, Y. F., Duarte, V. & Lopes, C. A. First report of blackleg of potato caused by Dickeya solani in Brazil. Plant. Dis. 101, 243–243. https://doi.org/10.1094/pdis-07-16-1045-pdn (2017).
Khayi, S., Blin, P., Chong, T. M., Chan, K. G. & Faure, D. Complete genome anatomy of the emerging potato pathogen Dickeya solani type strain IPO 2222(T). Stand. Genom. Sci. 11, 87. https://doi.org/10.1186/s40793-016-0208-0 (2016).
Czajkowski, R., Ozymko, Z. & Lojkowska, E. Isolation and characterization of novel soilborne lytic bacteriophages infecting Dickeya spp. biovar 3 (‘D. solani’). Plant Pathol. 63, 758–772, doi:https://doi.org/10.1111/ppa.12157 (2014).
Czajkowski, R., Smolarska, A. & Ozymko, Z. The viability of lytic bacteriophage PhiD5 in potato-associated environments and its effect on Dickeya solani in potato (Solanum tuberosum L.) plants. PLoS ONE 12, e0183200, doi:https://doi.org/10.1371/journal.pone.0183200 (2017).
Adriaenssens, E. M. et al. A suggested new bacteriophage genus: “Viunalikevirus”. Arch Virol 157, 2035–2046. https://doi.org/10.1007/s00705-012-1360-5 (2012).
Adriaenssens, E. M. et al. T4-related bacteriophage LIMEstone isolates for the control of soft rot on potato caused by “Dickeya solani”. PLoS ONE 7, e33227. https://doi.org/10.1371/journal.pone.0033227 (2012).
Petrzik, K., Vacek, J., Brazdova, S., Sevcik, R. & Koloniuk, I. Diversity of limestone bacteriophages infecting Dickeya solani isolated in the Czech Republic. Arch Virol 166, 1171–1175. https://doi.org/10.1007/s00705-020-04926-7 (2021).
Ranjan, M. et al. Genomic diversity and organization of complex polysaccharide biosynthesis clusters in the genus Dickeya. PLoS ONE 16, e0245727. https://doi.org/10.1371/journal.pone.0245727 (2021).
Pedron, J., Chapelle, E., Alunni, B. & Van Gijsegem, F. Transcriptome analysis of the Dickeya dadantii PecS regulon during the early stages of interaction with Arabidopsis thaliana. Mol Plant Pathol 19, 647–663. https://doi.org/10.1111/mpp.12549 (2018).
Czajkowski, R. Bacteriophages of Soft Rot Enterobacteriaceae-a minireview. FEMS Microbiol. Lett. 363, fnv230, doi:https://doi.org/10.1093/femsle/fnv230 (2016).
Toth, I. K. et al. in Plant Diseases Caused by Dickeya and Pectobacterium Species (eds Frédérique Van Gijsegem, Jan M. van der Wolf, & Ian K. Toth) Ch. Chapter 2, 13–37 (Springer International Publishing, 2021).
Holt, K. E., Lassalle, F., Wyres, K. L., Wick, R. & Mostowy, R. J. Diversity and evolution of surface polysaccharide synthesis loci in Enterobacteriales. ISME J. 14, 1713–1730. https://doi.org/10.1038/s41396-020-0628-0 (2020).
Schnaitman, C. A. & Klena, J. D. Genetics of lipopolysaccharide biosynthesis in enteric bacteria. Microbiol. Rev. 57, 655–682. https://doi.org/10.1128/mr.57.3.655-682.1993 (1993).
Mangalea, M. R. & Duerkop, B. A. Fitness trade-offs resulting from bacteriophage resistance potentiate synergistic antibacterial strategies. Infect. Immun. 88, e00926-e1919. https://doi.org/10.1128/IAI.00926-19 (2020).
Bohannan, B. J. M. & Lenski, R. E. Linking genetic change to community evolution: insights from studies of bacteria and bacteriophage. Ecol. Lett. 3, 362–377. https://doi.org/10.1046/j.1461-0248.2000.00161.x (2000).
