
Abstract
ALDH2 is a key enzyme in alcohol metabolism that protects cells from acetaldehyde toxicity. Using iHS, iSAFE and FST statistics, we identified regulatory acting variants affecting ALDH2 gene expression under positive selection in populations of European ancestry. Several SNPs (rs3184504, rs4766578, rs10774625, rs597808, rs653178, rs847892, rs2013002) that function as eQTLs for ALDH2 in various tissues showed evidence of strong positive selection. Very large pairwise FST values indicated high genetic differentiation at these loci between populations of European ancestry and populations of other global ancestries. Estimating the timing of positive selection on the beneficial alleles suggests that these variants were recently adapted approximately 3000–3700 years ago. The derived beneficial alleles are in complete linkage disequilibrium with the derived ALDH2 promoter variant rs886205, which is associated with higher transcriptional activity. The SNPs rs4766578 and rs847892 are located in binding sequences for the transcription factor HNF4A, which is an important regulatory element of ALDH2 gene expression. In contrast to the missense variant ALDH2 rs671 (ALDH2*2), which is common only in East Asian populations and is associated with greatly reduced enzyme activity and alcohol intolerance, the beneficial alleles of the regulatory variants identified in this study are associated with increased expression of ALDH2. This suggests adaptation of Europeans to higher alcohol consumption.
Similar content being viewed by others

Introduction
The Neolithic transition from a hunter-gatherer lifestyle to an agriculturist one, about 9000–13,000 years ago, included substantial changes in food processing and dietary habits associated with plant and animal domestication1. One of the key questions in biological anthropology is whether these changes resulted in selective pressure, influencing the expression of genes in the human genome. Identifying such loci has the potential to detect the underlying genetic variants contributing to the risk for various human diseases such as autoimmune diseases, cancer, or cardiovascular disease1. Alcohol consumption and culture-related drinking behavior is probably one of the major changes in human dietary habits and lifestyle over the last 10,000 years. Production of larger amounts of alcoholic beverages had probably begun by the early Neolithic. A recent study reports archaeological evidence for cereal-based beer brewing by the semi-nomadic Natufians at Raqefet Cave (Mount Carmel in the north of Israel) dating back 11,700–13,700 years ago2. Today, large amounts of alcohol are consumed in many societies. Recent data from the World Health Organization (WHO) show that worldwide about 3 million deaths and 132.6 million disability-adjusted life years are attributable to the harmful use of alcohol3. In particular, Europe stands out in the WHO data as the region with the highest alcohol consumption and the highest burden of alcohol-related diseases. Large amounts of episodic drinking (binge drinking) as well as chronic alcohol consumption are associated with several very harmful effects such as alcoholic liver disease, intestinal inflammation, cancer, hypertension, brain damage including adverse behavioural changes, and decreased fertility4,5. While heavy alcohol consumption can cause complex negative physiological effects, positive effects of light to moderate alcohol consumption have also been reported. Light to moderate alcohol consumption has been associated with a reduced risk of some forms of cardiovascular disease and autoimmune diseases6,7,8,9,10. The most commonly ingested alcohol is ethanol (EtOH, CH3CH2OH) which is absorbed from the gastrointestinal tract by passive diffusion. EtOH is oxidized in the first step, mainly in the liver, to acetaldehyde by alcohol dehydrogenases (ADH). The genes ADH4, ADH1A, ADH1B and ADH1C, which are located in an array in the region of chromosome 4q23, encode closely related proteins and carry out most of the EtOH oxidation in liver. Cytochrome P450 2E1 (CYP2E1) and the enzyme catalase (CAT) also participate in this metabolic pathway, albeit to a lesser extent. EtOH is also metabolized in non-liver tissue tissues such as the brain, mainly by the microsomal EtOH oxidation system (MEOS), involving the CYP2E1 enzyme11. In the second step of EtOH metabolism, acetaldehyde, which is a chemically reactive and toxic compound, is oxidized by aldehyde dehydrogenases (ALDHs) to acetate12,13.
Several studies provide evidence that recent positive selection acts on the ADH1B locus in Asian, European and African populations14,15,16,17,18,19. At this locus, two missense substitutions play a role at the SNPs rs1229984 (G > A; p.Arg48His) and rs2066702 (C > T; p.Arg370Cys). They express three different isoforms. The ADH1B*1 isoform with arginine at both codon positions is the most common allele globally, except in populations of East Asia ancestry. In East Asia, the derived allele ADH1B*2 (rs1229984) presents the common allele (with a frequency of about 0.70)13. The ADH1B*3 allele (rs2066702) occurs only in individuals of African ancestry, with allele frequencies ranging from 0.09 to 0.2813. The two derived isoforms ADH1B*2 and ADH1B*3 metabolise EtOH at about 11 and 3 times the rate of ADH1B*1, respectively13. Several studies report that rs1229984 in the ADH1B locus is associated with a reduced risk of alcoholism in different ancestries20,21,22,23,24,25,26,27. The positive selection on the derived allele was estimated to have occurred about 7000–15,000 years ago16,28,29, which overlaps with the time-frame of the origin and expansion of Neolithic agriculture in East Asia. Nonetheless, it remains unclear whether the driving selective force acting on this genetic polymorphism emanates from the protective effect against alcohol dependence or from the higher efficiency of this polymorphism in metabolizing EtOH.
