
Abstract
Human papillomavirus (HPV) is detected in up to 96% of anal squamous cell cancers, where screening programs needed. However, the best methodology is still undetermined. Host DNA methylation markers CADM1, MAL and miR124 have been identified in cervical disease, but not anal disease. Anal swabs varying by disease grade were assessed for DNA methylation of CADM1, MAL and miR124-2. Each marker was compared across disease grades, stratified by HPV and HIV status. Receiver operating characteristic curves identified the predictive value of significant gene candidates. CADM1 methylation was significantly higher in high-grade squamous intraepithelial lesions (HSIL) compared with low-grade (LSIL) (p = 0.005) or normal (p < 0.001) samples with 67.2% correctly identified as HSIL. MAL methylation was significantly (p = 0.002) increased in HSIL compared with LSIL in HIV positive participants with 79.8% correctly indicated as HSIL. Gene miR124-2, showed no difference between disease grades. Biomarkers with established diagnostic value in cervical disease have limited utility in the prediction of anal disease, with CADM1 identified as a marker with screening potential in a gay and bisexual men (GBM) population and MAL in HIV positive GBM population. New markers specific to the anal mucosa are required to improve triage of high-risk individuals.
Similar content being viewed by others


Introduction
Human papillomavirus (HPV) causes approximately 88% to 96% of anal cancers1,2. The majority of anal cancers arise from the squamo-columnar junction of the anal canal and are predominantly anal squamous cell carcinomas (ASCC)3.
Rates of ASCC in the general population are low, at less than 2 per 100,000 person years, with slightly higher rates in women compared with men4. Incidence has been steadily increasing over the past three decades5 although, recent data has suggested ASCC in people with HIV have decreased over the past decade (2008–2012), thought to be due to improved HIV treatments6. Vaccination against high-risk HPV (HRHPV) genotypes that cause the majority of anal cancers, when administered prior to HPV exposure, are highly effective in reducing the incidence of the precursor lesion, anal high-grade squamous intraepithelial lesions (HSIL)7. However, the progress of introduction of gender neutral vaccine programs is slow8, as such a reduction in anal cancer rates will not be seen for many decades7,9. Among the high-risk groups predicted to benefit from effective screening, gay and bisexual men (GBM) are the most studied as HIV negative GBM are up to 20 times and HIV positive GBM are up to 100 times more likely to develop anal HSIL and/or ASCC than heterosexual men. Other high-risk populations with increased rates of ASCC include women and heterosexual men with HIV infection (12-fold higher), women with previous lower genital tract disease related to HPV, and transplant recipients5,10,11,12.
Current clinical screening algorithms are based on anal cytology assessment, followed by high-resolution anoscopy (HRA) guided biopsy and histology as a diagnostic tool. Performance of anal cytology as a screening tests for anal cancer prevention has been demonstrated to vary depending on the population and screening protocols to define risk of cancer development13. A recent meta-analysis evaluated the performance of anal cytology in women and men and identified an overall sensitivity and specificity of atypical squamous cells of undetermined significance or higher positive cytology for the detection of AIN2 or higher at 77.3% and 55.5%, respectively14. Furthermore, the use of HRA-guided biopsies are considered imperfect due to anal mucosal folds and coexisting pathology15. There is also a lack of treatment options available. Consequently, there are no universally accepted guidelines for anal screening14.
HRHPV DNA detection has been adopted for cervical screening in many countries being more objective and sensitive to cytology16. The application of HRHPV DNA detection as a primary test for anal screening in high-risk populations may be limited, due to the high prevalence of anal HRHPV and the presence of multiple HPV types, resulting in high sensitivity but poor specificity14,17,18,19.
Cellular biomarkers are indicative of underlying biological changes as a result of modulation of oncogenic gene regulation during carcinogenesis, and may be used as markers of neoplastic transformation of cells20. The methylation of CpG sites for cell adhesion molecule 1 (CADM1), T-lymphocyte maturation associated protein (MAL) and the micro RNA 124-2 (miR124-2) are promising biomarkers in HPV-related cervical intraepithelial neoplasia (CIN)21 but have had limited evaluations in HPV-related anal disease, with only a single study investigating miR124-222. These three genes have been previously identified as tumour suppressors, mediators of organised epithelial cell growth and potent immune system regulators23,24,25,26,27,28,29.
This study aimed to examine the performance of previously identified cervical cancer methylation markers (CADM1, MAL, and miR124-2) in anal HSIL, using samples collected as part of the Study of the Prevention of Anal Cancer (SPANC)5. This was used to determine how well these methylation markers predict anal HSIL, as a potential triage test alone and in combination with HRHPV positivity and HIV status.