Bartnik, P., Jafra, S., Narajczyk, M., Czaplewska, P. & Czajkowski, R. Pectobacterium parmentieri SCC 3193 mutants with altered synthesis of cell surface polysaccharides are resistant to N4-like lytic bacteriophage phiA38 (vB_Ppp_A38) but express decreased virulence in potato (Solanum tuberosum L.) Plants. Int J Mol Sci 22, 7346, doi:https://doi.org/10.3390/ijms22147346 (2021).
Evans, T. J., Ind, A., Komitopoulou, E. & Salmond, G. P. C. Phage-selected lipopolysaccharide mutants of Pectobacterium atrosepticum exhibit different impacts on virulence. J. Appl. Microbiol. 109, 505–514. https://doi.org/10.1111/j.1365-2672.2010.04669.x (2010).
Lukianova, A. A. et al. Morphologically Different Pectobacterium brasiliense bacteriophages PP99 and PP101: deacetylation of O-polysaccharide by the tail spike protein of phage PP99 accompanies the Infection. Front. Microbiol. 10, 3147. https://doi.org/10.3389/fmicb.2019.03147 (2019).
Kim, H. et al. Colanic acid is a novel phage receptor of Pectobacterium carotovorum subsp. carotovorum phage POP72. Front. Microbiol. 10, 143, https://doi.org/10.3389/fmicb.2019.00143 (2019).
Costerton, J. W., Irvin, R. T. & Cheng, K. J. The role of bacterial surface structures in pathogenesis. Crit. Rev. Microbiol. 8, 303–338. https://doi.org/10.3109/10408418109085082 (1981).
Beveridge, T. J. & Graham, L. L. Surface layers of bacteria. Microbiol. Rev. 55, 684–705. https://doi.org/10.1128/mr.55.4.684-705.1991 (1991).
D’Haeze, W. & Holsters, M. Surface polysaccharides enable bacteria to evade plant immunity. Trends Microbiol. 12, 555–561. https://doi.org/10.1016/j.tim.2004.10.009 (2004).
Li, J. & Wang, N. The gpsX gene encoding a glycosyltransferase is important for polysaccharide production and required for full virulence in Xanthomonas citri subsp. citri. BMC Microbiol. 12, 31 (2012).
Santaella, C., Schue, M., Berge, O., Heulin, T. & Achouak, W. The exopolysaccharide of Rhizobium sp. YAS34 is not necessary for biofilm formation on Arabidopsis thaliana and Brassica napus roots but contributes to root colonization. Environ. Microbiol. 10, 2150–2163, doi:https://doi.org/10.1111/j.1462-2920.2008.01650.x (2008).
Morona, J. K., Miller, D. C., Morona, R. & Paton, J. C. The effect that mutations in the conserved capsular polysaccharide biosynthesis genes cpsA, cpsB, and cpsD have on virulence of Streptococcus pneumoniae. J. Infect. Dis. 189, 1905–1913. https://doi.org/10.1086/383352 (2004).
Lawlor, M. S., Handley, S. A. & Miller, V. L. Comparison of the host responses to wild-type and cpsB mutant Klebsiella pneumoniae infections. Infect. Immun. 74, 5402–5407. https://doi.org/10.1128/IAI.00244-06 (2006).
Geider, K. et al. in Advances in Molecular Genetics of Plant-Microbe Interactions Vol. 1 Current Plant Science and Biotechnology in Agriculture (eds Hauke Hennecke & Desh Pal S. Verma) Ch. Chapter 14, 90–93 (Springer Netherlands, 1991).
Mohamed, K. H. et al. Deciphering the dual effect of lipopolysaccharides from plant pathogenic Pectobacterium. Plant Signal Behav. 10, e1000160. https://doi.org/10.1080/15592324.2014.1000160 (2015).
Katzen, F. et al. Xanthomonas campestris pv. campestris gum mutants: effects on xanthan biosynthesis and plant virulence. J. Bacteriol. 180, 1607–1617. https://doi.org/10.1128/JB.180.7.1607-1617.1998 (1998).
Whitfield, C., Wear, S. S. & Sande, C. Assembly of bacterial capsular polysaccharides and exopolysaccharides. Annu. Rev. Microbiol. 74, 521–543. https://doi.org/10.1146/annurev-micro-011420-075607 (2020).