The mitochondrial enzyme ALDH2 plays the key role in the second step of EtOH metabolism by converting acetaldehyde into acetate. ALDH2 is not only a major detoxification enzyme for EtOH-derived acetaldehyde, but is also involved in detoxifying reactive aldehydes derived from reactive oxygen species (ROS). Aldehydes are toxic molecules that can form genotoxic DNA- and protein-adducts in cells30. Accumulation of high levels of acetaldehyde can be mutagenic, carcinogenic31,32,33 and may negatively affects the immune system34. In contrast to the ADH genes, ALDH2 is expressed in most human tissues, with high levels in the liver, heart, kidney, and muscle tissues35. In the coding region of ALDH2, the missense variant rs671 (G > A; p.Glu504Lys) expresses the isoforms ALDH2*1 and ALDH2*2. The ALDH2*2 variant is found only in individuals of East Asian ancestry, reaching frequencies of up to 40% in some East Asian populations such as Han Chinese and Japanese13,36,37. This allele significantly affects alcohol metabolism because it results in an inactive enzyme and thus an excess of the toxic acetaldehyde in cells, even with moderate alcohol consumption. The symptoms are severe facial flushing, nausea, headache and tachycardia38. East Asians homozygous for ALDH2*2 have a very low risk for alcohol dependency26,38,39. The ALDH2 enzyme plays a central role in protecting cells from EtOH toxicity by metabolizing acetaldehyde (and other endogenous aldehyde products), is anti-inflammatory40, and functions in myocardial protection41,42,43. Accordingly, this gene is of great biomedical interest. ALDH2 is located at the human chromosomal region 12q24.12. Several genome-wide association studies (GWAS) have found this genomic region to be associated with multiple human diseases such as rheumatoid arthritis44, systemic lupus erythematosus45, type 1 diabetes46, hypertension47 and coronary artery disease48. This region, approximately 0.6 Mbp in size (according to the human reference genome), encompasses in addition to ALDH2 the genes CUX2, FAM109A, SH2B3, ATXN2, BRAP, ACAD10 and MAPKAPK5, as well as the uncharacterized transcript ENST00000546840.3 (UniProt F8VP50—Aldedh domain-containing protein), which partially overlaps with the genes ACAD10 and ALDH2. High FST values at linked sites at the ALDH2 locus point to some form of selection for this genomic region36. A recent study analysing rare singletons in the Japanese population identified the SNP rs3782886, which is in linkage disequilibrium (LD) with the missense SNP rs671 in the 12q24.12 region, as under recent positive selection49. In this study, we applied population genetic models of natural selection and included functional genetic data to identify the targets of positive selection in this genomic region. Several lines of evidence indicate that recent positive selection is acting on regulatory variants that influence ALDH2 gene expression in populations of European ancestry.
Results
Positive selection in populations of European ancestry
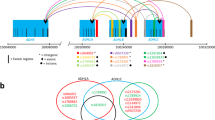
The iHS analysis shows evidence that the human chromosomal region 12.q24.12 is under positive selection in populations of European ancestry. Figure 1a plots the iHS scores in the European population GBR; Fig. 1b shows the pairwise FST values for GBR vs. the African population LWK across 12.q24.12. The red and green lines indicate significant (p < 0.01 and after Bonferroni correction p < 1 × 10–5, respectively) iHS scores and the genome-wide threshold (95% confidence level) for FST outlier loci (FST > 0.3).

(a) iHS p-values plotted across the human chromosomal region 12.q24.12 for the population GBR (European genetic ancestry); red/green lines: threshold for significant (p < 0.01; Bonferroni correction p < 1 × 10–5) iHS scores; (b) pairwise FST (GBR–LWK); red line: significant outlier loci with FST > 0.3. Bottom: position of genes and SNPs from Table 1.
Positive selection acts on regulatory variants of ALDH2
From the GTEx database, we obtained in total 1591 cis-QTLs that influence ALDH2 gene expression (Supplementary Table 1); of these cis-QTLs, we identified 204, 217 and 53 eQTLs that had significant (p < 0.01) iHS scores in the European samples GBR, TSI and FIN, respectively (Supplementary Table 2). We also obtained cis-eQTLs (1970 in total) for the other protein-coding genes located in this genomic region (CUX2, FAM109A, SH2B3, ATXN2, BRAP, ACAD10, MAPKAPK5) (Supplementary Table 3). In contrast to the eQTLs for ALDH2, we did not obtain significant iHS values for these SNP eQTLs, except for SNPs that also function as eQTLs for ALDH2. We further identified seven SNPs (rs3184504, rs4766578, rs10774625, rs597808, rs653178, rs847892, rs2013002) that are under positive selection in European populations that have very large global locus-specific FST values > 0.3, i.e. are outlier loci (Table 1). The corresponding EHH plots and pairwise FST values of these SNPs can be found in Supplementary Fig. 1 and Supplementary Table 4, respectively. The pairwise FST values for these SNPs comparing populations of European ancestries vs. African, East Asian and South Asian ancestries ranged from 0.253 to 0.691. The iSAFE analysis also identified several SNPs in the chr12q24.12 region under positive selection (Supplementary Table 5). Moreover, the identified seven SNPs under positive selection (by the iHS and FST statistics) were also identified by iSAFE as top-ranked mutations with iSAFE scores > 0.304, i.e., above the significant threshold (Fig. 2). These SNPs function as eQTLs for ALDH2, and the beneficial alleles are associated with increased ALDH2 gene expression in various human tissues (according to the set of tissues represented in GTEx) such as esophagus—mucosa, skin, muscle—skeletal, brain—nucleus accumbens, artery—tibial, artery—aorta, and thyroid. The average allele frequencies for these SNPs are given in Table 1; for most of these SNPs the frequency of the derived beneficial alleles reaches almost 50% in the European populations. In contrast, the derived alleles are very rare (< 0.3%) in African and East Asian ancestries and at low frequency in populations of South Asian ancestry (< 7% with the exception of rs847892). We also compared the allele frequency at these loci with ancient Eurasians, including ancient hunter-gatherers (8.2–7.5 kya) from the study of50. The allele frequency data from the latter study show for the SNPs rs3184504, rs4766578, rs10774625, and rs653178 that the ancestral alleles were fixed in ancient European hunter-gatherers. As expected, the Neanderthal and Denisovan data on the UCSC Genome Browser also show only ancestral alleles at these loci. In contrast, early European farmers (8.4–4.2 kya) and individuals with steppe ancestry (5.4–3.6 kya) had frequencies between 8 and 25% of the derived alleles at these loci. The analysis of the selection coefficient (s) revealed that s ranged from 0.04 to 0.1, suggesting very strong positive selection acting on these SNP eQTLs (Table 1). The corresponding allele trajectory plots, inferred by the Clues method, are presented in Supplementary Figure S2. In the European sample GBR we estimated the timing of positive selection (using the method Startmrca) of the derived beneficial alleles to be from about 3.0 to 3.7 kya with the exception of SNP rs847892, which we date at 6.0 kya (Table 1). This range of estimates are very similar to the TMRCA estimates calculated for the other European samples (TSI and FIN) (Supplementary Table 6). We also calculated the TMRCA for the derived allele of the East Asian-specific polymorphism (missense variants) at rs671-A/G and rs3782886-C/T in the East Asian population CHB, which yielded an estimation of 5.8 kya (CI: 4.8–6.7) and 5.4 kya (CI: 3.3–6.5), respectively. In addition, we used the method Clues to obtaining allele ages for the seven SNPs under positive selection. Clues calculated a similar timing of selection (2.6 kya to 4.5 kya) for the SNPs rs3184504, rs4766578 and rs10774625 as the Startmrca method (Supplementary Table S7). However, the timing of selection for the SNPs rs597808, rs653178 and rs2013002 was estimated to much older ages ranging in time frames from 7.4 kya to 14.1 kya; rs847892 between 21.3 to 30.1 kya.

