
Abstract
Biological nitrogen fixation (BNF) is the reduction of N2 into NH3 in a group of prokaryotes by an extremely O2-sensitive protein complex called nitrogenase. Transfer of the BNF pathway directly into plants, rather than by association with microorganisms, could generate crops that are less dependent on synthetic nitrogen fertilizers and increase agricultural productivity and sustainability. In the laboratory, nitrogenase activity is commonly determined by measuring ethylene produced from the nitrogenase-dependent reduction of acetylene (ARA) using a gas chromatograph. The ARA is not well suited for analysis of large sample sets nor easily adapted to automated robotic determination of nitrogenase activities. Here, we show that a reduced sulfonated viologen derivative (S2Vred) assay can replace the ARA for simultaneous analysis of isolated nitrogenase proteins using a microplate reader. We used the S2Vred to screen a library of NifH nitrogenase components targeted to mitochondria in yeast. Two NifH proteins presented properties of great interest for engineering of nitrogen fixation in plants, namely NifM independency, to reduce the number of genes to be transferred to the eukaryotic host; and O2 resistance, to expand the half-life of NifH iron-sulfur cluster in a eukaryotic cell. This study established that NifH from Dehalococcoides ethenogenes did not require NifM for solubility, [Fe-S] cluster occupancy or functionality, and that NifH from Geobacter sulfurreducens was more resistant to O2 exposure than the other NifH proteins tested. It demonstrates that nitrogenase components with specific biochemical properties such as a wider range of O2 tolerance exist in Nature, and that their identification should be an area of focus for the engineering of nitrogen-fixing crops.
Similar content being viewed by others

Introduction
Although almost 80% of the atmosphere is composed of nitrogen gas (N2), crop productivity in modern agriculture is limited by biologically available nitrogen such as oxidized (e.g. NO3−) or reduced (e.g. NH4+) species1. Crop yield is increased using synthetic N-based fertilizers that are costly both economically and ecologically, due to the consumption of non-renewable energy resources, production of greenhouse gasses, and water and air pollution2. On the other hand, biological nitrogen fixation (BNF) is performed by selected prokaryotes (bacteria and archaea), named diazotrophs for their capacity to grow using N2 as the sole N source3. A diverse range of diazotrophs are found in Nature and can be classified according to their lifestyle as free living, symbiotic (mainly bacteria living within root nodules of legume plants, including pulse crops), and those that live in associative or endophytic relationship with other organisms. Three biotechnological approaches are currently being explored to reduce the application of N-based fertilizers to cereal crops by enhancing their access to BNF4,5. In the first strategy, bacteria naturally associated with cereals are engineered to improve their colonization ability, N2-fixing capabilities or NH3 release. In the other two strategies, the plants are instead genetically engineered to either generate new symbiotic relationships between the non-legume plant and N2-fixing bacteria, thus mimicking the legume-rhizobium natural symbiosis, or by direct transfer of the prokaryotic N2 fixation genes into the plant, to create a crop capable of fixing N2 without the requirement for symbiotic associations. Both approaches are ambitious and challenging. The new symbiotic relationship requires molecular signaling between the bacteria and plants to avoid an immune response, the formation of a nodule-like structure with a low-O2 environment and the productive exchange of nutrients between the plant and the bacteria. On the other hand, the transfer of the N2 fixation capability is complicated by the estimated number of required genes (ca. 10–20), the sensitivity of their products towards O2, the need to perform time consuming functional validations, and difficulty to troubleshoot pathway engineering in plants6.
Diazotrophs harbor a protein complex called nitrogenase that converts nitrogen (N2) into ammonia (NH3) in an intricate process requiring a large amount of energy in the form of ATP and low potential electrons7. Nitrogenase has two protein components: an α2β2 heterotetrameric dinitrogenase formed by the nitrogen fixation (nif) nifD and nifK gene products, and a nifH-encoded homodimeric dinitrogenase reductase. During N2 to NH3 reduction, one NifH homodimer binds to each αβ half of the NifDK protein and, in an ATP-dependent reaction, transfers the electrons needed to break the N2 triple bond8. Nitrogenase requires a minimum of 16 ATP molecules and 8 electrons to convert one molecule of N2 into two molecules of NH39. The electrons are funneled through three [Fe-S] clusters, starting at a [Fe4S4] cluster bridging the two subunits of NifH, via the P-cluster ([Fe8S7]) and finally to the iron molybdenum cofactor (FeMo-co, [MoFe7S9C-(R)-homocitrate]). The latter two clusters are located at each αβ half of the NifDK heterodimer9. All three metalloclusters are extremely O2-sensitive, which makes engineering nitrogenase in plants especially challenging.
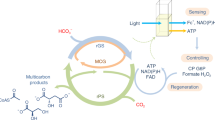
The activity of nitrogenase can be determined in vitro (e.g. using pure protein components), in vivo (e.g. in free-living cells) or in situ (e.g. bacteria associated to plants) using various techniques. One direct method is the quantitative measurement of the ammonia produced using its natural substrate N210, or by 15N enrichment or 15N natural abundance methods11,12,13. Furthermore, nitrogenase activity can be indirectly measured as nitrogenase can also reduce protons into H214,15, but also other double- and triple-bonded substrates such as acetylene, nitrite, nitrous oxide, and azide16. This promiscuity is often used by researchers to study nitrogenase in the laboratory. The most-used method is the acetylene reduction assay (ARA) in which acetylene is reduced to ethylene, which is detected using gas chromatography17,18,19,20. However, the ARA has several drawbacks. Firstly, the number of samples that can be measured is low due to the manual work involved. The manual steps include exchange of the gas phase in the reaction vial using inert argon to prevent N2 reduction; injection of acetylene to start the reaction; incubation in a water-bath, injection of EDTA or NaOH to stop the reaction; and finally injection of gas from the vial headspace into the gas chromatograph. Secondly, acetylene reduction cannot be easily monitored in real time, and only the end concentration of ethylene after a defined time is determined. This second limitation of the ARA was recently overcome by the development of a viologen-based electron donor to nitrogenase21. In that method a reduced sulfonated viologen derivative (1,1′-bis(3-sulfonatopropyl)-4,4′-bipyridinium radical, hereafter referred to as S2Vred) replaces the function of sodium dithionite (DTH) as electron donor to NifH in vitro. Upon nitrogenase activity and turnover, this deeply violet-colored substrate is converted into an oxidized colorless form (S2Vox) with greatly diminished absorbance at 600 nm (Fig. 1). The decrease in absorbance over time is therefore linear with nitrogenase activity. However, the exposure of the violet-colored S2Vred to an oxidizing agent such as O2 (or other reactive oxygen species), or to other natural electron acceptors, will lead to its conversion into the oxidized and colorless form (S2Vox), limiting the use of S2Vred to in vitro measurements under anaerobic conditions.

Schematic overview of nitrogenase activity and activity determination using S2Vred. The flow of electrons from S2Vred to protons (H+) or N2 via NifH and NifDK is shown. At NifDK, H2 or NH4+ (the dominating form at pH 7) is produced. ATP is regenerated from ADP and phosphocreatine by the action of creatine phosphokinase. Nitrogenase activity is determined by measuring the decrease in absorbance at 600 nm.