Results
Study participants
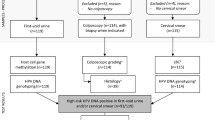
From the 617 SPANC participants with collected baseline anal cytology samples, 454 were included in this analysis (4 participants without consent, 86 participants were lost to follow-up, 17 samples with unassessable baseline HPV results and 56 samples with insufficient remaining sample). The median age was 49 years (range 35–75 years) and 36% were HIV positive. Of the total, 429 (94.5%) samples had valid host DNA methylation results (Table 1) and 315 (73.4%) tested positive for HRHPV. The highest degree of abnormality diagnosed was HSIL n = 117 (27%), LSIL n = 26 (6%) and normal n = 286 (66.7%) (Table 1).
Methylation of the gene CADM1 differentiates between HSIL and LSIL/normal samples
CADM1 methylation was significantly different across all samples when stratified by disease (p < 0.001) (Fig. 1, panel A1). A further group specific analysis (Wilcoxon), also revealed that CADM1 methylation from anal swab samples from men with HSIL was significantly different compared to those with both LSIL (p = 0.005) and normal samples (p < 0.001) (Fig. 1, panel A1). There was also a significant difference between HSIL and LSIL (p = 0.024) and HSIL and normal (p = 0.002) when the analysis was limited to HRHPV positive samples only (Fig. 1, panel A2).

Percentage Methylation of gene CADM1. All samples represented by HRHPV positivity and HIV status. Comparisons assessed by the nonparametric Wilcoxon test, with error bars corresponding to the first and third quartiles (the 25th and 75th percentiles).
The AUROC for the methylation of the gene CADM1 was 67.2% for HSIL over LSIL and 60.4% for HSIL over normal (Fig. 2A). Also, a comparison of HRHPV positive samples indicated an AUROC of 67.3% for HSIL over LSIL and 61.8% for HSIL over normal (Fig. 2B). Furthermore, similar AUROC (between 60 and 62%) were determined for comparisons between HSIL and normal for HIV negative and positive participants including all samples (Fig. 2A) and HRHPV positive samples only (Fig. 2B). Most significantly, in HIV positive men, CADM1 methylation distinguished HSIL from LSIL with an AUROC of 73.3% (Fig. 2A) and in HIV positive men with HRHPV infections, the AUC was 78.2% (Fig. 2B).

Receiver Operating Characteristic (ROC) curve for the percentage methylation of gene CADM1 for all significant (p, ≤ 0.05) comparison combinations identified in the nonparametric Wilcoxon test. Comparisons include HSIL versus LSIL (black), HSIL versus Normal (red), HSIL versus Normal HIV negative (blue), HSIL versus LSIL HIV positive (green) and HSIL versus Normal HIV positive (grey). (A) HRHPV positive and negative samples (B) HRHPV positive samples only.
Methylation of the gene MAL differentiates between HSIL and LSIL for HIV positive men
An overall analysis (Kruskal–Wallis) in the percentage methylation of MAL identified a significant difference in HIV positive only samples p = 0.041 respectively; Fig. 3, panel C1). Further individual analysis in all samples identified HSIL as significantly different to LSIL (p = 0.0065). This was also observed in HIV positive group (p = 0.0023).

Percentage Methylation of gene MAL. All samples represented by HRHPV positivity and HIV status. Comparisons assessed by the nonparametric Wilcoxon test, with error bars corresponding to the first and third quartiles (the 25th and 75th percentiles).
Further comparative analysis of each group (all samples and HRHPV positive samples) stratified by HIV status identified that only HIV positive men had significant differences (p = 0.016) (Fig. 3, panel C1). Within the HIV positive group, methylation in HSIL was significantly higher compared with LSIL in all samples (p = 0.002) (Fig. 3, panel C1) and HRHPV samples (p = 0.017) (Fig. 3, panel C2), however there was no difference when compared with normal samples (Fig. 3, panels C1 and C2); (as observed in the overall group). The AUROC for the methylation of the gene MAL was 67.6% for HSIL over LSIL (Fig. 4). Furthermore, HSIL compared with LSIL in HRHPV, and HIV positive anal swab samples indicated an AUROC of 78.9%.

Receiver Operating Characteristic (ROC) curve for the percentage methylation of gene MAL for all significant (p, ≤ 0.05) comparison combinations identified in the nonparametric Wilcoxon test. Comparisons include HSIL versus LSIL in all samples (black), HSIL versus LSIL in HIV positive samples (red), HSIL versus LSIL in all HRHPV positive samples (blue) and HSIL versus LSIL in all HIV and HRHPV positive samples (green).
The methylation of gene miR124-2 is not predictive of anal HSIL in GBM
The methylation of the promoter region for miR124-2 compared with composite histologically and cytologically defined anal disease identified no significant differences by anal disease grade between any of the subgroups analysed, including HRHPV positive samples or HIV status (Fig. 5).

Percentage Methylation of gene miR124-2. All samples represented by HRHPV positivity and HIV status. Comparisons assessed by the nonparametric Wilcoxon test, with error bars corresponding to the first and third quartiles (the 25th and 75th percentiles).