iSAFE scores plotted for SNPs surrounding the chr12q24.12 region (5.6 Mbp window) for the population GBR (European genetic ancestry); also indicated are the SNPs identified by the iHS statistics as being under positive selection; the top-ranked SNPs are above the threshold sores iSAFE > 0.304.
We further included in the analysis the ALDH2 promoter variant rs886205-A/G, which is located − 360 bp from the ATG start codon of the ALDH2 gene51. This promoter variant shows very large genetic differentiation with global locus-specific FST = 0.378 (s.d. = 0.055). In the 1000 Genomes data the derived allele A is the common allele in European and South Asian populations with average frequencies of about 83% and 71%, respectively. In contrast, in populations of African and East Asian ancestry the common allele is the ancestral allele G with frequencies of about 78% and 84%, respectively. For the ALDH2 promoter variant, a study showed (in vivo and in vitro experiments) that the—360G (ancestral) allele has a significantly lower basal transcriptional activity than the − 360A (derived) allele52. Our LD analysis revealed that the positively selected SNPs are in complete LD (D’ = 1) with the ALDH2 promoter variant rs886205 (Table 2). The chromatin state data from RegulomeDB showed that the identified SNPs are associated with active transcription start site (TSS), enhancers and strong transcription in different tissues (Table 3). Importantly, the positively selected SNPs rs4766578 and rs847892 are located in the binding motif for the transcription factor hepatocyte nuclear factor 4 alpha (HNF4A). This transcription factor is an important regulatory element of ALDH2. The mapped phenotypes (Table 3) show that the positively selected SNPs are associated with various traits and diseases, in particular with blood pressure, cardiovascular disease, cholesterol level and autoimmune diseases. The variants rs597808 and rs2013002 are also associated with alcohol drinking and physiological traits such as blood pressure53. We pooled related traits (Supplementary Table 8) into four main trait category namely autoimmune diseases (AIS), blood pressure (BP), cardiovascular disease (CDS) and cancer to test the null hypothesis that the traits and the allele state are independent. We found a significant (χ2 = 28.828, df = 3, p value = 2.4e−06) relationship between the allele state and trait; the derived beneficial alleles are positively associated with AIS, BP and CDS whereas the ancestral alleles with cancer.
Discussion
This study provides evidence of positive selection across the human chromosomal region 12.q24.12. This finding is in line with two previous studies55,56. We identified seven SNPs (rs3184504, rs4766578, rs10774625, rs597808, rs653178, rs847892, rs2013002) that are under positive selection and show very large global locus-specific FST values (> 0.3), indicating high genetic differentiation between populations of European ancestry and populations from other global ancestries (Table 1). The GTEx data show that these SNPs function primarily as eQTLs for the ALDH2 gene. We further found that this genomic region is enriched in eQTLs that influence ALDH2 gene expression. A high number of these SNP eQTLs had significant iHS scores in the populations of European ancestry. In contrast, cis-eQTLs of the other genes located at chr12q24.12 showed no significant iHS values. In addition, the iHS results are supported by the iSAFE analysis which ranked the identified SNPs (eQTLs) as top-ranked mutations, with iSAFE scores > 0.304. This indicates that the target of positive selection are regulatory acting variants that influence ALDH2 gene expression. The derived beneficial alleles at these SNP eQTLs are associated with increased expression of ALDH2 in multiple human tissues. However, in the GTEx database, no ALDH2 cis-eQTLs are reported for the liver tissue. Nonetheless, the two positive selected SNPs, rs4766578 and rs847892, are located in binding sequences for transcription factor HNF4. That transcription factor is considered to be a master regulator of liver-specific gene expression57 and is an important regulatory element of ALDH2 gene expression35,58. Positive selection leads to changes in the allele frequencies at the transcription factor binding sites, which could potentially lead to significant changes in the binding specificity of the liver-specific transcription factor HNF4. Therefore, we are inclined to hypothesize that individuals carrying the positively selected haplotypes will have higher basal expression of ALDH2 than individuals lacking the positively selected haplotypes. In addition, the RegulomeDB data indicate that the positively selected SNPs are located in active enhancer histone marks in different tissues including the liver. Moreover, the positively selected SNP eQTLs are in complete LD with the ALDH2 promoter variant rs847892. This promoter polymorphism influences individual differences in acetaldehyde elimination. The ancestral allele G, the common allele in populations of African and East Asian ancestry, has a lower basal transcriptional activity than the derived allele A, the common allele in populations of European and South Asian ancestry52. These results suggest that higher transcriptional activity and increased ALDH2 expression in individuals of European ancestry represent a form of genetic adaptation to increased alcohol consumption, possibly enabling faster detoxification of acetaldehyde.