Ormeno-Orrillo, E., Rosenblueth, M., Luyten, E., Vanderleyden, J. & Martinez-Romero, E. Mutations in lipopolysaccharide biosynthetic genes impair maize rhizosphere and root colonization of Rhizobium tropici CIAT899. Environ. Microbiol. 10, 1271–1284. https://doi.org/10.1111/j.1462-2920.2007.01541.x (2008).
Touze, T., Goude, R., Georgeault, S., Blanco, C. & Bonnassie, S. Erwinia chrysanthemi O antigen is required for betaine osmoprotection in high-salt media. J. Bacteriol. 186, 5547–5550. https://doi.org/10.1128/JB.186.16.5547-5550.2004 (2004).
Bowden, M. G. & Kaplan, H. B. The Myxococcus xanthus lipopolysaccharide O-antigen is required for social motility and multicellular development. Mol. Microbiol. 30, 275–284. https://doi.org/10.1046/j.1365-2958.1998.01060.x (1998).
Andrianopoulos, K., Wang, L. & Reeves, P. R. Identification of the fucose synthetase gene in the colanic acid gene cluster of Escherichia coli K-12. J. Bacteriol. 180, 998–1001. https://doi.org/10.1128/JB.180.4.998-1001.1998 (1998).
Islam, R., Brown, S., Taheri, A. & Dumenyo, C. K. The gene encoding NAD-dependent epimerase/dehydratase, wcaG, affects cell surface properties, virulence, and extracellular enzyme production in the soft rot phytopathogen Pectobacterium carotovorum. Microorganisms 7, 172. https://doi.org/10.3390/microorganisms7060172 (2019).
Qimron, U., Marintcheva, B., Tabor, S. & Richardson, C. C. Genomewide screens for Escherichia coli genes affecting growth of T7 bacteriophage. Proc. Natl. Acad. Sci. USA 103, 19039–19044. https://doi.org/10.1073/pnas.0609428103 (2006).
Pagnout, C. et al. Pleiotropic effects of rfa-gene mutations on Escherichia coli envelope properties. Sci. Rep. 9, 9696. https://doi.org/10.1038/s41598-019-46100-3 (2019).
Montanaro, L. & Arciola, C. R. in Handbook of Bacterial Adhesion: Principles, Methods, and Applications (eds Yuehuei H. An & Richard J. Friedman) 331–343 (Humana Press, 2000).
Berne, C., Ellison, C. K., Ducret, A. & Brun, Y. V. Bacterial adhesion at the single-cell level. Nat. Rev. Microbiol. 16, 616–627. https://doi.org/10.1038/s41579-018-0057-5 (2018).
Brown, M. R. & Williams, P. The influence of environment on envelope properties affecting survival of bacteria in infections. Annu. Rev. Microbiol. 39, 527–556. https://doi.org/10.1146/annurev.mi.39.100185.002523 (1985).
Czajkowski, R. et al. Genome-Wide identification of Dickeya solani transcriptional units up-regulated in response to plant tissues from a crop-host Solanum tuberosum and a weed-host Solanum dulcamara. Front. Plant. Sci. 11, 580330. https://doi.org/10.3389/fpls.2020.580330 (2020).
Meaden, S. & Koskella, B. Exploring the risks of phage application in the environment. Front. Microbiol. 4, 358. https://doi.org/10.3389/fmicb.2013.00358 (2013).
Reverchon, S., Muskhelisvili, G. & Nasser, W. in Progress in Molecular Biology and Translational Science Vol. 142 (eds Michael San Francisco & Brian San Francisco) 51–92 (Academic Press, 2016).
Jiang, X. et al. Global transcriptional response of Dickeya dadantii to environmental stimuli relevant to the plant infection. Environ. Microbiol. 18, 3651–3672. https://doi.org/10.1111/1462-2920.13267 (2016).
Li, Y. et al. LPS remodeling is an evolved survival strategy for bacteria. Proc. Natl. Acad. Sci. USA 109, 8716–8721. https://doi.org/10.1073/pnas.1202908109 (2012).