The value of different screening algorithms to predict histologically confirmed HSIL
The utilisation of percentage methylation for gene CADM1 and MAL in potential screening algorithms was assessed in combination with and without HRHPV positivity and/or cytological HSIL. Overall, the addition of cytology results had limited effect on the AUROC value of gene methylation (Fig. 6). Cytology alone had the highest AUROC within histologically-confirmed HSIL (68.8%) (Fig. 6A). Using multiple screening markers, CADM1 in combination with HRHPV positivity had an AUROC value of 62.3% for histologically-confirmed HSIL (Fig. 6A). Within the HIV positive population, the highest AUROC was cytology alone (70.1%) (Fig. 6B). Using a combination of different markers lowered the AUROC value with all markers only achieving 56.8% in histology confirmed HSIL (Fig. 6B). Cut-off values for each methylation marker were also assessed, however no combination tested improved the AUROC percentage above cytology alone (Supplementary Fig. 1).

Difference in predictive power for different screening algorithms for anal cancer in men to identify histology defined HSIL from LSIL and normal combined. Algorithms include combinations of gene methylation and/or HRHPV positivity and/or cytology defined HSIL for (A) gene CADM1 in all men and (B) gene MAL in HIV positive men only. Comparisons for each gene are identical and include gene methylation in men with HRHPV and HSIL cytology (black), gene methylation in men with HSIL cytology (red), gene methylation only (blue), gene methylation in men with HRHPV (green), HRHPV positivity only (grey) and cytology defined HSIL only (orange).
Discussion
In the current study, men with HSIL had an increase in the host DNA methylation of CpG sites within promoter regions for the gene CADM1 and, to a lesser extent, MAL. Among HIV positive individuals only, gene MAL had significantly less gene methylation in men whose highest grade of disease was LSIL compared with both normal histology and HSIL. This limits the utility of MAL as a biomarker of disease detection.
The methylation of CpG sites for both CADM1 and MAL have previously been indicated as potential biomarkers in HPV-related cervical intraepithelial neoplasia (CIN) with reports of 78% specificity and 70% sensitivity in women with HRHPV infections for histological CIN3+30. This is the first report of these markers being associated with anal HSIL in men and identifies their potential to be included in combination with other biomarkers for anal cancer screening programs.
The predictive value of CADM1 methylation for the determination of HSIL indicated that approximately 67% of anal samples would be correctly diagnosed as HSIL with this single marker, with no difference between HIV or HRHPV status.
Other studies of gene methylation markers from anal HSIL and cancer in HIV positive and negative men include p16, Ki-67, ASCL1, SST, ZIC1, ZNF582 and the HPV E4 gene where significant differences have been reported from tissue samples31,32,33. The methylation of the CpG promoter region of gene MAL was observed to be significantly increased only in HIV positive GBM with HSIL (p, ≤ 0.01) as compared with those with LSIL, but not when compared to normal samples or in HIV negative participants. Changes to the expression of the MAL gene have previously been identified in several different cancers with early reports identifying MAL as a tumour suppressor34. Subsequent reports have also identified decreases in MAL expression observed in cervical, colon, breast, stomach, bladder, salivary gland, head and neck and non-small cell lung cancers35. In the current report, the significant increase in MAL methylation in HSIL compared with LSIL in HIV positive individuals is perplexing, due to the similar methylation patterns between normal and HSIL samples. This unusual progression of changing methylation between normal, low and high grade disease in HIV positive GBM could be due to the MAL protein acting as a raft for other oncogenic proteins, as proposed by Lara–Lemus35. However, the limitation of this change to HIV-positive individuals could also be due to HIV-related changes in T-lymphocytes. Individuals infected with HIV experience a sustained hyper immune activation, triggered by changes in T cell dynamics and function36. It is possible that during acute infections of HPV, HIV-positive individuals have a varied response to acute HPV infection compared with HIV negative individuals.
Although, the significant findings of ZNF582 with a 93% specificity for ≥ AIN3 does show promise for the diagnostic potential of methylation markers in the anal mucosa32. In addition to these strictly male gender assigned at birth and currently identifying as male studies, further analysis of the gene EPB41L3 and HPV16 in anal samples from HIV positive and negative men and women identified a combined sensitivity of 90.6%, specificity of 50.7% and AUROC of 0.82 in identifying HSIL and cancer from normal tissue samples37. It is important to note that these anal specific studies all utilised tissue samples whereas the current study focuses on swab samples. This difference is an important point of difference. If a methylation marker is to be utilised as a screening tool it needs to be sensitive enough to detect disease without the collection of tissue samples, which require highly specialised equipment and personnel. By only focusing on tissue samples the application of the findings are limited until the relevant sample has been assessed. This is where the current study is unique compared to all other anal methylation studies.
However, it should also be noted that determining small changes for low levels of disease is a difficult task which is compounded by utilising swab samples, which contain a high diversity of cellular material, to measure lesion specific changes. Our result indicating higher methylation levels in normal versus LSIL samples could be a consequence of the sample utilised and not differences between disease presentations.