The derived beneficial alleles of these loci reach almost 50% in the European population, whereas in African and East Asian populations the frequencies are very low (< 0.003). The ancestral alleles at these positively selected loci appear to be fixed in ancient European hunter-gatherers, but in early farmers and individuals with steppe ancestry the frequencies of the derived alleles already range between 8 and 25%50. The applied Clues method found for the SNPs rs3184504, rs4766578 and rs10774625 evidence of very strong positive selection with s = 0.1, corresponding to an allele age of about 2.6 kya to 4.5 kya (Table 1). This is in line with the estimated timing of positive selection on the beneficial alleles in the European population GBR calculated by the Startmrca method which ranges from about 3.0 kya to 3.7 kya (except for rs847892 for which TMRCA was estimated to 6.0 kya). However, in contrast to the Startmrca method, the Clues method estimated the allele age for the other SNPs much further back in time to about 7.4 kya to 14.1 kya (again with the exception for rs847892 for which the allele age was estimated to about 21.3 kya to 30.1 kya). Nevertheless, the strong putative selection (s = 0.1) acting on several SNPs indicates that at these loci the alleles are much more intensely under positive selection than for example the lactase persistence locus SNP rs4988235 for which s = 0.0161 were calculated59. We further calculated the TMRCA for the East Asian-specific derived alleles rs671-A and rs3782886-C (using the Startmrca method), yielding an estimation of 5.8 kya (CI: 4.8–6.7) and 5.4 kya (CI: 4.3–6.5), respectively. Rs3782886, which is in LD with rs671, shows signals of very recent selection for the past 2000–3000 years in the Japanese population as reported in a recent study49. Noteworthy, rs671-A and rs1229984-A (ADH1B locus) were found in a subsequent study to be significantly associated with better survival in the Japanese population60. The estimated TMRCA for the derived alleles in our study suggests that these alleles spread in East Asia at a much earlier time than the beneficial alleles in populations of European ancestry. Archaeological evidence indicates early production of fermented alcohol in China61. Analysis of starch granules, phytoliths and fungi in food residues adhering to 8000–7000 year-old alcohol-making pottery vessels suggests that, in East Asia in the early Neolithic, alcoholic beverages were already being produced62. For Europe, archaeologically recognizable brewing material in Central European lakeside settlements show that alcoholic beverages were being produced in this region in the late Neolithic period about 6000 years ago63. A recent study suggests that extensive fermented alcoholic beverages such as beer were already consumed in Central Europe during the Iron Age64. Later, in Greek-Roman antiquity, a richly developed viticulture with high wine production was achieved and, in this period, wine became part of the daily diet of many people65. Alcohol consumption has apparently increased steadily since then in Europe, especially in the nineteenth century. In Germany, for example, the high level of consumption, in particular of strong spirits, in the early nineteenth century was—in analogy to the plague—referred to as Branntweinpest (brandy plague). Since the rs671-A allele leads to an inactive enzyme and thus to an excess of toxic acetaldehyde in cells with its negative physiological effects, we suggest that this allele may explain the differences in the signature of positive selection between populations of European and East Asian ancestry.
The ALDH2 enzyme plays a critical role in the detoxification of both acetaldehyde and ROS-generated aldehyde adducts such as 4-hydroxy-2-nonenal and malondialdehyde. This enzyme thus has cytoprotective effects reducing oxidative stress66,67. In particular, the ALDH2*2 variant (rs671-A/A), which is common only in individuals of East Asian ancestry, has been intensively studied in East Asians. While individuals with the rs671-A allele have a reduced risk of developing alcoholism, it increases their cancer risk68,69. Nevertheless, this allele was found to be associated in Japanese population with better survival60. In European populations this allele is virtually absent. In our study, however, the identified variants that are under recent positive selection in European populations act as regulatory variants and are associated with increased ALDH2 gene expression in various human tissues. This suggests that individuals carrying these beneficial alleles should be more quickly able to detoxify the body from higher amounts of acetaldehyde and ROS-generated aldehyde adducts. However, a recent study reports higher methylation in alcohol-dependent patients compared to controls in the ALDH2 promoter region70. Furthermore, that study suggests that positive and negative regulatory elements interact at the ALDH2 promoter to induce genotype-mediated epigenetic changes, leading to differential transcriptional activity of this gene. In addition, a GWAS reported that the SNPs rs597808 and rs2013002, which were found in this study under positive selection, are associated with alcohol consumption and risk of developing hypertension53. We therefore suggest that individuals carrying the positively selected alleles may be able to consume more alcohol (over longer time periods), but may also have a higher likelihood of becoming heavy drinkers and alcohol dependent. This, then, could lead to increased methylation of the ALDH2 promoter, resulting in decreased ALDH2 gene expression. Accordingly, the protective effects of ALDH2 against oxidative damage through acetaldehyde would be lost, resulting in increased risk of numerous oxidative stress-related diseases such as cancer, diabetes, inflammatory disorders and cardiovascular conditions such as hypertension and stroke. Indeed, we found that the derived beneficial alleles are positively associated with AIS, BP and CDS whereas the ancestral alleles with cancer.
To conclude, we found that very strong positive selection (with s ranging between 0.04 and 0.1) acts on regulatory variants affecting ALDH2 gene expression in populations of European ancestry. Estimation of the timing of positive selection on the beneficial alleles suggests that these variants were recently adapted, approximately 3000 to 3700 years ago. The timing of selection and the signals of very strong selection make the chromosomal region chr12q24.12 one of the most intensely selected regions in the genomes of individuals of European ancestry. In contrast to the known functional consequence of the ALDH2*2 variant (rs671) in East Asians, which is associated with alcohol intolerance, in Europeans the beneficial derived alleles are associated with increased ALDH2 gene expression. This suggests local adaptation to higher alcohol consumption in Europeans. We further hypothesize that the beneficial effects of higher ALDH2 expression leads to an increased detoxification capacity for acetaldehyde, but possibly also to increased likelihood of chronic alcohol abuse, leading to decreased ALDH2 expression and thus increased cell toxicity from EtOH-derived acetaldehyde as well as from ROS-generated aldehydes.
