
Abstract
The epigenetic reader, bromodomain-containing 4 (BRD4), is overexpressed in hepatocellular carcinoma (HCC), and BRD4 inhibition is considered as a new therapeutic approach. The BRD inhibitor JQ1 is known to inhibit the enrichment of BRD4 at enhancer sites. Gene network analyses have implicated long non-coding RNAs (lncRNAs) in the effects of JQ1, but the precise molecular events remain unexplored. Here, we report that in HepG2 cells, JQ1 significantly reduced various proliferation-related lncRNAs, but up-regulated the known liver tumor marker, MALAT1. Using ChIP-sequencing data, ChIP-qPCR, luciferase reporter assays, and chromatin conformation capture (3C), we characterized the MALAT1 gene locus. We found that JQ1 elicited a rearrangement of its chromatin looping conformation, which involved the putative enhancers E1, E2, E3, the gene body, and the promoter. We further found that the forkhead box protein A2 (FOXA2) binds to E2 and the promoter; suppression of FOXA2 expression resulted in MALAT1 up-regulation and increased cell proliferation. These results suggest that the inhibition of MALAT1 may improve the effect of BET inhibitors as an anti-cancer therapy and that FOXA2 would be a suitable target for that approach.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer. HCC is prevalent cancer globally and a leading cause of cancer-related death1,2. Significant epigenetic alteration exists in HCC3. Therefore, epigenetic transcriptional regulators may be considered as potential therapeutic targets for anti-cancer treatment4. The epigenetic reader, BRD4, a member of the bromodomain and extraterminal (BET) proteins (BRD2, BRD3, BRD4, and BRDt) family, recognizes acetylated lysine residues of H3 tails with two tandem bromodomains (BD1 and BD2). Accumulation of BRD4 in hyper-acetylated chromatin regions, promoters, and enhancers facilitates their interaction and activates transcription5. In HCC, BRD4 is overexpressed and promotes gene expression related to cell migration, invasion, and apoptosis6,7. For example, BRD4 is closely associated with the overexpression of the key oncogene MYC; thus, inhibition of BRD4 is considered as a therapeutic strategy8,9,10. JQ1, a pan-bromodomain inhibitor with a high affinity to BRD4, enables the study of the antitumor effect of BRD4 inhibition11,12. Previous studies showed that JQ1 inhibits cancer cell proliferation and promotes apoptosis in various cancer cells by inhibiting BRD4 binding to super-enhancers of target genes13. Several studies were performed on transcriptome analysis to identify mechanisms and potential targets of BET inhibitors in the treatment of cancer13. More generally, the inhibition of BET proteins has been highlighted as a new therapeutic strategy for cancer, neurological, and inflammatory disease14,15
lncRNAs play diverse roles in regulating gene transcription, translation, post-transcriptional, and epigenetic modification16. Notably, lncRNAs play a role in tumor suppression (e.g., GAS5, LINC-PINT, MEG3) and tumorigenesis (e.g., HOTAIR, RCAR4, MALAT1). The abnormal expression of lncRNAs affects the malignity, growth, proliferation, and migration of cancer cells17. Thus, a role for lncRNAs in cancer has been established. However, the underlying mechanisms are poorly understood. Most reports are limited to genetic changes, mainly related to MYC18,19, while epigenetic mechanisms have received comparatively less attention. Here, we explored the mechanism of tumor-related lncRNA expression by inhibiting the BET protein, BRD4, in HepG2 cells, an established model for HCC.
Results
JQ1 treatment leads to the upregulation of MALAT1
To study the role of BRD4 in the HepG2 cells, we treated them with JQ1. This led to a significantly reduced proliferation within 24 h, and the effect increased further until at least 72 h (Fig. 1A; Supplementary Fig. S1A). We used the 24 h-time point for RNA-seq analysis. Of a total of 856 differentially expressed lncRNAs (DElncRNAs), 333 were up-regulated and 523 down-regulated by JQ1 (Fig. 1B). Heatmaps of the top 40 up- and down-regulated DElncRNAs are shown in Fig. 1C (numerical values are listed in Supplementary Table S1).

Differential lncRNA expression in JQ1-treated HepG2 cells. (A) HepG2 cells were treated with JQ1 (5 µM) or vehicle (DMSO) for the indicated durations, and cell proliferation was determined using a WST-1 assay. The data represent three biological indenpendent experiments. **p < 0.01. (B) Pie chart displaying the number of up-regulated (yellow) and down-regulated (green) lncRNAs. (C) Heat map representing the top 40 up- and down-regulated DElncRNAs. (D) Log2 fold changes of 9 selected lncRNAs affected by JQ1 and previously known to be over-activated in HCC. (E) qRT-PCR analysis of 6 selected DElncRNAs levels. The data represent three independent experiments. The values are mean ± SD of triplicate wells. **p < 0.01.
At least some of the downregulated lncRNAs (Fig. 1D, E) were previously found to be highly expressed in liver cancer and to promote proliferation and metastasis (AOC4P, PVT1, DANCR, DBH-AS1, HOXD-AS1, HNF1A-AS1, ANRIL)20,21,22,23,24. These lncRNAs probably are also important for HepG2 cells: When we randomly subjected one of them (DANCR) to RNA interference (Supplementary Fig. S2A), this resulted in a markedly decreased number and proportion of EdU-positive HepG2 cells (Supplementary Fig. S2B,C), in line with the known oncogenic role of DANCR.