Methylation-based silencing of precursor micro RNAs of microRNA 124 have previously been indicated in various human neoplasms, including the cervix38,39, where a significant inverse correlation was observed in the methylation of the promoter region and the presence of miR124-2 RNA. The same study also demonstrated in-vitro, that silencing of miR124-2 is functionally involved in cervical cancer development38. However, in the current study, methylation of the promoter region of miR124-2 was not found to be different between men with HSIL, LSIL or normal samples. The absence of this important biomarker could be due to the nature of the samples collected, as anal swab samples contain a mixture of normal and diseased cells. Other explanations could be that this is a phenotype specific to the anal canal. Further analyses utilising specific tissue samples from laser capture microdissection techniques and investigations into other precursor micro RNAs for miR124 are required to define any roles in the development of HSIL.
Combination algorithms utilising CADM1, HRHPV detection and cytology diagnosis marginally improved the predictive value (from 61.2% alone to 61.6% combined) with cytology alone outperforming all other combinations at 68.8% AUROC in differentiating HSIL from LSIL and normal samples. Similar findings were also observed utilising MAL, in HIV positive participants. These findings indicate that complex screening algorithms may be required to achieve similar results to those seen in the cervix. For example, if all samples are screened by cytology to filter out all normal samples then CADM1 and HRHPV testing can be added for disease grade diagnosis (between LSIL, HSIL and ASCC) then the predictive value increases to 78.2% (similar to findings in the cervix at 78% specificity and 70% sensitivity in women with HRHPV infections)30.
Limitations to this study were the use of single promoter regions for methylation measurements and the absence of mRNA measurements to assess the level of gene silencing at a transcriptional level. Future studies should also assess the mRNA effects due to methylation of all possible promoter regions. The methylation patterns of CADM1 and MAL will be assessed for changes in longitudinal studies of HPV persistence utilising the SPANC cohort of samples. The detection cut off for the internal control was also increased from the published > 32 to > 40 cycle threshold which is a considerably low threshold. However, through modifications to the PCR reactions and confirmation through serial dilutions of the SiHa cell line (inter and intra assays) we confirmed that our modifications increased the assays sensitivity allowing for the increased cycle threshold cut-off.
In conclusion, this study demonstrates methylation of the promoter region for gene CADM1, when used in a panel of biomarkers, is a promising candidate for use as part of an anal screening algorithm for HSIL in HRHPV positive GBM. Furthermore, in HIV positive GBM, methylation of the promoter region for gene MAL may be important in HIV related immune modulation. It is clear from these findings that biomarkers utilised in the cervix are of limited use within other mucosa tissue sites highlighting the need to further explore new genes of interest specific for HPV related disease of the anal mucosa.
Methods
Study population
Anal specimens were obtained as part of the SPANC study. SPANC was a prospective cohort study based in Sydney, Australia, that followed HIV-negative and HIV-positive GBM aged 35 years and older at five visits over a period of three years. Overview of study design, recruitment and protocols have been previously published5. Participants were eligible for inclusion if they met the following criteria: had given consent to the use of their samples for further research; had sufficient residual sample remaining after routine SPANC tests were completed; had completed the first three study visits; had baseline samples with satisfactory HPV testing. In this substudy, only baseline samples were utilised.
Diagnosis and disease classification
Laboratory methods have been previously described5. Briefly, cytology was graded as per the Bethesda System criteria40 and terms used for cytology reporting. Histology reporting was in accordance with the Lower Anogenital Squamous Terminology (LAST) Project41. Final diagnosis for each sample was a composite of cytology and histology reporting (disease results were derived by histology findings from HRA guided biopsy collection. In cases of normal or LSIL cytology grades where biopsies were not collected, or histology unavailable cytology results were utilised). Where multiple samples were collected during HRA, the highest-grade histology findings were used. Composite end points were as follows; All reports of negative were reported as normal, all reports of LSIL was reported as LSIL, all reports of AIN2 and AIN3 were reported as HSIL.
HPV genotype testing
Baseline residual anal cytology specimens were tested for HPV DNA (including HRHPV types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66 and 68) as previously described42, using the Roche Linear Array HPV genotyping test (Roche Molecular Systems, Alameda, CA, United States). As it has been established that Linear Array has a lower sensitivity for several HRHPV genotypes (HPV40, 42, 54 and 68)43, a secondary assay was utilised with a higher sensitivity (for these specific genotypes). The Anyplex™ II HPV HR Detection (Seegene, Seoul, South Korea)44, a multiplex real-time PCR method that detects 14 HRHPV genotypes (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68), was utilised for all specimens. Specimens were considered positive for any HPV type that was detected on either Linear Array or Anyplex™ to achieve a high sensitivity for all HPV genotypes.