Materials and methods
Genomic data
We downloaded the phased genomic datasets from the 1000 Genomes project (phase 3; ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/)71. Only 1000 Genomes data were used in this study according to the Declaration of Helsinki. We obtained SNP data from 12 human populations: three representative populations each from African ancestry (AFR), European ancestry (EUR), South Asian ancestry (SAS) and East Asian ancestry (EAS) (populations names in accordance with the 1000 Genomes project—see Supplementary Table 9). We excluded related individuals and did not include the admixed populations from the datasets because of the underlying statistical principle of the method used to detect positive selection. We used the software program PLINK 1.972 (https://www.cog-genomics.org/plink/) and VCFtools v0.1.1473 (https://vcftools.github.io/index.html) to process the variant call format (VCF) files. We used the following filter parameters in VCFtools: –maf 0.05 (include only sites with a Minor Allele Frequency (MAF) greater than 0.05), –minQ 30 (include only sites with quality value above this threshold) and –remove-indels (exclude sites that contain an indel). Furthermore, we excluded all SNPs that deviated from Hardy–Weinberg equilibrium (with p-value < 1e−6) using PLINK –hwe midp threshold filter. We further excluded potential duplicated SNPS using bcftools version 1.10.2, (https://github.com/samtools/bcftools/) using the parameter norm –Ov –check-ref w –fasta-ref human_g1k_v37.fasta (ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/). SNP positions are in accordance with the human genome version GRCh37/hg19 (https://genome-euro.ucsc.edu/).
Population genetic analyses
To detect positive selection in phased genomic population data, we used the integrated Haplotype Score (iHS) approach74, which is implemented in the software programme selscan version 1.2.0a75 (https://github.com/szpiech/selscan). All scans, with default selscan model parameters, were run on phased whole chromosome data (except the Y-chromosome) with a genetic map from HapMap phase II b3776. The iHS approach compares extended haplotype homozygosity (EHH) values between alleles at a given SNP. It is based on the model of a selective sweep, in which a de novo adaptive mutation arises on a haplotype that is rapidly fixed in the population, thereby reducing genetic diversity around that locus74. The unstandardized iHS scores were normalized in default frequency bins across the entire genome using the script ‘norm’ provided by the selscan programme. Negative iHS values (iHS score < − 2.0) indicate unusually long haplotypes carrying the derived allele, and significant positive values (iHS score > 2.0) are associated with long haplotypes carrying the ancestral allele74. We used the Ensembl Variant Effect Predictor programme package (https://github.com/Ensembl/ensembl-vep)77 to map genetic information such as gene symbol and biotype to the analysed SNPs. We calculated empirical p-values for the obtained iHS scores across all chromosomes using R programme version 4.1.078 (https://www.r-project.org/). In this study we report only results for the human chromosomal region 12q24.12, the genomic location of the ALDH2 gene. We considered statistically significant (p < 0.01) iHS scores > 2.4 or < − 2.4; however, we also applied Bonferroni correction, which yields p-values p < 1 × 10–5 (= iHS scores > 4.2 or < − 4.2). We used the script 'colormap.plotting.R' provided by the selscan package to display the EHH plots for the SNPs that are under positive selection. Pairwise FST were calculated using Weir & Cockerham FST calculation implemented in VCFtool73. Negative FST values were set to zero. We calculated empirical p-values for the FST values (across all chromosomes) to obtain the significant threshold (p < 0.05) of outlier loci. In addition, locus-specific FST values and standard deviations (sd) across all analysed populations were calculated for SNPs that were detected to be under positive selection with the Genetix programme version 4.0579 (https://kimura.univ-montp2.fr/genetix/) applying the jackknife resampling procedure. We used the R package ggplot280 to plot iHS and FST values. Allele frequency data, SNP information and ancestral/derived allele state were obtained from the Ensembl genome browser (https://www.ensembl.org/index.html)81. We used LDlink, a web-based application (https://analysistools.cancer.gov/LDlink/?tab=home)82, to explore population-specific linkage disequilibrium (LD); we report D’ and goodness-of-fit statistics (chi-square statistics).
iSAFE analysis
The iSAFE programme (https://github.com/alek0991/iSAFE)56 was used to identify beneficial mutations in the genomic region of chr12q24.12. iSAFE exploits coalescent-based signals in the surroundings of a candidate region under positive selection to rank all mutations based on their contribution to the selection signal. We used as target population under selection the European population GBR and as control populations the populations ESN (African ancestry), BEB (South Asian ancestry) and CHB (East Asian ancestry). We used the default settings for analysing a 5.6 Mbp window (9060 SNPs) surrounding the chr12q24.12 region (hg19; chr12:109,200,001–114,800,000). The developer of iSAFE showed that iSAFE scores higher than 0.304 are considered to have p value < 1.34 × 10–8. We used this cut-off as the significant threshold.