Hendrick, C. A. & Sequeira, L. Lipopolysaccharide-defective mutants of the wilt pathogen Pseudomonas solanacearum. Appl. Environ. Microbiol. 48, 94–101. https://doi.org/10.1128/aem.48.1.94-101.1984 (1984).
Berry, M. C., McGhee, G. C., Zhao, Y. & Sundin, G. W. Effect of a waaL mutation on lipopolysaccharide composition, oxidative stress survival, and virulence in Erwinia amylovora. FEMS Microbiol. Lett. 291, 80–87. https://doi.org/10.1111/j.1574-6968.2008.01438.x (2009).
Czajkowski, R., Ozymko, Z., Zwirowski, S. & Lojkowska, E. Complete genome sequence of a broad-host-range lytic Dickeya spp. bacteriophage phiD5. Arch. Virol. 159, 3153–3155, doi:https://doi.org/10.1007/s00705-014-2170-8 (2014).
Lisicka, W. et al. Oxygen availability influences expression of Dickeya solani genes associated with virulence in potato (Solanum tuberosum L.) and chicory (Cichorium intybus L.). Front. Plant. Sci. 9, 374, doi:https://doi.org/10.3389/fpls.2018.00374 (2018).
Czajkowski, R., Marcisz, M. & Bartnik, P. Fast and reliable screening assay developed to preselect candidate Soft Rot Pectobacteriaceae Tn5 mutants showing resistance to bacteriophage infection. Eur. J. Plant Pathol. 155, 671–676. https://doi.org/10.1007/s10658-019-01786-z (2019).
Aziz, R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genom. 9, 75. https://doi.org/10.1186/1471-2164-9-75 (2008).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Kurowski, M. A. & Bujnicki, J. M. GeneSilico protein structure prediction meta-server. Nucl. Acids Res. 31, 3305–3307. https://doi.org/10.1093/nar/gkg557 (2003).
Altschul, S. F. & Koonin, E. V. Iterated profile searches with PSI-BLAST-a tool for discovery in protein databases. Trends Biochem. Sci. 23, 444–447. https://doi.org/10.1016/s0968-0004(98)01298-5 (1998).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Letunic, I., Yamada, T., Kanehisa, M. & Bork, P. iPath: interactive exploration of biochemical pathways and networks. Trends Biochem. Sci. 33, 101–103. https://doi.org/10.1016/j.tibs.2008.01.001 (2008).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucl. Acids Res. 47, D607–D613. https://doi.org/10.1093/nar/gky1131 (2019).
Czajkowski, R., Ozymko, Z. & Lojkowska, E. Isolation and characterization of novel soilborne lytic bacteriophages infecting Dickeya spp. biovar 3 (‘D. solani’). Plant Pathol. 63, 758–772, https://doi.org/10.1111/ppa.12157, (2014).
Shao, Y. & Wang, I. N. Bacteriophage adsorption rate and optimal lysis time. Genetics 180, 471–482. https://doi.org/10.1534/genetics.108.090100 (2008).
Czajkowski, R., Kaczyńska, N., Jafra, S., Narajczyk, M. & Lojkowska, E. Temperature-responsive genetic loci in pectinolytic plant pathogenic Dickeya solani. Plant. Pathol. 66, 584–594. https://doi.org/10.1111/ppa.12618 (2017).
Roth, V. Doubling time computing, application available from: http://www.doubling-time.com/compute.php, (2006).
Krzyzanowska, D. M. et al. Compatible mixture of bacterial antagonists developed to protect ootato tubers from soft rot caused by Pectobacterium spp. and Dickeya spp. Plant. Dis. 103, 1374–1382, doi:https://doi.org/10.1094/PDIS-10-18-1866-RE (2019).
Czajkowski, R., de Boer, W. J., van Veen, J. A. & van der Wolf, J. M. Characterization of bacterial isolates from rotting potato tuber tissue showing antagonism to Dickeya sp. biovar 3 in vitro and in planta. Plant Pathology 61, 169–182, https://doi.org/10.1111/j.1365-3059.2011.02486.x (2012).