Isolation of nucleic acids for methylation PCR
A 1 mL aliquot taken from a 20 mL cytology sample in Thin Prep PreservCyt medium (Hologic, Inc. Marlborough, MA, USA) was centrifuged at 17000g for 15 min, the supernatant removed, and the pellet resuspended in 200 µl of PBS. DNA was extracted on an automated MagNAPure 96 (Roche Diagnostics GmbH, Penzberg, Germany) using the DNA and Viral Nucleic Acid Small Volume Kit (Roche Diagnostics) with a sample volume of 200 µl and an elution volume of 50 µl, according to the manufacturer’s instructions. The concentration of double-stranded DNA was measured with a Qubit® Fluorometer (Life technologies, California, USA).
Bisulphite DNA modification and quantitative methylation-specific PCR
Extracted DNA underwent bisulphite conversion using the Methylamp™ DNA Modification Kit (Epigentek, New York, USA). Quantitative methylation-specific PCR (qMSP) targeting CpG sites in promoter regions of CADM1 (promoter region M9), MAL (promoter region M1) and miR124-2 (promoter region 2) were performed using the primer sets and methods previously described45, with minor modifications (single-plex PCR reactions, and an increase in magnesium chloride concentration from 3 to 4 mM MgCl2). PCR mixtures for the modified protocol contained 2.5 μl of bisulfite modified DNA, primers at 417 nM, probes at 208 nM (labelled with 5’-FAM and 3’-BHQ1), 1 mM of added MgCl2, and 1 × Bioline Sensifast Probe Mastermix (containing 3 mM MgCl2 per reaction). A segment of the beta actin gene (ACTB) was targeted as an internal reference and for quantitation of input DNA46,47,48. A standardised sample of bisulphite-treated DNA extracted from cervical cancer cell line SiHa, (American Type Culture Collection, Manassas, Virginia, USA) containing a single copy of HPV16 per cell, was included as a positive control in each qMSP run. PCR thermocycler conditions were unchanged from the previously published method and were performed on the LC480 LightCycler technology (Roche Diagnostics). The human cell line A549 was utilised as a negative control. SiHa cell line was used as a positive control for variations of each bisulphite modification assay performed and validation of the qPCR methylation analysis for each gene. Briefly, each PCR run had an old bisulphite modified SiHa cell line that had been previously validated for methylation analysis and a new (identical DNA concentration) SiHa cell line that was bisulphite modified at the same time and with the same bisulphite reagents that the samples to be analysed by qPCR that day (in general 9 samples). The objective of this procedure was to analyse the variation in the bisulphite modification procedure between assays and to validate the methylation analysis between different runs (reproducibility of % of methylation of the positive control for each gene). Our results showed very high reproducibility in the % of methylation in the SiHa cell lines for each gene between runs (data not shown).
Data analysis
A sample was deemed satisfactory if the crossing threshold value for ACTB was < 40. Any sample that was negative for ACTB was not considered for analysis regardless of methylation marker status. The percentage methylation in each individual sample (% meCpG) was calculated as described based on the house keeping gene ACTB.
Percentage methylation for each marker (CADM1, MAL and miR124-2), for each composite disease grade (HSIL, LSIL and normal) was compared using two non-parametric comparative analysis, Wilcoxon test for between groups and Kruskal–Wallis for overall analysis: this was visualised using box and whisker plots. These analyses were further stratified by HRHPV positivity and HIV status.
The diagnostic performance of each methylation marker was evaluated by receiver operating characteristics (ROCs) analyses. Area under the curve (AUROC) was the main measure used to assess the ability of the genes to distinguish/differentiate HSIL from LSIL/normal by using the best cut-off with the maximum sum of sensitivity and specificity. Different screening algorithms were also assessed utilising gene methylation for markers that were significantly associated with HSIL detection, within a sub cohort of HRHPV positive samples and anal cytological HSIL, to predict histologically confirmed HSIL. All %meCpG calculations were performed in Microsoft Excel v16.39 (Microsoft, Redmond, Washington, USA). The results were then analysed and figures produced using the statistical platform R studio (v4.0.1)49 and programs ggplots2 (v3.3.2)50, ggpubr (v0.4)51 and pROC (v1.16.2)52 and cutpointr (v1.1.1)53 with p values ≤ 0.05 considered significant.
All mentioned methods were performed in accordance with the relevant guidelines and regulations.
Ethics approval and consent to participate
Ethics approval for the Study of the Prevention of Anal Cancer (SPANC) was granted by the St Vincent’s Hospital (SVH, Sydney, Australia) Human Research Ethics Committee (File Number 09/203).
Consent for publication
Informed consent was granted by each participant included in the trial through the SPANC recruitment process and detailed in the human research ethics approval (File Number 09/203).
Data availability
All data and materials are available within this document and the supplementary files.