Estimating timing of positive selection and selection coefficient (s)
We estimated the timing of selection on a beneficial allele using the R package Startmrca29. The method applies a Markov chain Monte Carlo simulation (MCMC) that samples over the unknown ancestral haplotype to generate a sample of the posterior distribution for the time to the most recent common ancestor (TMRCA). The model takes advantage of both the length of the ancestral haplotype on each chromosome and the accumulation of derived mutations on the ancestral haplotype to generate a sample of the posterior distribution for the TMRCA. The model requires a sample (panel) containing the haplotypes with the selected allele and a reference panel of haplotypes without the selected allele. In this study, populations of European ancestry were used as samples, and populations of the other analysed genetic ancestries were used as reference panels. Because the calculated TMRCA estimates for the populations of European ancestry were very similar regardless of the reference panels used, we report in this study only the TMRCA estimates calculated for the European populations, using the European populations both as sample panel and as reference panel. We also estimated TMRCA for the East Asian-specific functional variants rs671 (ALDH2 locus) and rs3782886 (BRAP locus)49 using the East Asian population CHB as sample and reference panel (see Supplementary Table 9 for the corresponding population names). After normalising the TMRCA data we calculated 95% credible intervals (CI = 0.95) for the timing estimates using the equal-tailed interval method implemented in the R package bayestestR83. We used recombination rates from the sex-averaged recombination map from deCODE to model recombination rate variation across the human genome. We analysed 1 Mb regions up- and downstream of the genetic variants under selection with an assumed mutation rate of 1.6 × 10–8. We ran three independent MCMC chains, each with 25,000 iterations. We discarded the first 9000 iterations (burn–in), retaining the remaining iterations. We assumed 25 years as generation time. We further used the programme Clues (https://github.com/35ajstern/clues59) to estimate timing of selection and the selection coefficient (s) of SNPs that were identified to be under positive selection by the iHS statistics. The method relies on MCMC samples of the gene tree at the SNPs of interest. We used the programme Relate v1.1.8 (https://myersgroup.github.io/relate/84) to obtain this tree and to extract coalescence times. We calculated coalescence rates and effective population sizes using the integrated script EstimatePopulationSize.sh (obtaining the .col file); we then applied the SampleBranchLengths.sh module with the following parameters: –num_samples (number of times branch lengths sampled) 100, –m (mutation rate) 1.25 × 10–8, target region chr12q24.12 with –first_bp 111,700,001 –last_bp 112,300,000 and –coal (the previously obtained population size file). The output file (resample.timeb) was then used to run the Clues programme (inference.py script) with the option –coal (.coal file) in order to account again for population size changes and with –tCutoff 1000 (time to ‘cut off’ the coalescence process). We used Clues also to calculate for each SNP the selection coefficient (s) and the corresponding likelihood ratio (logLR) statistics59.
GTEx and RegulomeDB functional data
We utilized expression quantitative trait loci (eQTLs) (accessed between May and July 2021 (dbGaP Accession phs000424.v8.p2) from GTEx Portal V8 Release (https://www.gtexportal.org/home/)85 to test whether any of the potential SNPs that are under positive selection function as eQTL. We included cis-eQTL variants within a 1 Mb window of analysed genes. The RegulomeDB database (https://regulomedb.org/)54 was used to obtained chromatin states; this database comprises known classes of genomic elements such as promoters, enhancers, transcription start sites, and transcription factor (TF) binding motifs. Additionally, mapped phenotype data were obtained from the NHGRI-EBI GWAS catalogue (https://www.ebi.ac.uk/gwas/)86 (accessed between May and July 2021).
Consent to participate/Consent to publish
Not applicable. The 1000 Genomes data are publicly available.
Data availability
The gnomic data can be obtained from 1000 Genomes database. The generated iHS and Fst dataset are available from the corresponding author on reasonable request.
Change history
10 October 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41598-023-44342-w
References
Ye, K. X. & Gu, Z. L. Recent advances in understanding the role of nutrition in human genome evolution. Adv. Nutr. 2, 486–496 (2011).
Liu, L. et al. Fermented beverage and food storage in 13,000 y-old stone mortars at Raqefet Cave, Israel: Investigating Natufian ritual feasting. J. Archaeol. Sci. Rep. 21, 783–793 (2018).
World Health Organization. Global status report on alcohol and health 2018. World Health Organization (WHO) (Geneva, 2018).
Rocco, A., Compare, D., Angrisani, D., Zamparelli, M. S. & Nardone, G. Alcoholic disease: Liver and beyond. World J. Gastroenterol. 20, 14652–14659 (2014).
Ricci, E. et al. Semen quality and alcohol intake: a systematic review and meta-analysis. Reprod. Biomed. Online 34, 38–47 (2017).
Ronksley, P. E., Brien, S. E., Turner, B. J., Mukamal, K. J. & Ghali, W. A. Association of alcohol consumption with selected cardiovascular disease outcomes: A systematic review and meta-analysis. BMJ Br. Med. J. 342, d671 (2011).
Lu, B., Solomon, D. H., Costenbader, K. H. & Karlson, E. W. Alcohol consumption and risk of incident rheumatoid arthritis in women a prospective study. Arthr. Rheumatol. 66, 1998–2005 (2014).
Fernandez-Sola, J. Cardiovascular risks and benefits of moderate and heavy alcohol consumption. Nat. Rev. Cardiol. 12, 576–587 (2015).
Barbhaiya, M. & Costenbader, K. H. Environmental exposures and the development of systemic lupus erythematosus. Curr. Opin. Rheumatol. 28, 497–505 (2016).
Aguet, F. et al. Genetic effects on gene expression across human tissues. Nature 550, 204-+ (2017).
Heit, C. et al. The role of CYP2E1 in alcohol metabolism and sensitivity in the central nervous system. Subcell. Biochem. 67, 235–247 (2013).
Cederbaum, A. I. Alcohol metabolism. Clin. Liver Dis. 16, 667-+ (2012).
Edenberg, H. J. & McClintick, J. N. Alcohol dehydrogenases, aldehyde dehydrogenases, and alcohol use disorders: a critical review. Alcohol. Clin. Exp. Res. 42, 2281–2297 (2018).
Han, Y. et al. Evidence of positive selection on a class I ADH locus. Am. J. Hum. Genet. 80, 441–456 (2007).
Li, H. et al. Geographically separate increases in the frequency of the derived ADH1B*47His allele in eastern and western Asia. Am. J. Hum. Genet. 81, 842–846 (2007).
Peng, Y. et al. The ADH1B Arg47His polymorphism in East Asian populations and expansion of rice domestication in history. BMC Evolut. Biol. 10, 15 (2010).