In contrast, we could not make an obvious physiological link for an up-regulated lncRNA (Fig. 1D, E; MALAT1 and TUC338). Interestingly, one of them was MALAT1 (Fig. 2A, B), which appeared paradoxical because MALAT1 is known to be highly expressed in liver cancer25, in line with our own bioinformatics analysis using The Atlas of non-coding RNA in Cancer (TANRIC; https://ibl.mdanderson.org/tanric/design/basic/main.html)26 (Supplementary Fig. S3). However, the stimulation of MALAT1 expression was observed not only with JQ1 but also with other BET inhibitors (OTX015 and ABBV-075) (Fig. 2A, B, obtained by RNA-seq and qRT-PCR, respectively). Furthermore, an antisense oligonucleotide (ASO) directed against MALAT1 (Supplementary Fig. S4) increased the anti-proliferative effect of JQ1, although the oligo alone did not affect cell proliferation (Fig. 2C). This result indicated that the up-regulation of MALAT1 dampened the anti-proliferative effect of JQ1 (Fig. 1A). We, therefore, decided to take a closer look at the MALAT1 gene regulation in JQ1-treated HepG2 cells.

MALAT1 expression and putative MALAT1 enhancers in JQ1-treated HepG2 cells. (A) RNA-seq read densities (left) and corresponding log2 fold changes (right) of the MALAT1 gene transcripts in BET inhibitor-treated vs. control HepG2 cells. (B) qRT-PCR analysis of MALAT1 levels. The data represent three independent experiments. The values are mean ± SD of triplicate wells. **p < 0.01. (C) Cell proliferation was determined using WST-1 assay in MALAT1 ASO- and/or JQ1-treated HepG2 cells. The data represent three biologically independent experiments. *p < 0.05 and **p < 0.01. (D) USCS genome browser view of the GRO-seq peaks, H3K27ac enrichment, p300 binding sites, and BRD4 binding sites along the MALAT1 locus (chr11: 65,468,400–65,509,628). The potential MALAT1 enhancer regions E1, E2, and E3 upstream of the MALAT1 gene are denoted. MALAT1 expression from RNA-seq read densities is represented with black (untreated) and green (JQ1-treated) peaks. (E) Verification of putative MALAT1 enhancers by luciferase reporter gene assays. The data represent three independent experiments. **p < 0.01.
Identification of putative MALAT1 enhancers
We examined ENCODE ChIP-seq and global run-on sequencing (GRO-seq) data to localize the potential MALAT1 enhancers (Fig. 2D). Using the GRO-seq peaks (GSE92375), H3K27ac ChIP-seq peaks (GSE29611), and p300 ChIP-seq peaks (GSE32465) at the UCSC Genome browser, we analyzed the upstream regions of MALAT1. In region (chr11: 65,487,241–65,488,714), we found enrichment for H3K27ac that co-localized with the lncRNA gene LINC02736, whose expression was decreased by JQ1 (Supplementary Fig. S5). In addition, we identified three putative enhancer loci (E1, E2, and E3) further upstream (Fig. 2D; Table 1). We observed an approximately 20-fold increased luciferase reporter gene expression by the E2 region but not the E1 or E3 regions (Fig. 2E). From these results, we hypothesized that the increased MALAT1 expression in JQ1-treated HepG2 cells might be regulated by enhancer E2.
FOXA2, but not FOS, is involved in MALAT1 expression and HepG2 cell proliferation
Next, we searched for potential regulators, especially transcription factors (TFs), that might be involved in the JQ1-caused MALAT1 gene upregulation. Using RNA-seq, we found that 274 mRNAs were up-regulated and 737 down-regulated by JQ1 (Supplementary Fig. S6A). The heatmaps of the top 40 up- and down-regulated differentially expressed mRNAs (DEmRNAs) are shown in Fig. 3A (numerical values are listed in Supplementary Table S2). The DEmRNAs were associated with cancer, hepatic system disease, cell death and survival, and cellular growth and proliferation (Fig. 3B). Many down-regulated genes were related to angiogenesis and negative regulation of apoptosis (Supplementary Fig. S6B). IPA network analysis highlighted known tumor cell apoptosis-related genes (Fig. 3C), some of which we validated by qRT-PCR (Fig. 3D). More to the point, we found that several TFs were also altered, including the apoptosis-associated genes of Fig. 3C (Fig. 3E). Of these, we validated four up-regulated (FOS, EGR1, ZFP36, ID2, JUND) and two down-regulated (FOSL1 and FOXA2) TFs by qRT-PCR (Fig. 3F).

Expression of DEmRNAs and selected TFs in JQ1-treated vs. control HepG2 cells. (A) Heat map representing the top 40 up- and down-regulated DEmRNAs (p-value < 0.05, log2-fold change ≥ 1.5, log2-fold change ≤ − 1.5). (B) Disease and biofunction analysis of differentially expressed genes using IPA. (C) IPA network analysis of tumor cell apoptosis-related genes in JQ1-treated cells. The DEmRNAs are colored according to their predicted activation state following JQ1 treatment, activated (red) or suppressed (green). The arrows indicate predicted relationships: Red leads to activation; yellow, findings inconsistent with the state of downstream molecule. (D) qRT-PCR analysis of selected DEmRNAs. The data represent three independent experiments. The values are the mean ± SD of triplicate wells. **p < 0.01. (E) Heat map showing expression of eight selected TF mRNAs in JQ1-treated vs. control cells, each in triplicate. (F) Effect of JQ1 on the mRNA levels of FOXA2 and other TFs (qRT-PCR). The values are the mean ± SD of triplicate wells. **p < 0.01.