In this work, we have adapted the S2Vred method to determine nitrogenase activity in 96-well microtiter plates with the aim to screen distinct NifH variants expressed in the mitochondria of the yeast Saccharomyces cerevisiae for functionality. The development of screening methods allows us to find Nif components with improved properties desirable for its expression in plant organelles. We demonstrate that: (1) the results obtained using S2Vred are in accordance with those seen when using the standard ARA; (2) the method is compatible with NifH proteins originating from different prokaryotic origins; and (3) many samples and assay conditions can be tested in parallel. We used S2Vred to determine the activity of nine distinct NifH proteins. NifH variants that were compatible with the Azotobacter vinelandii NifDK were tested for NifM dependency and O2 sensitivity, two properties of importance to engineer nitrogenase in crop plants.
Results
Adaptation and use of S2Vred for screening and activity determination of NifH proteins using 96 well microtiter plates
To confirm that S2Vred-dependent electron donation to nitrogenase is not unique to NifH isolated from A. vinelandii (hereafter denoted as NifHAv), as previously shown21, but is also suitable for functional screening of novel NifH variants, we combined the Hydrogenobacter thermophilus NifH protein previously isolated from S. cerevisiae22 with NifDKAv. NifH variants expressed in yeast are hereafter denoted as ScNifHXx where Sc and Xx indicates S. cerevisiae and the species from which the NifH sequence was obtained, respectively. Robust, although slower, oxidation of S2Vred was observed using ScNifHHt compared to NifHAv (Fig. 2). This was expected from the lower specific activity of ScNifHHt when combined with NifDKAv determined by ARA in previous studies22. We next tested functionality of the S2Vred assay using NifHAv and NifDKAv in 96 well microtiter plates sealed in the glove box (95% N2 and 5% H2). Several NifDK concentrations (0.05–0.2 µM) and NifH to NifDK molar ratios (2x-40x) were tested to identify conditions that provided a robust and linear rate constant (kobs) over an extended time-period (to facilitate sample preparations and plate handling), and that minimized the amount of NifH and NifDK proteins required for the assay. Supplementary Fig. S1 shows the decrease in absorbance over time under the tested experimental conditions. As expected, higher NifDK concentrations resulted in a faster S2Vred oxidation (Supplementary Fig. S1d). Supplementary Figure S2 shows the corresponding kobs variation over time. The fast S2Vred oxidation observed at high NifDK concentration resulted in lower kobs (Supplementary Fig. S2c), presumably because the concentration of S2Vred was already suboptimal by the time the plate was being scanned (after sealing in the glove box and transfer to the plate reader). The maximum activity observed under the conditions tested here (kobs of ca. 12 s−1) was obtained when the mixture contained 2 µM NifHAv and 0.05 µM NifDKAv (corresponding to a NifH:NifDK ratio of 40) (Fig. 2b), and was almost identical to that reported when performed under argon atmosphere and using cuvettes21. Importantly, the decrease in absorbance was linear under these conditions for more than 10 min (Supplementary Fig. S1b) (R2 = 0.9997, t1 = 1 to t2 = 10 min), which provided sufficient time for microtiter plate preparation, transfer and reading. These experimental conditions were therefore implemented for subsequent use of S2Vred.

Adaptation and optimization of S2Vred for nitrogenase activity determination using 96-well microtiter plates. (a) Nitrogenase activity (kobs (s−1)) measured in a cuvette using ScNifHHt and NifDKAv at 0–60 × molar ratios in reactions containing 0.4 µM NifDKAv. Mean and SD is shown. n = 2 technical replicates. (b) Nitrogenase activity (kobs (s−1)) measured in a 96-well microtiter plate using NifHAv and NifDKAv at 0–40 × molar ratios in reactions containing 0.05 µM (blue dots), 0.1 µM (red squares) or 0.2 µM (green triangles) NifDKAv. Mean and SD is shown. n = 2 technical replicates.
Solubility screening of NifH variants targeted to the mitochondria of yeast
To obtain proof-of-concept of S2Vred suitability to quickly screen the function of engineered Nif proteins, we used a library of 35 mitochondria-targeted NifH proteins in yeast (Supplementary Tables S1 and S2). The bulk of the NifH variants originated from a library that had previously been tested in Nicotiana benthamiana22. The NifH variants were expressed as N-terminally TwinStrep (TS)-tagged proteins with a Cox4 mitochondria targeting signal23,24. The purpose of the TS-tag was to enable equal detection of the distinct NifH variants and their subsequent isolation using Strep-tag affinity chromatography (STAC). The NifH proteins were co-expressed with NifM, NifU and NifS using galactose inducible promoters. NifM has been proposed to be necessary for proper folding of the NifH polypeptide25, whereas NifU and NifS provide NifH with its [Fe4S4] cluster7. All the accessory proteins originated from A. vinelandii and were equipped with a Su9 leader sequence24,26. The functionality of the Cox4 and the Su9 sequences for mitochondrial targeting of each respective protein in yeast has been shown previously22,27,28.
With the assumption that Nif components must accumulate as soluble proteins to be relevant for engineering nitrogenase in eukaryotes, we first tested the NifH variant solubility. Accumulation of ScNifHXx variant polypeptides, co-expressed with NifM, NifU and NifS, was confirmed by immunoblot analysis of total protein extracts (Supplementary Fig. S3). Nine out of the 35 ScNifH variants were detectable at noticeable levels in soluble extracts (Fig. 3). These nine NifH variants originated from Roseiflexus sp. (strain RS-1), H. thermophilus (strain TK-6), Geobacter sulfurreducens (strain PCA), Ruminococcus albus (strain SY3), Methanothermobacter marburgensis (strain Marburg), Methanocaldococcus infernus (strain ME), Firmicutes bacterium CAG:536, Leptolyngbya boryana (strain Dg5) and Dehalococcoides ethenogenes (strain 195). Of these nine, the NifH variants from H. thermophilus, M. marburgensis and M. infernus were shown to be soluble in a previous study from our group22.

NifH library solubility screening. (a,b) Presence of ScNifHXx (see Supplementary Table S2 for full species names) in soluble (a) and total (b) protein extracts was determined using antibodies detecting the TwinStrep-tag (α-Strep). Two different exposure times (s.e., short exposure (above) and l.e., long exposure (below)) are shown for the analysis of total extracts.
Isolation and activity measurements of soluble ScNifH variants
Three of the nine soluble ScNifH variants (from H. thermophilus, M. marburgensis and M. infernus) had already been purified in our laboratory22. The remaining ScNifH proteins (together with ScNifHHt that was reisolated for further work described in this study) were isolated using STAC under anaerobic conditions (Fig. 4a–c, Supplementary Fig. S4). The yield varied from 9–35 mg ScNifH per 100 g cell paste for six of the variants (Table 1), in line to what was previously reported for ScNifHMm and ScNifHMi. Only ScNifHLb was isolated at much lower level. This variant was excluded from further analysis. These ScNifH proteins all presented color and an UV–vis absorbance spectrum characteristic of [Fe–S] cluster containing proteins (Fig. 4d, Supplementary Fig. S5). Iron quantification suggested that six ScNifH variants had a similar amount of bound [Fe-S] cluster as NifHAv purified from its native host29 (Table 1), while ScNifHMi and ScNifHMm were isolated with much lower Fe content.