Galinsky, K. J. et al. Fast principal-component analysis reveals convergent evolution of ADH1B in Europe and East Asia. Am. J. Hum. Genet. 98, 456–472 (2016).
Gu, S. et al. Recent selection on a class I ADH locus distinguishes southwest Asian populations including Ashkenazi Jews. Genes 9, e452 (2018).
Johnson, K. E. & Voight, B. F. Patterns of shared signatures of recent positive selection across human populations. Nat. Ecol. Evol. 2, 713–720 (2018).
Toth, R. et al. ADH1B*2 allele is protective against alcoholism but not chronic liver disease in the Hungarian population. Addiction 105, 891–896 (2010).
Craddock, N. et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464, 713-U786 (2010).
Bierut, L. J. et al. ADH1B is associated with alcohol dependence and alcohol consumption in populations of European and African ancestry. Mol. Psychiatry 17, 445–450 (2012).
Gelernter, J. et al. Genome-wide association study of alcohol dependence: significant findings in African-and European-Americans including novel risk loci. Mol. Psychiatry 19, 41–49 (2014).
Way, M. et al. Genetic variants in or near ADH1B and ADH1C affect susceptibility to alcohol dependence in a British and Irish population. Addict. Biol. 20, 594–604 (2015).
Xu, K. et al. Genomewide association study for maximum number of alcoholic drinks in European Americans and African Americans. Alcohol. Clin. Exp. Res. 39, 1137–1147 (2015).
Jorgenson, E. et al. Genetic contributors to variation in alcohol consumption vary by race/ethnicity in a large multi-ethnic genome-wide association study. Mol. Psychiatry 22, 1359–1367 (2017).
Thompson, A. et al. Functional validity, role, and implications of heavy alcohol consumption genetic loci. Sci. Adv. 6, eaay034 (2020).
Peter, B. M., Huerta-Sanchez, E. & Nielsen, R. Distinguishing between selective sweeps from standing variation and from a de novo mutation. PLoS Genet. 8, E1003011 (2012).
Smith, J., Coop, G., Stephens, M. & Novembre, J. Estimating time to the common ancestor for a beneficial allele. Mol. Biol. Evol. 35, 1003–1017 (2018).
Rodriguez-Zavala, J. S., Calleja, L. F., Moreno-Sanchez, R. & Yoval-Sanchez, B. Role of aldehyde dehydrogenases in physiopathological processes. Chem. Res. Toxicol. 32, 405–420 (2019).
Chen, C. H., Ferreira, J. C. B., Gross, E. R. & Mochly-Rosen, D. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol. Rev. 94, 1–34 (2014).
Zhou, Y. et al. Alcoholic beverage consumption and chronic diseases. Int. J. Environ. Res. Public Health 13, 522 (2016).
Lee, D. J., Lee, H. M., Kim, J. H., Park, I. S. & Rho, Y. S. Heavy alcohol drinking downregulates ALDH2 gene expression but heavy smoking up-regulates SOD2 gene expression in head and neck squamous cell carcinoma. World J. Surg. Oncol. 15, 163 (2017).
Ceni, E., Mello, T. & Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 20, 17756–17772 (2014).
Stewart, M. J. et al. Binding and activation of the human aldehyde dehydrogenase 2 promoter by hepatocyte nuclear factor 4. Biochim. Biophys. Acta Gene Struct. Exp. 1399, 181–186 (1998).
Oota, H. et al. The evolution and population genetics of the ALDH2 locus: random genetic drift, selection, and low levels of recombination. Ann. Hum. Genet. 68, 93–109 (2004).
Li, H. et al. Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann. Hum. Genet. 73, 335–345 (2009).
Macgregor, S. et al. Associations of ADH and ALDH2 gene variation with self report alcohol reactions, consumption and dependence: an integrated analysis. Hum. Mol. Genet. 18, 580–593 (2009).
Quillen, E. E. et al. ALDH2 Is associated to alcohol dependence and is the major genetic determinant of “daily maximum drinks” in a GWAS study of an isolated rural Chinese sample. Am. J. Med. Genet. B Neuropsych. Genet. 165, 103–110 (2014).
Pan, C. et al. Aldehyde dehydrogenase 2 inhibits inflammatory response and regulates atherosclerotic plaque. Oncotarget 7, 35562–35576 (2016).
Ma, H., Guo, R., Yu, L., Zhang, Y. M. & Ren, J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur. Heart J. 32, 1025–1038 (2011).
Zhang, H., Gong, D. X., Zhang, Y. J., Li, S. J. & Hu, S. S. Effect of mitochondrial aldehyde dehydrogenase-2 genotype on cardioprotection in patients with congenital heart disease. Eur. Heart J. 33, 1606–1614 (2012).
Panisello-Rosello, A. et al. Role of aldehyde dehydrogenase 2 in ischemia reperfusion injury: An update. World J. Gastroenterol. 24, 2984–2994 (2018).
Coenen, M. J. H. et al. Common and different genetic background for rheumatoid arthritis and coeliac disease. Hum. Mol. Genet. 18, 4195–4203 (2009).
Bentham, J. et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47, 1457-+ (2015).
Auburger, G. et al. 12q24 locus association with type 1 diabetes: SH2B3 or ATXN2?. World J. Diabetes 5, 316–327 (2014).
Yasukochi, Y. et al. Longitudinal exome-wide association study to identify genetic susceptibility loci for hypertension in a Japanese population. Exp. Mol. Med. 49, 589 (2017).
Wild, P. S. et al. Large-scale genome-wide analysis identifies genetic variants associated with cardiac structure and function. J. Clin. Investig. 127, 1798–1812 (2017).
Okada, Y. et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 9, 1631 (2018).
Mathieson, I. et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528, 499-+ (2015).
Chou, W. Y. et al. An A/G polymorphism in the promoter of mitochondrial aldehyde dehydrogenase (ALDH2): Effects of the sequence variant on transcription factor binding and promoter strength. Alcohol. Clin. Exp. Res. 23, 963–968 (1999).