Bioinformatics analysis (by IPA) suggested that one of the upregulated TFs, FOS, regulates MALAT1 (Supplementary Fig. S7A), in line with DNA sequence analysis that revealed the co-localization of FOS binding sites and putative MALAT1 enhancers (Supplementary Fig. S7B). However, both in the absence and presence of JQ1, the levels of MALAT1 were not significantly changed by a FOS siRNA, neither was the JQ1-caused increment of MALAT1 expression (Fig. 4A; Supplementary Fig. S7C). Furthermore, JQ1 did not increase the binding of FOS to the promoter and putative enhancer regions of MALAT1 (Supplementary Fig. S7D). These results indicate that contrary to expectation, FOS is not involved in the regulation of MALAT1 in the HepG2 cells.

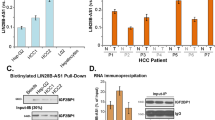
Effects of FOXA2 on MALAT1 transcription and proliferation. (A) qRT-PCR analysis of MALAT1 levels in JQ1- and FOS siRNA-treated HepG2 cells. The values are the mean ± SD of triplicate wells. **p < 0.01. (B) Analysis of FOS (GSM2797520) and FOXA2 (ENCSR066EBK) binding to the MALAT1 locus. H3K27ac enrichment and TFs binding (gray lines) from published ChIP-seq data in HepG2 cells. (C) ChIP-qPCR analysis of FOXA2 binding in JQ1-treated HepG2 cells. X-P, X-1, X-2, X-3, amplicons (B red lines). Enrichment was calculated relative to input DNA from three independent experiments. The values are the mean ± SD of triplicate experiments. *p < 0.05 and **p < 0.01. (D) qRT-PCR analysis of MALAT1 levels in FOXA2 siRNA-treated cells. The values are the mean ± SD of triplicate wells. **p < 0.01. (E) Cell proliferation assay of FOXA2 siRNA-treated cells. The data represent three biologically independent experiments. **p < 0.01.
Next, we focused on the down-regulated TF, FOXA2 (Fig. 3E, F). Bioinformatics analysis of published HepG2 ChIP-seq data indicates that FOXA2 binds to the putative enhancer E2 (X-3) and the promoter (X-P) regions of the MALAT1 gene (Fig. 4B), as validated by our ChIP-qPCR analysis. These bindings were significantly reduced by JQ1 (Fig. 4C). In contrast, the binding of FOXA2 to X-1 and X-2 did not co-localize with E1 or E3 (Fig. 4B), and the JQ1 treatment did not elicit a statistically significant change of FOXA2 binding to X-1 and X-2 (Fig. 4C). These results suggest that the direct binding of FOXA2 to the MALAT1 promoter and enhancer E2, but not E1 or E3, interferes with the transcription of MALAT1, thus mirroring the effect of E2, but not E1 or E3, on luciferase reporter gene expression (compare with Fig. 2E).
The reduction of FOXA2 mRNA (Supplementary Fig. S7E) and protein (Supplementary Fig. S7F) by RNA interference led to a significant increase of MALAT1 expression (Fig. 4D) and an increase in the proliferation of the HepG2 cells (Fig. 4E). Of note, we observed the same reciprocal relationship between FOXA2 and MALAT1 in Huh7 cells, another human HCC line (Supplementary Fig. S8). This result suggests that MALAT1 expression stimulates cell proliferation under negative control by FOXA2.
JQ1 treatment reconfigures the MALAT1 locus
To better understand the mechanism of how JQ1 affects MALAT1 expression, we performed a 3C assay. Using the promoter region as the anchor (P), we assessed the relative positions of E1, E2, and E3 in the absence and presence of JQ1. Figure 5A shows that in the absence of JQ1, E2 (amplicon C2-P3) and the gene body M (amplicon M-P2), but neither E1 (amplicon C1-P3) nor E3 (amplicon C3-P1), associated with the promoter. Upon the addition of JQ1, all three putative enhancers became associated with the promoter, while the gene body was no longer associated (Fig. 5A). These interactions were confirmed by sequencing the agarose gel bands (Fig. 5B).

JQ1 reconfigures the MALAT1 gene locus. (A) 3C analysis of MALAT1 gene locus in JQ1-treated and control HepG2 cells. Top, diagram of the MALAT1 gene locus showing the restriction enzyme sites used for 3C. Bottom, PCR results of 3C experiment. E1, E2, E3, putative enhancers; M, P1, P2, P3, C1, C2, C3, primers. (B) Sequencing of the MALAT1 enhancer and promoter intrachromosomal loop products. Green and red lines indicate the sequences of each fragment marked on the right. (C) Schematic interpreting the 3C analysis. In untreated HepG2 cells (left), the E2 enhancer and FOXA2 and BRD4 are key components of the promoter-associated chromatin complex, in contrast with E1 and E3. In JQ1-treated HepG2 cells (right), the reconfigured chromatin complex also involves E1 and E3 but loses FOXA2 and a significant portion of the BRD4. These changes result in an increased expression of MALAT1.
Discussion
This study found that when HepG2 cells were treated with JQ1, the long non-coding RNA MALAT1, which has been positively correlated with malignancy, was up-regulated. Our data suggest the down-regulation of the transcription factor FOXA2 and a reconfiguration of the associated chromatin complex as an underlying mechanism.