Functionality of soluble ScNifHXx candidates. (a) Example of the STAC-purification process of ScNifHXx (represented here by ScNifHRa). TE, total extract after yeast cell breakage using high-pressure homogenizer; CFE, cell-free extract after centrifugation and filtering of the TE; FT, flow-through after passing the CFE through the STAC column; W, wash fraction; E, final concentrated and desalted elution fraction. (b) Example of concentrated and desalted elution fraction (here represented by ScNifHFb, Supplementary Fig. S4d), 7 ml final volume. (c) Coomassie staining of soluble ScNifHXx variants isolated from soluble yeast extracts using STAC. Approximately 3 µg protein was loaded per sample. More details of the purification process are shown in Supplementary Fig. S4. The uncropped Coomassie stained gel is shown in Fig. S11. (d) Example of UV–vis absorption spectra of as-isolated and air-exposed ScNifHXx (represented here by ScNifHRa). (e) Nitrogenase activities with increasing concentrations of ScNifHXx proteins using S2Vred as electron donor and NifDKAv as electron acceptor. Mean and SD is shown. n = 2 technical replicates. (f) Nitrogenase activities using ScNifHXx proteins and NifDKAv (at a 40:1 molar ratio) as determined by ARA (ethylene production, left Y-axis, red bars) or using S2Vred (kobs(s-1), right Y-axis, blue bars). Mean and SD is shown. n = 3 technical replicates (ARA) and n = 4 (S2Vred).
We then performed nitrogenase assays with the different ScNifH proteins using S2Vred as the electron donor to NifH, and NifDKAv as its electron acceptor (Fig. 4e). Three variants, namely those originating from H. thermophilus (ScNifHHt), G. sulfurreducens (ScNifHGs) and D. ethenogenes (ScNifHDe), accelerated S2Vred oxidation when the ScNifH:NifDKAv ratio was increased, which is expected from a functional and NifDKAv-compatible NifH variant. These ScNifH proteins showed acetylene reduction activities consistent with those obtained using S2Vred (Fig. 4e,f). Interestingly, ScNifHRs (and to some extent ScNifHRa) could act as reductase for NifDKAv during ARA (i. e. when using DTH as electron donor), but not in the S2Vred assay (Fig. 4f). This divergence could potentially originate from different reduction potential requirements among the NifH variants, as S2Vred has a potential of − 0.40 V vs Normal Hydrogen Electrode (NHE)21 and DTH has a potential of − 0.66 V vs NHE30. Whether this discrepancy indicates a different mechanism or requirement for NifHRs activity remains to be investigated in future studies.
Inhibition of ScNifH by O2
The NifDKAv compatible ScNifH variants were assayed for their sensitivity to O2, which represents a major barrier to engineer nitrogenase in plants. As S2Vred itself is oxidized by O2, and O2-destruction of the [Fe4S4]-cluster at NifH is extremely fast (the half-life of NifH upon O2 exposure is reported to be about 30–45 s)31,32, we were not able to design an experiment using S2Vred to study the effect of O2 on the activity. We therefore measured the ScNifH variants capacity to support acetylene reduction upon exposure to O2 following a previously reported method31. In short, DTH present in the buffer of the isolated ScNifH protein was first removed using a desalting column inside an anaerobic glove box. The ScNifH protein was then added to anaerobic buffer in a glass vial containing argon in the headspace (representing t = 0). Then, O2 was injected into the headspace to a final concentration of 20% and the vial was incubated with rigorous shaking. At distinct time points, ScNifH was extracted using a Hamilton syringe and transferred to an open vial containing anaerobic buffer supplemented with DTH to quench the O2. Finally, NifDKAv and an ATP-regenerating mixture was added before the standard ARA. Similar to the Klebsiella pneumoniae NifH protein31, the half-life for NifHAv was less than one minute (Fig. 5). While ScNifHHt and ScNifHDe presented similar kinetics regarding the inhibition from O2 exposure as NifHAv, the ScNifHGs retained 50% activity for about 4 min, and about 25% activity after 10 min O2 exposure.

Sensitivity of ScNifHXx variants to O2. Nitrogenase activity of ScNifHHt (green squares), ScNifHGs (blue triangles) and ScNifHDe (red dots) was measured by ARA upon exposure to oxygen. NifHAv was used as NifH control protein (black stars). The molar ratio of NifH:NifDKAv was 40:1. Nitrogenase activity is shown in relation to the activity obtained prior to oxygen exposure at t0 (for ScNifHHt 532 ± 30 units (nmol ethylene formed per min and mg of NifDKAv), for ScNifHGs 159 ± 37 units, for ScNifHDe 188 ± 14 units and for NifHAv 1673 ± 51 units). Mean and SD is shown. n = 5 or 6 technical replicates.
NifM-dependency for ScNifH solubility and functionality
Co-expression of the A. vinelandii nifM gene with nifHAv is required for the accumulation of functional NifHAv protein in the mitochondria of S. cerevisiae33. Several of the NifH sequences in this study were selected because of the absence of a nifM orthologue in the organism’s genome. To test whether NifM was required for the solubility of the eight ScNifH variants, we compared their accumulation in total and soluble protein extracts when co-expressed with NifUAv and NifSAv, but not NifMAv, in yeast. Surprisingly, six of the eight ScNifH variants (Roseiflexus sp., R. albus, M. marburgensis, M. infernus, Firmicutes bacterium, and D. ethenogenes) showed no obvious decrease in solubility when NifMAv was absent (Fig. 6a). To test whether functionality could be affected although solubility was not, we isolated ScNifHDe from the yeast strain not expressing NifMAv (Fig. 6b, Supplementary Fig. S6a). The UV–vis spectrum suggested no apparent difference in [Fe-S] cluster content (Supplementary Fig. S6b), and the specific activity was similar to ScNifHDe protein isolated from cells co-expressing NifMAv (Fig. 6c).

Effect of NifMAv on ScNifHXx solubility and functionality. (a) Immunoblot analysis of the levels of ScNifHXx variants in total yeast extracts (TE) and the soluble fractions (SN) when expressed in the absence of NifMAv. The uncropped immunoblots and membranes are shown in Fig. S12. (b) ScNifHDe isolated from yeast cells not expressing NifMAv. The uncropped Coomassie stained gel is shown in Fig. S13. (c) Comparison of the specific activity of ScNifHDe isolated from yeast cells expressing (+ NifM) or not expressing NifM (− NifM) using ARA. The molar ratio of ScNifHDe to NifDKAv is indicated. Mean and SD is shown. n = 2 technical replicates. (d) Alignment of NifHAv with the eight ScNifHXx variants analyzed in (a). The C-terminal domain containing Pro259 in NifHAv (indicated by a black arrow) proposed to be the target of NifM action is shown. The full sequence alignment can be found in Supplementary Fig. S7.
Seven of our eight ScNifH variants contained a proline residue at the site corresponding to Pro259 (when including the methionine) in A. vinelandii (Fig. 6d, Supplementary Fig. S7), which is thought to be the target of NifM prolyl isomerase activity25. The NifH protein from Firmicutes bacterium is shorter and terminates before this proline. Interestingly, the only genome of the eight selected NifH variants that contained a gene with high similarity to NifMAv was G. sulfurreducens (Supplementary Table S2). ScNifHGs was also the variant that was least soluble when NifMAv was not co-expressed (Fig. 6a). The only other protein that showed reduced solubility in the absence of NifMAv was ScNifHHt. The genome of H. thermophilus harbors a gene encoding a hypothetical protein with a PPIC-type PPIASE domain and with moderate similarity to NifMAv. Interestingly, isolation of the soluble population of ScNifHHt that was produced in the absence of NifMAv resulted in a protein with identical specific activity to ScNifHHt isolated from yeast cells co-expressing of NifMAv (Supplementary Fig. S8). Therefore, the direct action of NifM with regards to NifH is not clear and to some extent in disagreement with the published literature7, and should be the topic of future studies.