Kimura, Y. et al. A promoter polymorphism in the ALDH2 gene affects its basal and acetaldehyde/ethanol-induced gene expression in human peripheral blood leukocytes and HepG2 cells. Alcohol Alcohol. 44, 261–266 (2009).
Feitosa, M. F. et al. Novel genetic associations for blood pressure identified via gene-alcohol interaction in up to 570K individuals across multiple ancestries. PLoS ONE 13, e0198166 (2018).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 (2012).
Barreiro, L. B. & Quintana-Murci, L. From evolutionary genetics to human immunology: How selection shapes host defence genes. Nat. Rev. Genet. 11, 17–30 (2010).
Akbari, A. et al. Identifying the favored mutation in a positive selective sweep. Nat. Methods 15, 279-+ (2018).
Bolotin, E. et al. Integrated approach for the identification of human hepatocyte nuclear factor 4 alpha target genes using protein binding microarrays. Hepatology 51, 642–653 (2010).
You, M., Fischer, M., Cho, W. K. & Crabb, D. Transcriptional control of the human aldehyde dehydrogenase 2 promoter by hepatocyte nuclear factor 4: Inhibition by cyclic AMP and COUP transcription factors. Arch. Biochem. Biophys. 398, 79–86 (2002).
Stern, A. J., Wilton, P. R. & Nielsen, R. An approximate full-likelihood method for inferring selection and allele frequency trajectories from DNA sequence data. PLoS Genet. 15, 589 (2019).
Sakaue, S. et al. Functional variants in ADH1B and ALDH2 are non-additively associated with all-cause mortality in Japanese population. Eur. J. Hum. Genet. 28, 378–382 (2020).
McGovern, P. E. et al. Fermented beverages of pre- and proto-historic China. Proc. Natl. Acad. Sci. USA 101, 17593–17598 (2004).
Liu, L. et al. The origins of specialized pottery and diverse alcohol fermentation techniques in Early Neolithic China. Proc. Natl. Acad. Sci. USA 116, 12767–12774 (2019).
Heiss, A. G. et al. Mashes to mashes, crust to crust. Presenting a novel microstructural marker for malting in the archaeological record. PLoS ONE 5, 589 (2020).
Maixner, F. et al. Hallstatt miners consumed blue cheese and beer during the Iron Age and retained a non-Westernized gut microbiome until the Baroque period. Curr. Biol. 31, 1–14 (2021).
Retief, F. & Cilliers, L. Wine in Graeco-Roman antiquity with emphasis on its effect on health. AASCIT J. Health 2(5), 44–49 (2015).
Budas, G. R., Disatnik, M. H. & Mochly-Rosen, D. Aldehyde dehydrogenase 2 in cardiac protection: a new therapeutic target?. Trends Cardiovasc. Med. 19, 158–164 (2009).
Guo, J. M. et al. ALDH2 protects against stroke by clearing 4-HNE. Cell Res. 23, 915–930 (2013).
Crabb, D. W., Matsumoto, M., Chang, D. & You, M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc. Nutr. Soc. 63, 49–63 (2004).
Zhang, H. & Fu, L. W. The role of ALDH2 in tumorigenesis and tumor progression: Targeting ALDH2 as a potential cancer treatment. Acta Pharm. Sin. B 11, 1400–1411 (2021).
Pathak, H. et al. Promoter polymorphism rs886205 genotype interacts with DNA methylation of the ALDH2 regulatory region in alcohol dependence. Alcohol Alcohol. 52, 269–276 (2017).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Voight, B. F., Kudaravalli, S., Wen, X. Q. & Pritchard, J. K. A map of recent positive selection in the human genome (vol 4, pg 154, 2006). PLoS Biol. 4, 659–659 (2006).
Szpiech, Z. A. & Hernandez, R. D. selscan: An efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol. 31, 2824–2827 (2014).
Altshuler, D. M. et al. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58 (2010).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
R Core Team, 2021. R: a language and environment for statistical computing, Vienna, Austria. https://www.R-project.org/.
Belkhir, K., Borsa, P., Chikhi, L., Raufaste, N. & Bonhomme, F. GENETIX4. 05, logiciel sous Windows TM pour la génétiquedes populations. Laboratoire génome, populations, interactions, CNRS UMR 5000, 1996–2004 (2004).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. Ggplot2: Elegant Graphics for Data Analysis, 1–212 (2009).
Howe, K. L. et al. Ensembl 2021. Nucleic Acids Res. 49, D884–D891 (2021).
Machiela, M. J. & Chanock, S. J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015).
Makowski, D., Ben-Shachar, M. S., Chen, S. H. A. & Ludecke, D. Indices of effect existence and significance in the Bayesian framework. Front. Psychol. 10, 2767 (2019).
Speidel, L., Forest, M., Shi, S. N. & Myers, S. R. A method for genome-wide genealogy estimation for thousands of samples. Nat. Genet. 51, 1321-+ (2019).
Ardlie, K. G. et al. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660 (2015).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019).
Acknowledgements
We thank Michael Stachowitsch from the Department of Evolutionary Anthropology, University of Vienna and Franz Suchentrunk from the University of Veterinary Medicine Vienna for valuable comments on the manuscript. Open access funding provided by University of Vienna.
Author information
Authors and Affiliations
Contributions
H.S. conceived and designed this study; H.S., T.G. and D.M. performed statistical analyses. H.S. wrote the draft manuscript; all authors contributed to the results, edited, read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained a repeated error, where gene name “SH2B3” was incorrectly given as “SH2B2” in the Introduction and Results section.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schaschl, H., Göllner, T. & Morris, D.L. Positive selection acts on regulatory genetic variants in populations of European ancestry that affect ALDH2 gene expression. Sci Rep 12, 4563 (2022). https://doi.org/10.1038/s41598-022-08588-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-08588-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