Abbreviations
- HPV:
-
Human papillomavirus
- LSIL:
-
Low-grade squamous intraepithelial lesion
- HSIL:
-
High-grade squamous intraepithelial lesion
- ASCC:
-
Anal squamous cell carcinomas
- HRHPV:
-
High-risk HPV
- GBM:
-
Gay and bisexual men
- HRA:
-
High-resolution anoscopy
- ROC:
-
Receiver operating characteristic
- AUROC:
-
Area under the receiver operating characteristics
- SPANC:
-
Study of Prevention of Anal Cancer
- LAST:
-
Lower Anogenital Squamous Terminology
- qMSP:
-
Quantitative methylation-specific PCR
- CADM1 :
-
Cell adhesion molecule 1
- MAL :
-
T-lymphocyte maturation associated protein
- miR124 :
-
Micro RNA 124
- ACTB :
-
Beta actin gene
- CIN:
-
Cervical intraepithelial neoplasia
References
Alemany, L. et al. Human papillomavirus DNA prevalence and type distribution in anal carcinomas worldwide. Int. J. Cancer 136(1), 98–107 (2015).
Hillman, R. J. et al. Human papillomavirus (HPV) genotypes in an Australian sample of anal cancers. Int. J. Cancer 135(4), 996–1001 (2014).
Machalek, D. A. et al. Anal human papillomavirus infection and associated neoplastic lesions in men who have sex with men: A systematic review and meta-analysis. Lancet Oncol. 13(5), 487–500 (2012).
Pessia, B. et al. Squamous cell anal cancer: Management and therapeutic options. Ann. Med. Surg. 55, 36–46 (2020).
Machalek, D. A. et al. The Study of the Prevention of Anal Cancer (SPANC): Design and methods of a three-year prospective cohort study. BMC Public Health 13, 946 (2013).
Colón-López, V. et al. Anal cancer risk among people with HIV infection in the United States. J. Clin. Oncol. 36(1), 68–75 (2018).
Palefsky, J. M. et al. HPV vaccine against anal HPV infection and anal intraepithelial neoplasia. N Engl J Med. 365(17), 1576–1585 (2011).
Bruni, L. et al. HPV vaccination introduction worldwide and WHO and UNICEF estimates of national HPV immunization coverage 2010–2019. Prev. Med. 144, 106–399 (2021).
Giuliano, A. R. et al. Efficacy of quadrivalent HPV vaccine against HPV Infection and disease in males. N. Engl. J. Med. 364(5), 401–411 (2011).
Jin, F. et al. Trends in anal cancer in Australia, 1982–2005. Vaccine 29(12), 2322–2327 (2011).
Long, K. C., Menon, R., Bastawrous, A. & Billingham, R. Screening, surveillance, and treatment of anal intraepithelial neoplasia. Clin. Colon. Rectal. Surg. 29(1), 57–64 (2016).
Crum-Cianflone, N. F. et al. Anal cancers among HIV-infected persons: HAART is not slowing rising incidence. AIDS 24(4), 535–543 (2010).
Jin, F. et al. The performance of anal cytology as a screening test for anal HSILs in homosexual men. Cancer Cytopathol. 124(6), 415–424 (2016).
Clarke, M. A. & Wentzensen, N. Strategies for screening and early detection of anal cancers: A narrative and systematic review and meta-analysis of cytology, HPV testing, and other biomarkers. Cancer Cytopathol. 126(7), 447–460 (2018).
Mathews, W. C. et al. Estimating the accuracy of anal cytology in the presence of an imperfect reference standard. PLoS ONE 5(8), 122–84 (2010).
Chrysostomou, A. C., Stylianou, D. C., Constantinidou, A. & Kostrikis, L. G. Cervical cancer screening programs in Europe: The transition towards HPV vaccination and population-based HPV testing. Viruses 10(12), 66 (2018).
Castle, P. E. et al. A comparison of human papillomavirus genotype-specific DNA and E6/E7 mRNA detection to identify anal precancer among HIV-infected men who have sex with men. Cancer Epidemiol. Biomark. Prev. 22(1), 42–49 (2013).
Park, I. U., Introcaso, C. & Dunne, E. F. Human papillomavirus and genital warts: A review of the evidence for the 2015 Centers for Disease Control and prevention sexually transmitted diseases treatment guidelines. Clin. Infect. Dis. 61(Suppl 8), S849–S855 (2015).
Salit, I. E. et al. The role of cytology (Pap tests) and human papillomavirus testing in anal cancer screening. AIDS 24(9), 1307–1313 (2010).
Baylin, S. B. & Ohm, J. E. Epigenetic gene silencing in cancer—A mechanism for early oncogenic pathway addiction?. Nat. Rev. Cancer 6(2), 107–116 (2006).
Del Pino, M. et al. CADM1, MAL, and miR124 promoter methylation as biomarkers of transforming cervical intrapithelial lesions. Int. J. Mol. Sci. 20(9), 2262 (2019).
Lahiri, C. D. et al. Pilot study of markers for high-grade anal dysplasia in a southern cohort from the women’s interagency human immunodeficiency virus study. Clin. Infect. Dis. 70(6), 1121–1128 (2020).
Kuramochi, M. et al. TSLC1 is a tumor-suppressor gene in human non-small-cell lung cancer. Nat. Genet. 27(4), 427–430 (2001).
Yageta, M. et al. Direct association of TSLC1 and DAL-1, two distinct tumor suppressor proteins in lung cancer. Cancer Res. 62(18), 5129–5133 (2002).