Discussion
The transfer of prokaryotic nitrogenase activity into cereals could generate crops suited to grow well under limited nitrogen fertilizer. Although there are excellent reports on engineering of nitrogenase in heterologous (non-N2-fixing) bacterial hosts34,35,36,37,38,39,40, our experience is that it is very difficult to directly translate and transfer that knowledge to a eukaryotic system and expect comparable results, even in a relatively simple, unicellular eukaryote such as yeast41. A major challenge arises from the extremely complex biochemical requirements of the nitrogenase enzyme and its stepwise maturation involving several inter-dependent gene products7. Additionally, from a metabolic point of view, nitrogenase requires high levels of energy and reducing power in an environment that is low in O2 to protect its metalloclusters from oxidative damage.
In this study we have developed an important part of the nitrogenase engineering process, namely the analysis of NifH protein functionality in a high throughput assay. While the ARA is very precise, it requires training to generate consistent results, it is rather time-consuming and would be difficult to scale up for screening large numbers of samples and/or conditions. We optimized the S2Vred assay and showed that it fulfilled many of our main objectives, most importantly to be fast and simple to use, to require a lower amount of purified proteins (corresponding to about half of that used in the ARA as the reaction volume is smaller), and to not depend on expensive or sophisticated equipment. We also believe that this method is easily adaptable to automated robotic systems as the reactions are performed in microtiter plates. In addition, the S2Vred assay has two important advantages over the ARA. Firstly, activities can be monitored in real-time, which means that it is possible to directly study the effect of various effector molecules or reaction components on nitrogenase functionality. Secondly, the reduction potential of S2Vred (used as the electron donor to NifH) is much closer to that of ferredoxin or flavodoxin, the physiological reductants of nitrogenase42,43 than DTH. NifF for example, a flavodoxin in the diazotrophic free-living model-bacteria A. vinelandii donating electrons to NifH, harbors a flavin mononucleotide (FMN) cofactor with a redox potential in the semiquinone/hydroquinone state of − 0.483 V vs NHE44. The corresponding potentials for S2Vred is − 0.40 V vs NHE, compared to − 0.66 V vs NHE for DTH21,30. However, it is important to note that other reported flavodoxins and ferredoxins have lower reduction potentials, for example flavodoxin in A. chroococcum (− 522 mV)45 and ferredoxin in A. vinelandii (− 619 mV)46, and that S2Vred would not be a suitable electron donor to study nitrogenases requiring such strong reductants. Whether this could explain the lack of nitrogenase activity when combining ScNifHRs and ScNifHRa with NifDKAv in the S2Vred is not clear, as it is also possible that other steric or charge factors prelude productive electron transfer from S2Vred to these NifH variants. Other important drawback with using S2Vred is that it is not commercially available, and that it cannot be used directly with yeast extracts as it is effectively oxidized by unknown molecule(s) in the lysate (data not shown). Therefore, Nif proteins must be purified prior to the activity assay. Solving this limitation would further expand the use of the S2Vred.
Regarding the functional assessment of Nif proteins expressed in yeast and plants, we have observed that many of the essential Nif components have poor solubility, especially NifH and NifB22,24,27,47. This is critical as the structural components (NifH and NifDK) are needed at very high levels during nitrogen fixation. In N2-fixing A. vinelandii for example the NifH concentration within the cell can reach up to 100 μM48, whereas in K. oxytoca about 40% of the total protein is NifHDK49. For this, a simple protein solubility study is always the first experiment to perform before initiating more complex analyses22,27. From the 35 mitochondrial-targeted NifH variants expressed in this study, we obtained nine that were soluble in yeast mitochondria. The phyla from where these nine NifH variants originated were diverse, and so was their mechanisms of nutrition and relationship to oxygen. The only common factor we could observe was a bias towards coming from thermophilic organisms, as has been observed and discussed previously in works from our laboratory22,24,27.
To expand the analysis of these soluble NifH variants and to see if we could identify properties that would facilitate their functionality in future crops, we tested two aspects that are sought after for eukaryotic nitrogenase engineering; 1) simplification of the nitrogenase genetic machinery by minimizing the number of genes needed to transfer, and 2) identification of Nif components with better functionality in an environment containing oxygen. In this work, that meant (1) the identification of a NifH variant that did not depend on NifM for solubility and functionality, and (2) one NifH variant whose [Fe4S4] cluster was more resistant towards O2.
Although NifM is just one protein, each gene fewer to transfer will make the engineering of nitrogenase in plants less complex. When the K. pneumoniae NifH protein was expressed in Escherichia coli in the absence of NifM, the protein was much less stable and completely inactive50. This was in agreement with the low levels of NifHKp polypeptide and dinitrogenase reductase activity detected in nifM− strains of the native host51. Work in yeast has shown that NifM co-expression was required for homodimer formation and polypeptide stability of Rhizobium meliloti NifH52, and in tobacco NifM was required to prevent NifH aggregation in the mitochondria53. While not many studies have investigated how NifM acts on NifH, sequence analysis suggests NifM to be a member of the rotamase family (PF00639) containing a PPIC-type PPIASE domain54. Prolyl isomerases (also known as peptidylprolyl isomerases or PPIases) are enzymes that accelerate protein folding by catalyzing the cis–trans isomerization of prolyl peptide bonds. This annotation is consistent with work identifying Pro259 in NifH from A. vinelandii as the prime target for NifM action25. In this work, seven of the final eight variants contained a proline residue at the site corresponding to Pro259 in A. vinelandii. The only exception was NifH from Firmicutes bacterium, but this protein was significantly shorter than the other NifH variants and therefore lacking this proline. However, the only two NifH variants that showed NifM-dependent solubility (ScNifHGs and ScNifHHt) corresponded to those that originated from organisms containing genes with some similarity to nifMAv. Therefore, our work suggests that presence of a nifM homologue in the organism genome is a better indicator of NifM-dependency than presence of a proline at a site corresponding to Pro259 in A. vinelandii.
Equally important is the identification of more O2-resistant nitrogenase components, as O2-sensitivity is likely to be the major barrier to overcome to obtain a functional plant nitrogenase5. Active NifH could be expressed in the cytosol of anaerobically cultured yeast, while only the mitochondria could produce active protein under aerobic conditions33. This has been explained by the low O2 concentration in the mitochondria of actively respiring cells. Whether it is possible to obtain similarly O2-depleted conditions in plant mitochondria is not known. One scenario would be to limit nitrogenase expression to plant cells in hypoxic niches55. In any case, the identification of more O2-tolerable nitrogenase components would be a breakthrough for the engineering of nitrogenase in crops. In this regard, we were surprised to see increased resistance towards O2 by NifH from G. sulfurreducens. As all NifH proteins contain a [Fe4S4] cluster, we assumed that the variants tested in this study would also show similar O2-susceptibility. Whether the [Fe4S4] cluster in ScNifHGs is less exposed, or whether it is stabilized by other means, is not known but these are interesting questions for future work. Importantly, this study shows that Nif proteins with better properties for expression in eukaryotic cells exist in Nature, and that their identification could pave the way for the engineering of N2 fixing crops.