The JQ1-caused up-regulation of MALAT1 appears paradoxical because BET inhibitors are being considered as anti-cancer agents. However, MALAT1 is highly expressed in various cancers, including liver, lung, and breast cancer, and plays a role in cancer progression27. In addition, the MALAT1 expression level is negatively correlated with the survival rate in cancer patients28. MALAT1 induces cell proliferation and metastasis via the MAPK/ERK and PI3K/AKT signaling pathways in retinoblastoma and ovarian cancer, respectively29,30, and it is known to enable the high expression of the key oncogene MYC in thymic epithelial tumors31. Interestingly, in HepG2 cells, MALAT1 was also found in mitochondria, and its knockdown limited ATP synthesis and tumor cell invasion32. In addition, MALAT1 causes chemotherapy resistance by regulating miR-216b in HCC23. Taken together, literature strongly suggests that MALAT1 expression should be considered as an undesired feature of HCC and other tumors.
We have recently shown that JQ1 down-regulates MYC in HCC cells33, which is in line with the anti-cancer effects of JQ1 in other tumors. Similarly, JQ1 reduced the expression of pro-apoptotic BCL2L11 in HCC9. However, in prostate cancer, JQ1 inhibited the transcriptional repressor FOXA1, thereby increasing the expression of invasion genes34 or even activating the DNA damage response35. The increased expression of MALAT1 after the JQ1 treatment that we described here may also contribute to the unwanted effects of JQ1. These findings collectively emphasize the need to learn more about the mechanisms of BET inhibitors as potential anti-cancer agents. Hence, investigating the mechanisms regulating the overexpression of MALAT1 by JQ1 treatment may contribute to understanding the unwanted side effects of the BET inhibitors.
In our study, contrary to expectations36, the general TF FOS did not regulate MALAT1. Instead, we identified the lineage-specific TF FOXA2 as a candidate for the modulation of MALAT1 expression in HepG2 cells. The forkhead box (FOX) proteins are transcription factors related to cancer development and progression. FOXA1 is a well-studied regulator of estrogen receptor (ER) and androgen receptor (AR) activity in breast and prostate cancer37. In this context, FOX proteins play a crucial role in the rearrangement and reprogramming of super-enhancers38,39. FOXA1 and FOXA2 regulate the transcription of liver-specific genes and are known to complement each other40. In addition, the importance of FOXA2, particularly concerning liver disease, has been demonstrated41. Interestingly, FOXA1 and FOXA2 play dual roles as tumor suppressors and oncogenes42. FOXA1 is a transcriptional repressor and reduces the viability and motility in liver cancer cells43, while FOXA2 inhibits EMT in HCC, breast cancer, and lung cancer37,44,45. Hence, our data suggest that a focus on FOXA2 in HCC may help address the problem of JQ1’s and potentially other BET inhibitors’ detrimental effects in anti-cancer therapy.
In the present study, we associated the JQ1-promoted MALAT1 expression with decreased binding of FOXA2 to the promoter and E2 along with the formation of an (E1, E2, E3)-promoter complex, where E1, E2, and E3 are putative enhancers that we identified. JQ1 has been shown to directly bind to FOXA1, which neutralizes the repressor function of that TF34. Our finding that JQ1 reduced the binding of FOXA2 to the MALAT1 gene locus, along with an increase of MALAT1 expression, points to a similar mechanism.
Our data show that JQ1 affects MALAT1 expression by two mechanisms. The first mechanism is indirect and is mediated by the reduced expression of FOXA2, probably caused by the interference of JQ1 with the activity of BRD4 at the FOXA2 locus. This mechanism would be similar to the typical effects of JQ1 on other genes. It leads to the increased expression of MALAT1, as supported by our findings that FOXA2 binds to E2 and that a knockdown of FOXA2 increased the expression of MALAT1. These data reveal that FOXA2 is a repressor of the MALAT1 gene in the HepG2 cells. The second mechanism directly affects MALAT1 expression, as indicated by our finding (by ChIP-qPCR) of a reduced association of BRD4 with the MALAT1 promoter region upon JQ1 treatment. However, the outcome (stimulation versus inhibition of MALAT1 expression) is not yet certain. In general, one might expect that the reduced BRD4 availability reduces the expression of MALAT1 just like it reduces the expression of FOXA2 and other genes. Such a mechanism would counteract the indirect, FOXA2-mediated effect. However, our 3C analysis of the MALAT1 promoter and upstream region points to the opposite possibility. We found that JQ1 treatment, which implies a reduced BRD4 level, led to a re-organization of the enhancer-containing chromatin loops associated with the MALAT1 promoter. We note that even reduced levels of BRD4/mediators by BET inhibitors are sufficient to maintain enhancer-promoter interaction46. In addition to E2 (now free of its repressor), the putative enhancers E1 and E3 became directly associated with the promoter, suggesting the possibility of a stimulatory effect on MALAT1 gene expression. Future experiments will need to determine the direct effect of JQ1 on MALAT1 gene expression and the relative contributions of the indirect vs. direct mechanisms. It is worth mentioning that we observed the reciprocal relationship between the FOXA2 and MALAT1 also in the independently derived Huh7 human HCC cell line (Supplementary Fig. S8), indicating that the mechanistic relationships that we studied in the HepG2 cells are not a cell line-specific artifact.
In conclusion, our study suggests a regulatory model for the up-regulation of the lncRNA MALAT1 due to JQ1 treatment (Fig. 5C). The model predicts that manipulating MALAT1 expression could improve the therapeutic effect of BET inhibitors in HCC. Firstly, JQ1 inhibits the binding of FOXA2, a repressor of MALAT1 expression, to the MALAT1 enhancer E2 and the promoter. Secondly, alteration of chromatin looping recruits the enhancers E1 and E3 to the promoter site. Thus, further analysis of the MALAT1 promoter-associated chromatin looping is likely to suggest additional approaches to improve the BET-based therapy.