Shao, X., Xie, Y., Zhang, Y. & Deng, X. Biofilm formation assay in Pseudomonas syringae. Biol. Protoc. 9, e3237. https://doi.org/10.21769/BioProtoc.3237 (2019).
Dickey, R. S. Erwinia chrysanthemi: a comparative study of phenotypic properties of strains from several hosts and other Erwinia species. Phytopathol. 69, 324–329 (1979).
Perombelon, M. C. M. & van Der Wolf, J. M. Methods for the detection and quantification of Erwinia carotovora subsp. atroseptica (Pectobacterium carotovorum subsp. atrosepticum) on potatoes: a laboratory manual. Scottish Crop Research Institute Annual Report 10 (2002).
Py, B., Bortoli-German, I., Haiech, J., Chippaux, M. & Barras, F. Cellulase EGZ of Erwinia chrysanthemi: structural organization and importance of His98 and Glu133 residues for catalysis. Prot. Eng. 4, 325–333. https://doi.org/10.1093/protein/4.3.325 (1991).
Ji, J. W., Hugouvieux Cotte Pattat, N. & Robert Baudouy, J. Use of Mu-Lac insertions to study the secretion of pectate lyases by Erwinia chrysanthemi. J. Gen. Microbiol. 133, 793–802 (1987).
Schwyn, B. & Neilands, J. B. Universal chemical assay for the detection and determination of siderophores. Anal Biochem. 160, 47–56. https://doi.org/10.1016/0003-2697(87)90612-9 (1987).
Fiolka, M. J. et al. Antimycobacterial action of a new glycolipid-peptide complex obtained from extracellular metabolites of Raoultella ornithinolytica. APMIS 123, 1069–1080. https://doi.org/10.1111/apm.12466 (2015).
Sorroche, F. G., Rinaudi, L. V., Zorreguieta, A. & Giordano, W. EPS II-dependent autoaggregation of Sinorhizobium meliloti planktonic cells. Curr. Microbiol. 61, 465–470. https://doi.org/10.1007/s00284-010-9639-9 (2010).
Dorken, G., Ferguson, G. P., French, C. E. & Poon, W. C. Aggregation by depletion attraction in cultures of bacteria producing exopolysaccharide. J. R. Soc. Interface 9, 3490–3502. https://doi.org/10.1098/rsif.2012.0498 (2012).
Trunk, T., Khalil, H. S. & Leo, J. C. Bacterial autoaggregation. AIMS Microbiol. 4, 140–164. https://doi.org/10.3934/microbiol.2018.1.140 (2018).
Przepiora, T. et al. The periplasmic oxidoreductase DsbA is required for virulence of the phytopathogen Dickeya solani. Int. J. Mol. Sci. 23, 697 (2022).
Bauer, A. W., Kirby, W. M. M., Sherris, J. C. & Turck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 45, 493–500 (1966).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular cloning: a laboratory manual. (1989).
Tsai, C. M. & Frasch, C. E. A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal. Biochem. 119, 115–119. https://doi.org/10.1016/0003-2697(82)90673-x (1982).
Fikowicz-Krosko, J. & Czajkowski, R. Systemic colonization and expression of disease symptoms on bittersweet nightshade (Solanum dulcamara) infected with a GFP-tagged Dickeya solani IPO2222 (IPO2254). Plant. Dis. 102, 619–627. https://doi.org/10.1094/PDIS-08-17-1147-RE (2018).
Miller, W. G., Leveau, J. H. & Lindow, S. E. Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol. Plant. Microbe Interact 13, 1243–1250. https://doi.org/10.1094/MPMI.2000.13.11.1243 (2000).
Bloemberg, G. V., Wijfjes, A. H., Lamers, G. E., Stuurman, N. & Lugtenberg, B. J. Simultaneous imaging of Pseudomonas fluorescens WCS365 populations expressing three different autofluorescent proteins in the rhizosphere: new perspectives for studying microbial communities. Mol. Plant. Microbe Interact 13, 1170–1176. https://doi.org/10.1094/MPMI.2000.13.11.1170 (2000).