Materials and methods
NifH library design and assembly
The majority of the nifH genes originated from a previously published gene set22. To this nifH library the genes for expression of NifH originating from L. boryana, Frankia alni and D. ethenogenes were added. All yeast codon-optimized DNA sequences and their corresponding protein products can be found in Supplementary Table S2. The nifH genes originating from the previously published gene set22 were amplified as cox4-ts-nifH gene fusions by PCR using primers #2584 (5´-AATTTTTGAAAATTCGAATTCCTCTTGACCATGCTTTCAC-3’) and #2585 (5´-GAAGAATTGTTAATTAAGAGCTCGGGGAAATTCGAGCTGG-3´). These primers include 15 bp overhangs complementary to the pESC-HIS yeast expression plasmid (#217451, Agilent Technologies) when digested with SacI and EcoRI, and allowed for the insertion by an exonuclease and ligation-independent (ELIC) method56. The nifH genes originating from L. boryana, F. alni and D. ethenogenes were amplified using primers #2902 (5´-CACAATTTGAAAAAGGATCCATGTCTGACGAAAACATTAG-3´) and #2903 (5´-GGAAATTCGAGCTGGTCACCTTAAGCACCAGCCTTAGCCA-3´), #2904 (5´-CACAATTTGAAAAAGGATCCATGAGACAAATTGCTTTCTA-3´) and #2905 (5´-GGAAATTCGAGCTGGTCACCTTAAGCAACAGCAGCAGCCT-3´), #2906 (5´-CACAATTTGAAAAAGGATCCATGAGAAAGGTTGCTATTTA and #2907 (5´-GGAAATTCGAGCTGGTCACCTTAAGAAATAACACCAAATT-3´), respectively. The amplified nifH sequences were inserted by ELIC into pESC-HIS (cox4-ts-nifHB. japonicum) digested with NcoI and BstEII, replacing the B. japonicum nifH gene with nifH from L. boryana, F. alni or D. ethenogenes. The gene encoding for mitochondria-targeted SU9-NifMAv was amplified from plasmid pN2XJ16522 using primers #2478 (5´- CTCTACAAATCTATCTCTCTCGAGATGGCCTCCACTCGTG-3´) and #2479 (5´- ATTATGGAGAAACTCGAGTTAACCATGTGCTAAGTTTTCC-3´) and inserted into pESC-TRP (#217453, Agilent Technologies) digested with XhoI by ELIC. The pESC-URA plasmid for expression of mitochondria-targeted Su9-NifUAv and Su9-NifSAv has been previously described33. All DNA digestions were performed using enzymes from New England Biolabs. PCR amplifications were carried out using Phusion Hot Start II High-Fidelity DNA Polymerase (ThermoFisher Scientific). ELIC products with their corresponding digested target vectors were transformed using a molar ratio 1:4 (vector:insert) into chemically competent E. coli DH5α and selected on solid LB (Lysogenic broth) media supplemented with appropriate antibiotics. Plasmid preparations were performed using Qiaprep Spin Miniprep kit (QIAGEN) and correct cloning was confirmed by Sanger sequencing (Macrogen). Plasmids were transformed into S. cerevisiae W303-1a (MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15) according to the lithium acetate method57, and selected and grown in synthetic drop-out medium with the appropriate auxotrophic selection24.
Expression analysis and solubility screening of ScNifHXx variants
Small-scale yeast protein extracts were prepared from yeast grown in galactose induction media as previously described24. The YeastBuster protein extraction reagent (Merck) was used to prepare total and soluble yeast protein extracts. First, galactose-induced yeast was pelleted for 10 min at 3000×g. YeastBuster mixture supplemented with 25 μg/ml DNAse I and 1 mM phenyl-methylsulfonyl fluoride (PMSF) was added to the yeast pellets at a ratio of 9 μl per OD × ml in Eppendorf tubes, and then incubated on a Eppendorf shaker for 20 min at room temperature to lyse the cells. This sample was then divided in two equal parts. For total extracts, the resulting YeastBuster lysate was added to 2 × Laemmli buffer at a 1:1 (v/v) ratio. For soluble extracts, the YeastBuster lysate was centrifuged in a benchtop centrifuge at maximum speed for 20 min at 4 °C before the supernatant was added to 2 × Laemmli buffer at a 1:1 (v/v) ratio. Both samples were prepared for SDS-PAGE by heating for 5 min at 95 °C.
Following SDS-PAGE, proteins were either stained using Coomassie brilliant Blue R-250 (Sigma) or transferred to nitrocellulose membranes (Protran Premium 0.45 µm, GE Healthcare) membranes for immunoblotting. Nitrocellulose membranes were stained with Ponceau S (Sigma) to ensure equal loading control and successful transfer. The membranes were blocked with 5% non-fat milk in TBS-T (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.02% Tween-20) for 1 h at room temperature before incubation with primary antibodies overnight at 4 °C. Polyclonal antibodies detecting NifMAv (used at 1:2,000 in 5% BSA), NifUAv (used at 1:2000 in 5% BSA) and NifSAv (used at 1:1,000 in 5% BSA), were raised against purified preparations of the corresponding A. vinelandii proteins (generated in house). Strep-tag II antibody (“Strep-MAB”, IBA Lifesciences, 1:2000 in 5% BSA) was used for detection of all ScNifHXx variants. Secondary antibodies (Sigma) were diluted 1:20,000 in TBS-T supplemented with 2% non-fat milk and incubated for 2 h at room temperature. Membranes were developed using enhanced chemiluminescence and images were recorded digitally (iBright FL1000, ThermoFisher).
S. cerevisiae growth and NifH variants purification
The growth of yeast cultures, galactose-induced Nif expression and STAC-purification of soluble ScNifHXx variants followed the procedure previously described27. Cell pellets from 4 l fermenters stored in liquid N2 (typically 200–220 g) were resuspended in lysis buffer (100 mM Tris–HCl pH 8.8, 200 mM NaCl, 10% glycerol, 2 mM DTH, 1 mM PMSF, 1 μg/ml leupeptin, 5 μg/ml DNAse I) at a ratio of 1:2 (w/v) inside an anaerobic glovebox (Coy Laboratories). Total extracts (TE) were prepared by lysis of the cell suspensions under anaerobic atmosphere using an EmulsiFlex-C5 homogenizer (Avestin Inc.) operating at 20,000 psi. The TE was transferred to centrifuge tubes equipped with sealing closures (Beckman Coulter) and centrifuged at 50,000×g for 1 h at 4 °C (Avanti J-26 XP). The supernatant was filtered using filtering cups with a pore size of 0.2 μm (ThermoFisher), rendering cell-free extract (CFE) of soluble proteins that was loaded at 2.5 ml/min into a 5 ml Strep-Tactin XP column (IBA LifeSciences) attached to an ÄKTA FPLC (GE Healthcare) at O2-levels below 1 ppm in anaerobic chambers operating at 16 °C (MECAPLEX or MBraun). The column was washed overnight using about 120 ml wash buffer (100 mM Tris–HCl pH 8.0, 200 mM NaCl, 10% glycerol, 2 mM DTH). Strep-Tactin XP column-bound proteins were eluted with 15 ml washing buffer supplemented with 50 mM biotin (IBA LifeSciences). The elution fraction was concentrated using centrifugal filters with 30 kDa cutoff (Amicon, Millipore), loaded into PD-10 desalting columns (GE Healthcare) equilibrated with wash buffer to remove biotin and DTH, and then used to UV–Vis absorption spectrum analysis (see section below). The desalted eluate was supplemented with 2 mM DTH, further concentrated using centrifugal filters and finally snap-frozen as protein pellets in cryovials (Nalgene) and stored in liquid N2.