Experimental procedures
Cell culture and BET inhibitor treatment
The HCC cell line HepG2 was purchased from the Korean Cell Line Bank. HepG2 cells were cultured in Minimum Essential Medium supplemented with 10% fetal bovine serum (FBS) and penicillin (100 units/ml)/streptomycin (100 mg/ml) (Thermo Fisher Scientific, Waltham, MA, USA). The medium was replaced every 3–4 days. The cells were cultured in a humidified incubator at 37 °C with a 5% CO2 atmosphere. JQ1 was purchased from MedChemExpress (Monmouth Junction, NJ, USA). JQ1 was present at a concentration of 5 μM for 24 h.
Total RNA sequencing
RNA sequencing (RNA-seq) was performed as previously described47. Total RNA was extracted from HCC cells using RNAiso Plus (Takara, Shiga, Japan) and a Qiagen RNeasy Mini kit (Qiagen, Hilden, Germany). RiboMinus Eukaryote kit (Invitrogen, Carlsbad, CA, USA) was used for Ribosomal RNA (rRNA) depletion. An RNA library was created by a NEBNext Ultra directional RNA library preparation kit from Illumina (New England BioLabs, Ipswich, MA, USA). RNA library sequencing was performed on the Illumina HiSeq2500 platform (Macrogen, Seoul, Korea). Transcriptome sequencing was performed on independent RNA samples from DMSO-treated (3 samples) and JQ1-treated (3 samples) HepG2 cells in biological triplicate.
Differentially expressed genes analysis using RNA-seq data
For mRNA analysis, FASTQ files from RNA-seq were clipped and trimmed of adapters, and low-quality reads were removed using Trimmomatic48. These FASTQ files were aligned using STAR (version 2.7.8) aligner software with a UCSC hg38 reference49. Differentially expressed mRNAs (DEmRNAs) were analyzed using DESeq2 with the default parameters50. For lncRNA analysis, the raw data were trimmed with Trimmomatic (version 0.36)48 and processed using Bowtie2 (version 2.3.5)51 or STAR (version 2.7.8)49 aligner software with a GenCode GRCh38 reference (https://www.gencodegenes.org/human/) or an LNCipedia reference (https://lncipedia.org/; version 5.2)52. RNAs that exhibited an absolute log2-fold change larger than 1.5 or smaller than − 1.5 (log2-fold change ≥ 1.5 and log2-fold change ≤ −1.5, p-adjusted < 0.05) were designated as DEmRNAs or DElncRNAs. The dataset accession number GSE158552 was deposited in the Gene Expression Omnibus database53.
Gene and lncRNA expression analysis using quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was extracted from HepG2 cells using RNAiso Plus (Takara, Shiga, Japan) according to the manufacturer’s instructions. cDNA was synthesized by PrimeScript reverse transcriptase (Takara, Shiga, Japan) and amplified using gene-specific primers (Supplementary Table S4). The primers were designed by BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). qRT-PCR was performed with TBGreen Premix Ex Taq II (Takara, Shiga, Japan). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or RNU6-1 (U6) were used as an internal control. After performing qRT-PCR, the results were analyzed using the critical threshold (△CT) and the comparative critical threshold (△△CT) methods in ABI 7500 (Applied Biosystems, Foster City, CA, USA) software with the NormFinder and geNorm PLUS algorithms. The data represent three independent experiments (n = 3).
Cell proliferation assay
Cell proliferation was assessed using a premixed water-soluble tetrazolium salt (WST-1) cell viability test (Takara, Shiga, Japan) according to the manufacturer's instructions. The cells were seeded at a density of 5 × 103 cells per well and treated with JQ1 for different durations (0 h, 24 h, 48 h, and 72 h). WST-1 was added to each well. After an additional 4 h incubation, absorbances were measured at 450 nm. The data represent three independent experiments (n = 3).
Ethynyldeoxyuridine (EdU) analysis was performed using an EdU Cell Proliferation Assay kit (Invitrogen, CA, USA), following the manufacturer’s instructions. After that, the cells were washed with phosphate-buffered saline, mounted with a 4’,6-diamidino-2-phenylindole (DAPI)-containing mounting solution (Vectashield, Vector Laboratories, Burlingame, CA, USA), and imaged by microscopy (Nikon Eclipse 80i, Tokyo, Japan). The percentage of EdU-positive cells was assessed using ImageJ (Bethesda, MD, USA) software. The data represent three independent experiments (n = 3).
Knockdown of gene expression using siRNA treatment
Knockdown (KD) of gene expression was performed using small interfering RNA (siRNA). After seeding, the cells were transfected with siRNA constructs and scrambled siRNAs using the RNAiMax transfection agent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. FOS siRNA (siFOS-1 ID: 115631 and siFOS-2 ID: VHS41046), DANCR siRNA (siDANCR-1 ID: n505292 and siDANCR ID: n272702), FOXA2 siRNA (siFOXA2-1 ID: s6691 and siFOXA2-2 ID: s6692), and Silencer Negative Control siRNA (AM4611) were purchased from Thermo Fisher Scientific. The siRNAs were used at a concentration of 10 nM for 48 h in the growth medium.
Knockdown of MALAT1 expression using ASO treatment
Knockdown (KD) of MALAT1 gene expression was performed using locked nucleic acid (LNA)-modified antisense oligonucleotides (ASOs). After seeding the cells, transfection was performed using RNAiMax transfection agent according to the manufacturer’s instructions with ASO constructs and scrambled ASOs. MALAT1 antisense LNA GapmeR and LNA GapmeR Negative control B were purchased from Qiagen. MALAT1 siRNA and scrambled siRNA were used at 10 nM or 50 nM for 24 h or 48 h in the growth medium.