Takai, Y., Irie, K., Shimizu, K., Sakisaka, T. & Ikeda, W. Nectins and nectin-like molecules: roles in cell adhesion, migration, and polarization. Cancer Sci. 94(8), 655–667 (2003).
Boles, K. S., Barchet, W., Diacovo, T., Cella, M. & Colonna, M. The tumor suppressor TSLC1/NECL-2 triggers NK-cell and CD8+ T-cell responses through the cell-surface receptor CRTAM. Blood 106(3), 779–786 (2005).
Ma, R., Xu, Y. E., Wang, M. & Peng, W. Suppression of MAL gene expression is associated with colorectal cancer metastasis. Oncol. Lett. 10(2), 957–961 (2015).
Cai, Q. Q. et al. MiR-124 inhibits the migration and invasion of human hepatocellular carcinoma cells by suppressing integrin αV expression. Sci. Rep. 7(1), 40733 (2017).
Han, D., Dong, X., Zheng, D. & Nao, J. MiR-124 and the underlying therapeutic promise of neurodegenerative disorders. Front. Pharmacol. 10, 1555 (2020).
Overmeer, R. M. et al. Combined CADM1 and MAL promoter methylation analysis to detect (pre-)malignant cervical lesions in high-risk HPV-positive women. Int. J. Cancer 129(9), 2218–2225 (2011).
van der Zee, R. P. et al. DNA methylation markers have universal prognostic value for anal cancer risk in HIV-negative and HIV-positive individuals. Mol. Oncol. 15(11), 3024–3036 (2021).
van der Zee, R. P. et al. Host cell deoxyribonucleic acid methylation markers for the detection of high-grade anal intraepithelial neoplasia and anal cancer. Clin. Infect. Dis. 68(7), 1110–1117 (2019).
van der Zee, R. P. et al. Cancer risk stratification of anal intraepithelial neoplasia in human immunodeficiency virus-positive men by validated methylation markers associated with progression to cancer. Clin. Infect. Dis. 72(12), 2154–2163 (2020).
Cao, W. et al. Epigenetic silencing of MAL, a putative tumor suppressor gene, can contribute to human epithelium cell carcinoma. Mol. Cancer 9(1), 296 (2010).
Lara-Lemus, R. On the role of myelin and lymphocyte protein (MAL) in cancer: A puzzle with two faces. J. Cancer 10(10), 2312–2318 (2019).
Brenchley, J. M. et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12(12), 1365–1371 (2006).
Lorincz, A. T. et al. Methylation of HPV and a tumor suppressor gene reveals anal cancer and precursor lesions. Oncotarget 8(31), 65 (2017).
Wilting, S. M. et al. Methylation-mediated silencing and tumour suppressive function of hsa-miR-124 in cervical cancer. Mol. Cancer 9(1), 167 (2010).
Jia, X. et al. MicroRNA-124: An emerging therapeutic target in cancer. Cancer Med. 8(12), 5638–5650 (2019).
Darragh, T.M., & Pergon, J.M 2015. The Bethesda system for reporting cervical cytology. In: Ritu N, C. Dearge, W, editors. Springer; 2015.
Darragh, T. M. et al. The lower anogenital squamous terminology standardization project for HPV-associated lesions: Background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Int. J. Gynecol. Pathol. 32(1), 76–115 (2013).
Poynten IM, Jin F, Roberts JM, Templeton DJ, Law C, Cornall AM, et al. The Natural History of Anal High-grade Squamous Intraepithelial Lesions in Gay and Bisexual Men. Clin Infect Dis. 2020.
Estrade, C. & Sahli, R. Comparison of seegene anyplex II HPV28 with the PGMY-CHUV assay for human papillomavirus genotyping. J. Clin. Microbiol. 52(2), 607–612 (2014).
Cornall, A. M. et al. HPV genotype-specific concordance between EuroArray HPV, Anyplex II HPV28 and linear array HPV genotyping test in Australian cervical samples. Papillomavirus Res. 4, 79–84 (2017).
De Strooper, L. M. et al. CADM1, MAL and miR124-2 methylation analysis in cervical scrapes to detect cervical and endometrial cancer. J. Clin. Pathol. 67(12), 1067–1071 (2014).
Cornall, A. M. et al. Anal and perianal squamous carcinomas and high-grade intraepithelial lesions exclusively associated with “low-risk” HPV genotypes 6 and 11. Int. J. Cancer 133(9), 2253–2258 (2013).
Priest, D. et al. Neisseria gonorrhoeae DNA bacterial load in men with symptomatic and asymptomatic gonococcal urethritis. Sex. Transm. Infect. 93(7), 478–481 (2017).
Resnick, R. M. et al. Detection and typing of human papillomavirus in archival cervical cancer specimens by DNA amplification with consensus primers. J. Natl. Cancer Inst. 82(18), 1477–1484 (1990).
Team, RC. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2018.
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
Kassambara A. ggpubr: 'ggplot2' Based Publication Ready Plots; 2020.