Protein quantification, UV–Vis absorption spectrum and iron measurements
The concentrations of purified ScNifHXx variants were measured using the BCA protein assay (Pierce) in combination with iodoacetamide to eliminate the interfering effect of DTH58. ScNifHXx UV–Vis absorption spectra were recorded after removal of the DTH from the protein samples. The DTH-free protein samples were further diluted in wash buffer and transferred to Q6 spectroscopy cuvettes with sealing closures. Absorption (280 nm to 800 nm) was recorded using a UV-2600 spectrophotometer (Shimadzu). For recording of the air exposed ScNifHXx samples, the sealing closure was removed, and the protein sample was carefully exposed to air using a pipette equipped with a gel loading tip. Iron content of as isolated ScNifHXx preparations and NifHAv (used as [Fe4S4] containing control protein) was determined by atomic absorption using a graphite furnace installed in a ContrAA 800 AAS Spectrometer (Analytik Jena). Protein samples were denatured using 30% HNO3 for 1 h at 80 °C, and then diluted in metal-free ultra-pure water to a final concentration of 1.5% HNO3. Twenty µl of diluted sample were used for iron measurement according to the following protocol: I) sample drying at 100 °C for 25 s, II) pyrolysis at 350 °C for 10 s, III) pyrolysis at 1100 °C for 20 s, IV) gas adaptation at 1100 °C for 5 s, V) atomization at 2000 °C for 10 s, and finally VI) cleaning of the furnace at 2500 °C for 5 s. Each sample was measured in triplicates. An absorbance wavelength of 248.327 nm was selected for specific iron measurement. For quantification, an iron standard curve from 0 to 20 parts per billion (ppb) was prepared from a 1000 parts per million (ppm) iron standard solution (Inorganic Ventures). The spectrometer protocol was set up and controlled using the ASpect CS software (version 2.2.2.0).
Nitrogenase activity determination by S2Vred assay in cuvette
Nitrogenase activity of ScNifHHt expressed in yeast mitochondria and isolated by STAC22 was determined following a recently described spectrophotometric method21. Activity assays were performed in cuvette (600 µl final reaction volume) in the presence of ATP regenerating mixture (6.7 mM MgCl2, 5 mM ATP, 30 mM phosphocreatine, 0.2 mg/ml creatine phosphokinase, 1.3 mg/ml bovine serum albumin (BSA) in 100 mM MOPS pH 7.0) and 0.5 mM 1,1′-bis(3-sulfonatopropyl)-4,4′-bipyridinium (S2Vred). Nitrogenase activity was determined from the decrease in absorbance at 600 nm upon addition of 0.4 µM of NifDKAv and increasing concentrations of NifHAv or ScNifHHt. Absorbance was recorded using a USB 400-ISS-UV/VIS spectrophotometer (Ocean Optics) using a cuvette with a path length of 0.2 cm. Nitrogenase activity calculations were performed as previously described21.
Nitrogenase activity determination by S2Vred assay in 96-well microtiter plates
Nitrogenase activity determined by the S2V assay were scaled down to 200 µl reaction volume to be performed in a 96-well microtiter plate. Except for during the optimization of the method, the assay was performed using a mixture of 2 µM NifHAv and 0.05 µM NifDKAv (corresponding to a NifH:NifDK ratio of 40:1). Other reaction conditions such as buffer composition, ATP regenerating mixture and S2Vred concentration were identical to those described in the section above. The plate was prepared under anaerobic conditions inside a glovebox (Coy Laboratories) and sealed using PCR plate sealing films. Absorbance reading was performed using an absorbance plate reader (SPECTROstar Nano, BMG LABTECH) operating at 30 °C. The absorbance was recorded for 1 h, with measurements taken every 30 s. Nitrogenase activity calculations were performed in Excel (Microsoft) using an molar extinction coefficient of 9925 M−1 cm−1 at 600 nm21, and a path length of 5 mm. The slope was calculated using the range in which the decrease in absorbance was linear, normally for at least 10 min. The final calculation can be expressed in simplified form as kobs (s−1) =|m|/ ((ε∙l) ∙ [NifDKAv]), where |m| is the absolute value of the slope of the decrease in absorbance at 600 nm, ε is the molar extinction coefficient of S2Vred at 600 nm in M−1 cm−1 (9925), l is the path length in cm (0.5) and [NifDKAv] is the molar concentration of NifDKAv in the assay (normally 0.05e−6).
Nitrogenase activity determination by ARA
ARA were performed by combining isolated ScNifHXx variants (2.2 µM) with pure NifDKAv (0.055 µM) in an ATP regenerating mixture (1.23 mM ATP, 18 mM phosphocreatine disodium salt, 2.2 mM MgCl2, 3 mM DTH, 40 µg/mL creatine phosphokinase in 100 mM MOPS pH 7.0). The final reaction volume was 400 µl in 9 ml sealed vials. Vials were flushed with argon before the injection of 0.5 ml acetylene. After 15 min of incubation at 30 °C, reactions were stopped by the addition of 100 µl of 8 M NaOH. Ethylene produced was detected and quantified using a gas chromatograph (GC-2014, Shimadzu) fitted with a flame ionization detector. The separation column was a Porapak N 80/100 column (G3591-80072, Agilent technologies), using pure N2 as a column carrier gas (25 ml/min flow), and a mixture of H2/air for the flame.
Oxygen sensitivity assays
ScNifHXx samples and NifHAv were passed through PD-10 desalting columns (GE Healthcare) equilibrated with 100 mM MOPS (pH 7.5) to remove DTH. Oxygen sensitivity was tested inside an anaerobic glovebox (Coy Laboratories) as previously described31 with slight modifications. First, 2.5 ml of 17.4 μM NifH in 100 mM MOPS (pH 7.5) was prepared in a 13 ml sealed glass vial. The atmosphere in the headspace was exchanged for argon. Diluted NifH sample was removed (t0) before pure O2 was injected (0.2 atm final) using a 250 μl gastight syringe (Hamilton). The vial was incubated at room temperature with shaking (800 rpm) on a thermomixer (Eppendorf) with an adaptor for 13 ml vials. Air exposed NifH samples were removed after 1, 2, 5 and 10 min. Fifty ul was transferred to open 9 ml vials (three technical replicates) containing 150 μl of 100 mM MOPS (pH 7.5) supplemented with 4 mM DTH. Finally, 200 ul ATP mixture supplemented with 5 µg of NifDKAv was added before nitrogenase activity was measured following the protocol for ARA as described above.
Data availability
The authors declare that the data supporting the findings of this study are available within the article, its supplementary information and data, and upon request.
References
Chapin, F. S., Matson, P. A. & Vitousek, P. Principles of Terrestrial Ecosystem Ecology 2nd edn, 529 (Springer Science & Business Media, 2011).
Erisman, J. W. et al. Nitrogen: Too much of a vital resource. Sci. Brief https://doi.org/10.13140/RG.2.1.3664.8163 (2015).
Mus, F., Alleman, A. B., Pence, N., Seefeldt, L. C. & Peters, J. W. Exploring the alternatives of biological nitrogen fixation. Metallomics 10, 523–538. https://doi.org/10.1039/c8mt00038g (2018).
Oldroyd, G. E. & Dixon, R. Biotechnological solutions to the nitrogen problem. Curr. Opin. Biotechnol. 26, 19–24. https://doi.org/10.1016/j.copbio.2013.08.006 (2014).