Chromatin immunoprecipitation quantitative PCR (ChIP-qPCR)
The chromatin immunoprecipitation (ChIP) assay was performed as previously described54. Briefly, the HepG2 cell chromatin was incubated with antibodies against BRD4 (Bethyl; A301-985A50), FOS (SCBT; sc-166940x), FOXA2 (Abcam; ab256493) and then precipitated with Dynabeads Protein A beads (Invitrogen, CA, USA); normal rabbit IgG (CST; 2729) and normal mouse IgG (Santa Cruz; sc-2025) were used as controls. The immunoprecipitated DNA was analyzed by qRT-PCR, and the expression levels were normalized to the amounts of input DNA. The data represent three independent experiments (n = 3). Primers used for ChIP-qPCR are listed in Supplementary Table S5.
Genomic data analysis
We re-analyzed public H3K27ac ChIP-sequencing (seq) data sets in Gene Expression Omnibus (GEO) (GSE29611) as described previously55 and global run-on sequencing (GRO-seq) data sets in GEO (GSE92375). For the re-analysis, Trimmomatic (version 0.36)48 was used to trim the raw data and processed using Bowtie2 (version 2.3.5)51 or STAR (version 2.7.8)49 aligner software with a UCSC hg 38 reference. The ChIP-seq and GRO-seq peaks identified were analyzed with Homer (version 4.11)56 and visualized using UCSC Genome Browser (https://www.genome.ucsc.edu).
Western blotting assay
Cells were lysed with RIPA buffer for protein extraction after treatment. Proteins were separated using sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (Schleicher & Schuell Bioscience, Inc., Keene, NH, USA). The western blotting assay was performed using anti-β-actin (SCBT; sc-8432) and anti-FOXA2 (Abcam; ab256493) antibodies, both diluted at 1:1000.
Luciferase reporter assay
Putative enhancer regions (E1, E2, and E3) were amplified with LongAmp Taq 2X Master Mix (New England Biolabs, Ipswich, MA, USA), using forward and reverse primers that generated NheI and XhoI sites, respectively. These amplicons were cloned into the pGL4.26 construct (Promega, Madison, WI, USA). The primers used for cloning are listed in Supplementary Table S3. The cells were seeded into 24-well plates and transfected with Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA, USA). Luciferase activity was measured using the Dual-Glo Luciferase Assay kit (Promega, Madison, WI, USA). PRL-TK (Renilla luciferase expression construct; Promega) was used as an internal control. Luciferase activity was normalized to Renilla luciferase and the control (empty vector).
Chromosome conformation capture assay
Chromosome conformation capture (3C) assay was performed as previously described, with minor modifications57. HepG2 cells were cross-linked with 1% formaldehyde, and nuclei were prepared from approximately 1–2 × 106 cells. Five hundred units of BamHI, BglII, and EcoRI were used to digest the DNA overnight, followed by ligation and purification. The 3C products were quantified by Qubit assay kits (Thermo, Q32851) and amplified by PCR using TB Green Premix Ex Taq (Takara, BR420). The ligation of fragments was analyzed using agarose gel electrophoresis. Sequences of primers are presented in Supplementary Table S6.
Statistical analysis
Data are presented as the mean ± standard deviation (SD) of the mean. All statistical analyses were performed using the IBM SPSS Statistics 26.0 program (IBM corp., Armonk, NY). We used a one-way analysis of variance followed by Tukey’s honestly significant difference post hoc test. p-values < 0.05 were considered significant.
Data availability
The raw data of RNA-sequencing were deposited in the Gene Expression Omnibus (GEO) database with accession number GSE15855251.
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- DEmRNAs:
-
Differentially expressed mRNAs
- DElncRNAs:
-
Differentially expressed lncRNAs
- GRO-seq:
-
Global run-on sequencing
- H2K27ac:
-
Acetylated H3 lysine 27
- Chr:
-
Chromosome
- qRT-PCR:
-
Quantitative reverse transcription-polymerase chain reaction
- 3C:
-
Chromatin conformation capture
References
Balogh, J. et al. Hepatocellular carcinoma: A review. J. Hepatocell. Carcinoma 3, 41–53. https://doi.org/10.2147/JHC.S61146 (2016).
Yang, J. D. et al. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 16, 589–604. https://doi.org/10.1038/s41575-019-0186-y (2019).
Liu, M., Jiang, L. & Guan, X. Y. The genetic and epigenetic alterations in human hepatocellular carcinoma: A recent update. Protein Cell 5, 673–691. https://doi.org/10.1007/s13238-014-0065-9 (2014).
Duan, Y. et al. Targeting Brd4 for cancer therapy: Inhibitors and degraders. Medchemcomm 9, 1779–1802. https://doi.org/10.1039/c8md00198g (2018).
Shi, J. & Vakoc, C. R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 54, 728–736. https://doi.org/10.1016/j.molcel.2014.05.016 (2014).
Liu, S. et al. Autocrine epiregulin activates EGFR pathway for lung metastasis via EMT in salivary adenoid cystic carcinoma. Oncotarget 7, 25251–25263. https://doi.org/10.18632/oncotarget.7940 (2016).
Wang, Y. H. et al. BRD4 induces cell migration and invasion in HCC cells through MMP-2 and MMP-9 activation mediated by the Sonic hedgehog signaling pathway. Oncol. Lett. 10, 2227–2232. https://doi.org/10.3892/ol.2015.3570 (2015).