Robin, X. et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 12(1), 77 (2011).
Thiele, C. & Hirschfeld, G. cutpointr: Improved estimation and validation of optimal cutpoints in R. J. Stat. Softw. 98(11), 1–27 (2021).
Acknowledgements
We would like to acknowledge and thank the participants of the SPANC study. The SPANC study team includes Brian Acraman, Marjorie Adams, Claire Biro, Andrew Carr, Susan Carroll, David Cooper, Alyssa Cornall, Leonie Crampton, Deborah Ekman, Christopher Fairley, Annabelle Farnsworth, Lance Feeney, Eddie Fraissard, Marko Garcia, Suzanne Garland, Andrew Grulich, Richard Hillman, Kirsten Howard, Fengyi Jin, Johann Kolstee, Carmella Law, Matthew Law, Dorothy Machalek, Kirsten McCaffery, Ross McDonald, Patrick McGrath, Robert Mellor, Susan Pendlebury, Kathy Petoumenos, Piero Pezzopane, Samuel Phillips, Mary Poynten, Garrett Prestage, Adele Richards, Jennifer Roberts, Daniel Seeds, Sepehr Tabrizi, David Templeton, Julia Thurloe, Winnie Tong and Rick Varma. We would also like to acknowledge and thank the laboratory work of Kahli Cassells, Steph Atchison and Monica Molano.
Funding
This work was supported by National Health and Medical Research Council Program Grant (Sexually Transmitted Infections: causes, consequences, and interventions; grant no. 568971; to A.E. Grulich), Cancer Council New South Wales Strategic Research Partnership Program Grant (Preventing morbidity and mortality from anal cancer; Grant No. 13-11; to A.E. Grulich, I.M. Poynten, F. Jin, RJ Hillman, DJ Templeton, JM Roberts), and Cancer Council Victoria grant for evaluating molecular biomarkers of anal cancer risk (application no. APP1130507; to S.M. Garland, M. Molano, A.M. Cornall). The Kirby Institute is affiliated with the Faculty of Medicine, University of New South Wales and funded by the Australian Government of Health and Ageing. Cytologic testing materials were provided by Hologic (Australia) Pty Ltd.
Author information
Authors and Affiliations
Consortia
Contributions
S.P.: Methodology, completion of laboratory work, writing original draft, software analysis, data curation. K.C.: completion of laboratory work, methodology, writing original draft, review and editing. S.G. and G.M.: Supervision, funding acquisition, investigation, project administration, writing—review and editing. D.A.M.: visualization, methodology, writing–review and editing. J.M.R.: Resources, methodology, cytological and histological analysis writing—review and editing. D.J.T., F.J., I.M.P., R.J.H. and A.E.G.: Resources, funding acquisition, writing–review and editing. S.N.T.: Supervision, funding acquisition, investigation, project administration, writing—review and editing. M.M. and A.M.C.: Conceptualization, methodology, completion of laboratory work, supervision, project administration, writing review and editing.
Corresponding author
Ethics declarations
Competing interests
J.M. Roberts reports other from Hologic Australia (Thin Prep consumables) during the conduct of the study and other from Roche Australia (donated antibodies for further research in this field) outside the submitted work. I.M. Poynten reports other from Seqirus (travel funding) outside the submitted work. M. Molano reports grants from Cancer Council Victoria (project: evaluating molecular biomarkers of anal cancer risk, application no. APP1130507) during the conduct of the study. D.A. Machalek reports nonfinancial support from MSD and Seqirus, grants from Seqirus, and other from Roche Diagnostics (manuscript license sponsorship) outside the submitted work. F. Jin reports grants from the National Health and Medical Research Council and Cancer Council New South Wales during the conduct of the study. S.M. Garland reports grants (to institution for researcher-initiated grant for HPV young women's study), personal fees (lecture fees; work performed in personal time), and other from Merck (global advisory board member for HPV) outside the submitted work. A.E. Grulich reports grants from the National Health and Medical Research Council during the conduct of the study. A.M. Cornall reports grants from Cancer Council Victoria during the conduct of the study. No potential conflicts of interest were disclosed by the other authors.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Phillips, S., Cassells, K., Garland, S.M. et al. Gene methylation of CADM1 and MAL identified as a biomarker of high grade anal intraepithelial neoplasia. Sci Rep 12, 3565 (2022). https://doi.org/10.1038/s41598-022-07258-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-07258-5
This article is cited by
-
MAL expression downregulation through suppressive H3K27me3 marks at the promoter in HPV16-related cervical cancers is prognostically relevant and manifested by the interplay of novel MAL antisense long noncoding RNA AC103563.8, E7 oncoprotein and EZH2
Clinical Epigenetics (2024)
-
Unraveling Emerging Anal Cancer Clinical Biomarkers from Current Immuno-Oncogenomics Advances
Molecular Diagnosis & Therapy (2024)
-
Anogenitalwarzen – ein Update
Die Dermatologie (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