Curatti, L. & Rubio, L. M. Challenges to develop nitrogen-fixing cereals by direct nif-gene transfer. Plant Sci. 225, 130–137. https://doi.org/10.1016/j.plantsci.2014.06.003 (2014).
Buren, S. & Rubio, L. M. State of the art in eukaryotic nitrogenase engineering. FEMS Microbiol. Lett. 365, fnx274. https://doi.org/10.1093/femsle/fnx274 (2018).
Buren, S., Jimenez-Vicente, E., Echavarri-Erasun, C. & Rubio, L. M. Biosynthesis of nitrogenase cofactors. Chem. Rev. 120, 4921–4968. https://doi.org/10.1021/acs.chemrev.9b00489 (2020).
Bulen, W. A. & LeComte, J. R. The nitrogenase system from Azotobacter: Two-enzyme requirement for N2 reduction, ATP-dependent H2 evolution, and ATP hydrolysis. Proc. Natl. Acad. Sci. USA 56, 979–986. https://doi.org/10.1073/pnas.56.3.979 (1966).
Seefeldt, L. C., Hoffman, B. M. & Dean, D. R. Electron transfer in nitrogenase catalysis. Curr. Opin. Chem. Biol. 16, 19–25. https://doi.org/10.1016/j.cbpa.2012.02.012 (2012).
Corbin, J. L. Liquid chromatographic-fluorescence determination of ammonia from nitrogenase reactions: A 2-min assay. Appl. Environ. Microbiol. 47, 1027–1030. https://doi.org/10.1128/aem.47.5.1027-1030.1984 (1984).
Chalk, P. M. The strategic role of 15N in quantifying the contribution of endophytic N2 fixation to the N nutrition of non-legumes. Symbiosis 69, 63–80. https://doi.org/10.1007/s13199-016-0397-8 (2016).
Herridge, D. F. & Giller, K. E. In Working with Rhizobia (eds Howieson, J. G. & Dilworth, M. J.) 187–220 (Australian Centre for International Agricultural Research, 2016).
Smercina, D. N., Evans, S. E., Friesen, M. L. & Tiemann, L. K. Optimization of the 15N2 incorporation and acetylene reduction methods for free-living nitrogen fixation. Plant Soil 445, 595–611. https://doi.org/10.1007/s11104-019-04307-3 (2019).
Joe Hanus, F., Carter, K. R. & Evans, H. J. In Methods in Enzymology Vol. 69 (ed. Anthony, S. P.) 731–738 (Academic Press, 1980).
Simpson, F. B. & Burris, R. H. A nitrogen pressure of 50 atmospheres does not prevent evolution of hydrogen by nitrogenase. Science 224, 1095–1097. https://doi.org/10.1126/science.6585956 (1984).
Seefeldt, L. C. et al. Reduction of substrates by nitrogenases. Chem. Rev. 120, 5082–5106. https://doi.org/10.1021/acs.chemrev.9b00556 (2020).
Dilworth, M. J. Acetylene reduction by nitrogen-fixing preparations from Clostridium pasteurianum. Biochim. Biophys. Acta 127, 285–294. https://doi.org/10.1016/0304-4165(66)90383-7 (1966).
Stewart, W. D., Fitzgerald, G. P. & Burris, R. H. In situ studies on N2 fixation using the acetylene reduction technique. Proc. Natl. Acad. Sci. USA 58, 2071–2078. https://doi.org/10.1073/pnas.58.5.2071 (1967).
Shah, V. K. & Brill, W. J. Nitrogenase. IV. Simple method of purification to homogeneity of nitrogenase components from Azotobacter vinelandii. Biochim. Biophys. Acta 305, 445–454. https://doi.org/10.1016/0005-2728(73)90190-4 (1973).
Haskett, T. L., Knights, H. E., Jorrin, B., Mendes, M. D. & Poole, P. S. A simple in situ assay to assess plant-associative bacterial nitrogenase activity. Front. Microbiol. 12, 690439. https://doi.org/10.3389/fmicb.2021.690439 (2021).
Badalyan, A. et al. An efficient viologen-based electron donor to nitrogenase. Biochemistry 58, 4590–4595. https://doi.org/10.1021/acs.biochem.9b00844 (2019).
Jiang, X. et al. Exploiting genetic diversity and gene synthesis to identify superior nitrogenase NifH protein variants to engineer N2-fixation in plants. Commun. Biol. 4, 4. https://doi.org/10.1038/s42003-020-01536-6 (2021).
Vogtle, F. N. et al. Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell 139, 428–439. https://doi.org/10.1016/j.cell.2009.07.045 (2009).
Buren, S., Jiang, X., Lopez-Torrejon, G., Echavarri-Erasun, C. & Rubio, L. M. Purification and in vitro activity of mitochondria targeted nitrogenase cofactor maturase NifB. Front. Plant Sci. 8, 1567. https://doi.org/10.3389/fpls.2017.01567 (2017).
Gavini, N., Tungtur, S. & Pulakat, L. Peptidyl-prolyl cis/trans isomerase-independent functional NifH mutant of Azotobacter vinelandii. J. Bacteriol. 188, 6020–6025. https://doi.org/10.1128/JB.00379-06 (2006).
Westermann, B. & Neupert, W. Mitochondria-targeted green fluorescent proteins: Convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast 16, 1421–1427. https://doi.org/10.1002/1097-0061(200011)16:15%3c1421::AID-YEA624%3e3.0.CO;2-U (2000).
Buren, S. et al. Biosynthesis of the nitrogenase active-site cofactor precursor NifB-co in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 116, 25078–25086. https://doi.org/10.1073/pnas.1904903116 (2019).
Lopez-Torrejon, G., Buren, S., Veldhuizen, M. & Rubio, L. M. Biosynthesis of cofactor-activatable iron-only nitrogenase in Saccharomyces cerevisiae. Microb. Biotechnol. 14, 1073–1083. https://doi.org/10.1111/1751-7915.13758 (2021).
Georgiadis, M. M. et al. Crystallographic structure of the nitrogenase iron protein from Azotobacter vinelandii. Science 257, 1653–1659. https://doi.org/10.1126/science.1529353 (1992).
Mayhew, S. G. The redox potential of dithionite and SO-2 from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur. J. Biochem. 85, 535–547. https://doi.org/10.1111/j.1432-1033.1978.tb12269.x (1978).
Eady, R. R., Smith, B. E., Cook, K. A. & Postgate, J. R. Nitrogenase of Klebsiella pneumoniae. Purification and properties of the component proteins. Biochem. J. 128, 655–675. https://doi.org/10.1042/bj1280655 (1972).
Eady, R. R. & Postgate, J. R. Nitrogenase. Nature 249, 805–810. https://doi.org/10.1038/249805a0 (1974).
Lopez-Torrejon, G. et al. Expression of a functional oxygen-labile nitrogenase component in the mitochondrial matrix of aerobically grown yeast. Nat. Commun. 7, 11426. https://doi.org/10.1038/ncomms11426 (2016).
Temme, K., Zhao, D. & Voigt, C. A. Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. Proc. Natl. Acad. Sci. USA 109, 7085–7090. https://doi.org/10.1073/pnas.1120788109 (2012).
Wang, L. et al. A minimal nitrogen fixation gene cluster from Paenibacillus sp. WLY78 enables expression of active nitrogenase in Escherichia coli. PLoS Genet 9, e1003865. https://doi.org/10.1371/journal.pgen.1003865 (2013).