Czajkowski, R. et al. Virulence of ‘Dickeya solani’ and Dickeya dianthicola biovar-1 and -7 strains on potato (Solanum tuberosum). Plant. Pathol. 62, 597–610. https://doi.org/10.1111/j.1365-3059.2012.02664.x (2013).
Czajkowski, R., de Boer, W. J., Velvis, H. & van der Wolf, J. M. Systemic colonization of potato plants by a soilborne, green fluorescent protein-tagged strain of Dickeya sp. biovar 3. Phytopathology 100, 134–142, doi:https://doi.org/10.1094/PHYTO-100-2-0134 (2010).
Czajkowski, R., Grabe, G. J. & van der Wolf, J. M. Distribution of Dickeya spp. and Pectobacterium carotovorum subsp. carotovorum in naturally infected seed potatoes. Eur. J. Plant Pathol. 125, 263–275, doi:https://doi.org/10.1007/s10658-009-9480-9 (2009).
Czajkowski, R., de Boer, W. J., van Veen, J. A. & van der Wolf, J. M. Studies on the interaction between the biocontrol agent, Serratia plymuthica A30, with blackleg causing Dickeya sp. (biovar 3) in potato (Solanum tuberosum). Plant Pathol. 61, 677–688 (2012).
Shapiro, S. S. & Wilk, M. B. An analysis of variance test for normality (complete samples). Biometrika 52, 591–600. https://doi.org/10.2307/2333709 (1965).
Welch, B. L. The generalisation of student’s problems when several different population variances are involved. Biometrika 34, 28–35. https://doi.org/10.1093/biomet/34.1-2.28 (1947).
Box, G. E. P. Non-normality and tests on variances. Biometrika 40, 318–335. https://doi.org/10.2307/2333350 (1953).
Student. The probable error of a mean. Biometrika 6, 1–25, doi:https://doi.org/10.2307/2331554 (1908).
Shieh, G. & Jan, S. L. The effectiveness of randomized complete block design. Stat. Neerl. 58, 111–124 (2004).
Acknowledgements
This research was financially supported by the National Science Center, Poland (Narodowe Centrum Nauki, Polska) via a research grant OPUS 13 (2017/25/B/NZ9/00036) to Robert Czajkowski. The authors would like to express their gratitude to Prof. Steven E. Lindow (University of California -Berkeley, Berkeley, CA, United States) for his comments on the manuscript and his editorial work. The preprint version of this manuscript has been deposited at bioRxiv—the preprint server for biology as: Bartnik et al., (2022) “Resistance of Dickeya solani strain IPO 2222 to lytic bacteriophage vB_Dsol_D5 (ΦD5) results in fitness tradeoffs for the bacterium during infection” and can be accessed via https://www.biorxiv.org/content/10.1101/2022.02.23.481671v1.
Author information
Authors and Affiliations
Contributions
Conceptualization: P.B. and R.C., Methodology: P.B., K.L., M.F., P.C., M.N., R.C.. Investigation: P.B., K.L., M.F., P.C., M.N., R.C., Writing-original draft preparation: R.C., P.C., M.F., Writing-Review & Editing: R.C., Visualization: P.B., K.L., M.F., P.C., M.N., R.C., Supervision: R.C., Funding acquisition: R.C. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bartnik, P., Lewtak, K., Fiołka, M. et al. Resistance of Dickeya solani strain IPO 2222 to lytic bacteriophage ΦD5 results in fitness tradeoffs for the bacterium during infection. Sci Rep 12, 10725 (2022). https://doi.org/10.1038/s41598-022-14956-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-14956-7
This article is cited by
-
Bacteriophages: an overview of the control strategies against phytopathogens
Egyptian Journal of Biological Pest Control (2023)
-
Bacteriophage-loaded functional nanofibers for treatment of P. aeruginosa and S. aureus wound infections
Scientific Reports (2023)
-
Spontaneous mutations in hlyD and tuf genes result in resistance of Dickeya solani IPO 2222 to phage ϕD5 but cause decreased bacterial fitness and virulence in planta
Scientific Reports (2023)
-
Taxonomic and functional profiling of Indian smokeless tobacco bacteriome uncovers several bacterial-derived risks to human health
World Journal of Microbiology and Biotechnology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