Ba, M. et al. BRD4 promotes gastric cancer progression through the transcriptional and epigenetic regulation of c-MYC. J. Cell. Biochem. 119, 973–982. https://doi.org/10.1002/jcb.26264 (2018).
Li, G. Q. et al. Suppression of BRD4 inhibitis human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget 7, 2462–2474. https://doi.org/10.18632/oncotarget.6275 (2016).
Zuber, J. et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528. https://doi.org/10.1038/nature10334 (2011).
Bandopadhayay, P. et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 20, 912–925. https://doi.org/10.1158/1078-0432.CCR-13-2281 (2014).
Shu, S. et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529, 413–417. https://doi.org/10.1038/nature16508 (2016).
Jiang, G., Deng, W., Liu, Y. & Wang, C. General mechanism of JQ1 in inhibiting various types of cancer. Mol. Med. Rep. 21, 1021–1034. https://doi.org/10.3892/mmr.2020.10927 (2020).
Doroshow, D. B., Eder, J. P. & LoRusso, P. M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 28, 1776–1787. https://doi.org/10.1093/annonc/mdx157 (2017).
Muller, S., Filippakopoulos, P. & Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 13, e29. https://doi.org/10.1017/S1462399411001992 (2011).
Zhang, X. et al. Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int. J. Mol. Sci. 20, 66. https://doi.org/10.3390/ijms20225573 (2019).
Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 21, 1253–1261. https://doi.org/10.1038/nm.3981 (2015).
Bian, B. et al. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the BET bromodomain inhibitor JQ1: Implications for individualized medicine efforts. EMBO Mol Med 9, 482–497. https://doi.org/10.15252/emmm.201606975 (2017).
Mertz, J. A. et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 108, 16669–16674. https://doi.org/10.1073/pnas.1108190108 (2011).
Ding, C. H. et al. The HNF1alpha-regulated lncRNA HNF1A-AS1 reverses the malignancy of hepatocellular carcinoma by enhancing the phosphatase activity of SHP-1. Mol. Cancer 17, 63. https://doi.org/10.1186/s12943-018-0813-1 (2018).
Lu, S. et al. The noncoding RNA HOXD-AS1 is a critical regulator of the metastasis and apoptosis phenotype in human hepatocellular carcinoma. Mol. Cancer 16, 125. https://doi.org/10.1186/s12943-017-0676-x (2017).
Wang, F. et al. Oncofetal long noncoding RNA PVT1 promotes proliferation and stem cell-like property of hepatocellular carcinoma cells by stabilizing NOP2. Hepatology 60, 1278–1290. https://doi.org/10.1002/hep.27239 (2014).
Yuan, S. X. et al. Long noncoding RNA DANCR increases stemness features of hepatocellular carcinoma by derepression of CTNNB1. Hepatology 63, 499–511. https://doi.org/10.1002/hep.27893 (2016).
Hu, X. et al. A systematic review of long noncoding RNAs in hepatocellular carcinoma: Molecular mechanism and clinical implications. Biomed. Res. Int. 2018, 8126208. https://doi.org/10.1155/2018/8126208 (2018).
Malakar, P. et al. Long noncoding RNA MALAT1 promotes hepatocellular carcinoma development by SRSF1 upregulation and mTOR activation. Cancer Res. 77, 1155–1167. https://doi.org/10.1158/0008-5472.CAN-16-1508 (2017).
Li, J. et al. TANRIC: An interactive open platform to explore the function of lncRNAs in cancer. Cancer Res. 75, 3728–3737. https://doi.org/10.1158/0008-5472.CAN-15-0273 (2015).
Gutschner, T., Hammerle, M. & Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. (Berl.) 91, 791–801. https://doi.org/10.1007/s00109-013-1028-y (2013).
Amodio, N. et al. MALAT1: A druggable long non-coding RNA for targeted anti-cancer approaches. J. Hematol. Oncol. 11, 63. https://doi.org/10.1186/s13045-018-0606-4 (2018).
Jin, Y., Feng, S., Qiu, S., Shao, N. & Zheng, J.-H. LncRNA MALAT1 promotes proliferation and metastasis in epithelial ovarian cancer via the PI3K-AKT pathway. Eur. Rev. Med. Pharmacol. Sci. 21(14), 3176–3184 (2017).
Xie, S. J. et al. mascRNA and its parent lncRNA MALAT1 promote proliferation and metastasis of hepatocellular carcinoma cells by activating ERK/MAPK signaling pathway. Cell Death Discov. 7, 110. https://doi.org/10.1038/s41420-021-00497-x (2021).
Iaiza, A. et al. METTL3-dependent MALAT1 delocalization drives c-Myc induction in thymic epithelial tumors. Clin. Epigenet. 13, 173. https://doi.org/10.1186/s13148-021-01159-6 (2021).
Zhao, Y. et al. Aberrant shuttling of long noncoding RNAs during the mitochondria-nuclear crosstalk in hepatocellular carcinoma cells. Am. J. Cancer Res. 9, 999–1008 (2019).
Choi, H. I. et al. Targeting of noncoding RNAs encoded by a novel MYC enhancers inhibits the proliferation of human hepatic carcinoma cells in vitro. Sci. Rep. 12, 855. https://doi.org/10.1038/s41598-022-04869-w (2022).
Wang, L., Xu, M., Kao, C. Y., Tsai, S. Y. & Tsai, M. J. Small molecule JQ1 promotes prostate cancer invasion via BET-independent inactivation of FOXA1. J. Clin. Investig. 130, 1782–1792. https://doi.org/10.1172/JCI126327 (2020).