Wang, X. et al. Using synthetic biology to distinguish and overcome regulatory and functional barriers related to nitrogen fixation. PLoS ONE 8, e68677. https://doi.org/10.1371/journal.pone.0068677 (2013).
Smanski, M. J. et al. Functional optimization of gene clusters by combinatorial design and assembly. Nat. Biotechnol. 32, 1241–1249. https://doi.org/10.1038/nbt.3063 (2014).
Yang, J., Xie, X., Wang, X., Dixon, R. & Wang, Y. P. Reconstruction and minimal gene requirements for the alternative iron-only nitrogenase in Escherichia coli. Proc. Natl. Acad. Sci. USA 111, E3718-3725. https://doi.org/10.1073/pnas.1411185111 (2014).
Yang, J., Xie, X., Yang, M., Dixon, R. & Wang, Y. P. Modular electron-transport chains from eukaryotic organelles function to support nitrogenase activity. Proc. Natl. Acad. Sci. USA 114, E2460–E2465. https://doi.org/10.1073/pnas.1620058114 (2017).
Yang, J. et al. Polyprotein strategy for stoichiometric assembly of nitrogen fixation components for synthetic biology. Proc. Natl. Acad. Sci. USA 115, E8509–E8517. https://doi.org/10.1073/pnas.1804992115 (2018).
Burén, S. et al. Formation of nitrogenase NifDK tetramers in the mitochondria of Saccharomyces cerevisiae. ACS Synth. Biol. 6, 1043–1055. https://doi.org/10.1021/acssynbio.6b00371 (2017).
Ledbetter, R. N. et al. The electron bifurcating FixABCX protein complex from Azotobacter vinelandii: Generation of low-potential reducing equivalents for nitrogenase catalysis. Biochemistry 56, 4177–4190. https://doi.org/10.1021/acs.biochem.7b00389 (2017).
Alleman, A. B., Mus, F. & Peters, J. W. Metabolic model of the nitrogen-fixing obligate aerobe Azotobacter vinelandii predicts its adaptation to oxygen concentration and metal availability. mBio 12, e0259321. https://doi.org/10.1128/mBio.02593-21 (2021).
Segal, H. M., Spatzal, T., Hill, M. G., Udit, A. K. & Rees, D. C. Electrochemical and structural characterization of Azotobacter vinelandii flavodoxin II. Protein Sci. 26, 1984–1993. https://doi.org/10.1002/pro.3236 (2017).
Deistung, J. & Thorneley, R. N. Electron transfer to nitrogenase. Characterization of flavodoxin from Azotobacter chroococcum and comparison of its redox potentials with those of flavodoxins from Azotobacter vinelandii and Klebsiella pneumoniae (nifF-gene product). Biochem. J. 239, 69–75. https://doi.org/10.1042/bj2390069 (1986).
Chen, K. et al. Alteration of the reduction potential of the [4Fe-4S](2+/+) cluster of Azotobacter vinelandii ferredoxin I. J. Biol. Chem. 274, 36479–36487. https://doi.org/10.1074/jbc.274.51.36479 (1999).
Aznar-Moreno, J. A., Jiang, X., Burén, S. & Rubio, L. M. Analysis of nitrogenase Fe protein activity in transplastomic tobacco. Front. Agron. https://doi.org/10.3389/fagro.2021.657227 (2021).
Poza-Carrion, C., Jimenez-Vicente, E., Navarro-Rodriguez, M., Echavarri-Erasun, C. & Rubio, L. M. Kinetics of Nif gene expression in a nitrogen-fixing bacterium. J. Bacteriol. 196, 595–603. https://doi.org/10.1128/JB.00942-13 (2014).
Waite, C. J. et al. Resource allocation during the transition to diazotrophy in Klebsiella oxytoca. Front. Microbiol. 12, 718487. https://doi.org/10.3389/fmicb.2021.718487 (2021).
Howard, K. S. et al. Klebsiella pneumoniae nifM gene product is required for stabilization and activation of nitrogenase iron protein in Escherichia coli. J. Biol. Chem. 261, 772–778. https://doi.org/10.1016/s0021-9258(17)36161-6 (1986).
Roberts, G. P., MacNeil, T., MacNeil, D. & Brill, W. J. Regulation and characterization of protein products coded by the nif (nitrogen fixation) genes of Klebsiella pneumoniae. J. Bacteriol. 136, 267–279. https://doi.org/10.1128/jb.136.1.267-279.1978 (1978).
Petrova, N., Gigova, L. & Venkov, P. Dimerization of Rhizobium meliloti NifH protein in Saccharomyces cerevisiae cells requires simultaneous expression of NifM protein. Int. J. Biochem. Cell Biol. 34, 33–42. https://doi.org/10.1016/s1357-2725(01)00102-9 (2002).
Eseverri, A. et al. Use of synthetic biology tools to optimize the production of active nitrogenase Fe protein in chloroplasts of tobacco leaf cells. Plant Biotechnol. J. 18, 1882–1896. https://doi.org/10.1111/pbi.13347 (2020).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. https://doi.org/10.1093/nar/gkaa913 (2021).
Weits, D. A., van Dongen, J. T. & Licausi, F. Molecular oxygen as a signaling component in plant development. New Phytol. 229, 24–35. https://doi.org/10.1111/nph.16424 (2021).
Koskela, E. V. & Frey, A. D. Homologous recombinatorial cloning without the creation of single-stranded ends: Exonuclease and ligation-independent cloning (ELIC). Mol. Biotechnol. 57, 233–240. https://doi.org/10.1007/s12033-014-9817-2 (2015).
Gietz, R. D. & Schiestl, R. H. Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 35–37. https://doi.org/10.1038/nprot.2007.14 (2007).
Hill, H. D. & Straka, J. G. Protein determination using bicinchoninic acid in the presence of sulfhydryl reagents. Anal. Biochem. 170, 203–208. https://doi.org/10.1016/0003-2697(88)90109-1 (1988).
Acknowledgements
This work was supported, in whole or in part, by the Bill & Melinda Gates Foundation (INV-005889). Under the grant conditions of the Foundation, a Creative Commons Attribution 4.0 Generic License has already been assigned to the Author Accepted Manuscript version that might arise from this submission. R.T.G. and J.B were supported by Biotechnology and Biological Sciences Research Council, grant award BB/P012574/1. LCS and AB were supported by U.S. Department of Energy, Office of Science, Basic Energy Sciences (BES) award DE-SC0010687 to LCS. LP-T was recipient of FPU16/02284 from Ministerio de Educación, Cultura y Deporte. XJ is recipient of a doctoral fellowship from Universidad Politécnica de Madrid. SM-M was recipient of Becas de Colaboración (998142) from Ministerio de Educación y Formación Profesional. We thank Amanda Mpofu for helping with DNA constructs.
Author information
Authors and Affiliations
Contributions
L.P.-T., D.C., S.M.-M., A.B., R.T.G., X.J., G.L.-T. and S.B. carried out the experimental work. M.V. performed yeast fermentations. L.P.-T., X.J., R.T.G., L.C.S., J.B., S.B. and L.M.R. designed experiments, analyzed data, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Payá-Tormo, L., Coroian, D., Martín-Muñoz, S. et al. A colorimetric method to measure in vitro nitrogenase functionality for engineering nitrogen fixation. Sci Rep 12, 10367 (2022). https://doi.org/10.1038/s41598-022-14453-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-14453-x
This article is cited by
-
Functional expression of the nitrogenase Fe protein in transgenic rice
Communications Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