Bowry, A., Piberger, A. L., Rojas, P., Saponaro, M. & Petermann, E. BET inhibition induces HEXIM1- and RAD51-dependent conflicts between transcription and replication. Cell Rep. 25, 2061–2069e2064. https://doi.org/10.1016/j.celrep.2018.10.079 (2018).
Hirata, H. et al. Long noncoding RNA MALAT1 promotes aggressive renal cell carcinoma through Ezh2 and interacts with miR-205. Cancer Res. 75, 1322–1331. https://doi.org/10.1158/0008-5472.CAN-14-2931 (2015).
Lupien, M. et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 132, 958–970. https://doi.org/10.1016/j.cell.2008.01.018 (2008).
Fu, X. et al. FOXA1 upregulation promotes enhancer and transcriptional reprogramming in endocrine-resistant breast cancer. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.1911584116 (2019).
Parolia, A. et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 571, 413–418. https://doi.org/10.1038/s41586-019-1347-4 (2019).
Lee, C. S., Friedman, J. R., Fulmer, J. T. & Kaestner, K. H. The initiation of liver development is dependent on Foxa transcription factors. Nature 435, 944–947. https://doi.org/10.1038/nature03649 (2005).
Bochkis, I. M. et al. Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nat. Med. 14, 828–836. https://doi.org/10.1038/nm.1853 (2008).
Myatt, S. S. & Lam, E. W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 7, 847–859. https://doi.org/10.1038/nrc2223 (2007).
He, S., Zhang, J., Zhang, W., Chen, F. & Luo, R. FOXA1 inhibits hepatocellular carcinoma progression by suppressing PIK3R1 expression in male patients. J. Exp. Clin. Cancer Res. 36, 175. https://doi.org/10.1186/s13046-017-0646-6 (2017).
Wang, J. et al. FOXA2 suppresses the metastasis of hepatocellular carcinoma partially through matrix metalloproteinase-9 inhibition. Carcinogenesis 35, 2576–2583. https://doi.org/10.1093/carcin/bgu180 (2014).
Zhang, Z. et al. FOXA2 attenuates the epithelial to mesenchymal transition by regulating the transcription of E-cadherin and ZEB2 in human breast cancer. Cancer Lett. 361, 240–250. https://doi.org/10.1016/j.canlet.2015.03.008 (2015).
Crump, N. T. et al. BET inhibition disrupts transcription but retains enhancer-promoter contact. Nat. Commun. 12, 223. https://doi.org/10.1038/s41467-020-20400-z (2021).
Jung, K. H. et al. RNA sequencing reveals distinct mechanisms underlying BET inhibitor JQ1-mediated modulation of the LPS-induced activation of BV-2 microglial cells. J. Neuroinflamm. 12, 36. https://doi.org/10.1186/s12974-015-0260-5 (2015).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. https://doi.org/10.1093/bioinformatics/bts635 (2013).
Loven, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. https://doi.org/10.1016/j.cell.2013.03.036 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Volders, P. J. et al. LNCipedia 5: towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 47, D135–D139. https://doi.org/10.1093/nar/gky1031 (2019).
Choi, H. I. et al. BET inhibitor suppresses migration of human hepatocellular carcinoma by inhibiting SMARCA4. Sci. Rep. 11, 11799. https://doi.org/10.1038/s41598-021-91284-2 (2021).
Kim, S. H. et al. Transcriptome sequencing wide functional analysis of human mesenchymal stem cells in response to TLR4 ligand. Sci. Rep. 6, 30311. https://doi.org/10.1038/srep30311 (2016).
Kang, S. C. et al. Transcriptomic profiling and H3K27me3 distribution reveal both demethylase-dependent and independent regulation of developmental gene transcription in cell differentiation. PLoS ONE 10, e0135276. https://doi.org/10.1371/journal.pone.0135276 (2015).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. https://doi.org/10.1016/j.molcel.2010.05.004 (2010).
Kim, Y. W., Lee, S., Yun, J. & Kim, A. Chromatin looping and eRNA transcription precede the transcriptional activation of gene in the beta-globin locus. Biosci. Rep. https://doi.org/10.1042/BSR20140126 (2015).
Funding
This work was supported by the National Research Foundation of Korea (NRF) Grants 2017M3A9G7073033 and 2020R1A2C1014193 (to Y.G.C.), 2016R1D1A1B04934970 (to K.H.J.), and 2014M3C9A3064693 (to Y.S.L.), 2020R1F1A1063217 (to B.B.) from the Korean government.
Author information
Authors and Affiliations
Contributions
H.I.C. designed and performed experiments, analyzed and interpreted the data, and prepared the manuscript; A.G.Y., M.N.B., and E.Y.Y. designed and performed experiments; B.B. analyzed and interpreted the data, edited the manuscript; J.C.C. and Y.S.L. analyzed next-generation sequencing and bioinformatics data; K.H.J. designed experiments, secured financial support, analyzed and interpreted the data, prepared and edited the manuscript; Y.G.C. designed experiments, secured financial support, analyzed and interpreted next-generation sequencing and bioinformatics data, edited the manuscript and made the final approval of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Choi, H.I., An, G.Y., Yoo, E. et al. The bromodomain inhibitor JQ1 up-regulates the long non-coding RNA MALAT1 in cultured human hepatic carcinoma cells. Sci Rep 12, 7779 (2022). https://doi.org/10.1038/s41598-022-11868-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-11868-4
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
