
Abstract
Osteosarcoma (OS) is the most common type of primary malignant bone tumor. The high-throughput sequencing technology has shown potential abilities to illuminate the pathogenic genes in OS. This study was designed to find a powerful gene signature that can predict clinical outcomes. We selected OS cases with gene expression and survival data in the TARGET-OS dataset and GSE21257 datasets as training cohort and validation cohort, respectively. The univariate Cox regression and Kaplan–Meier analysis were conducted to determine potential prognostic genes from the training cohort. These potential prognostic genes underwent a LASSO regression, which then generated a gene signature. The harvested signature’s predictive ability was further examined by the Kaplan–Meier analysis, Cox analysis, and receiver operating characteristic (ROC curve). More importantly, we listed similar studies in the most recent year and compared theirs with ours. Finally, we performed functional annotation, immune relevant signature correlation identification, and immune infiltrating analysis to better study he functional mechanism of the signature and the immune cells’ roles in the gene signature’s prognosis ability. A seventeen-gene signature (UBE2L3, PLD3, SLC45A4, CLTC, CTNNBIP1, FBXL5, MKL2, SELPLG, C3orf14, WDR53, ZFP90, UHRF2, ARX, CORT, DDX26B, MYC, and SLC16A3) was generated from the LASSO regression. The signature was then confirmed having strong and stable prognostic capacity in all studied cohorts by several statistical methods. We revealed the superiority of our signature after comparing it to our predecessors, and the GO and KEGG annotations uncovered the specifically mechanism of action related to the gene signature. Six immune signatures, including PRF1, CD8A, HAVCR2, LAG3, CD274, and GZMA were identified associating with our signature. The immune-infiltrating analysis recognized the vital roles of T cells CD8 and Mast cells activated, which potentially support the seventeen-gene signature’s prognosis ability. We identified a robust seventeen-gene signature that can accurately predict OS prognosis. We identified potential immunotherapy targets to the gene signature. The T cells CD8 and Mast cells activated were identified linked with the seventeen-gene signature predictive power.
Similar content being viewed by others

Introduction
Osteosarcoma (OS) is a bone tumor that occurs predominantly in adolescents and young adults1,2,3. In the 0–24 age group, the incidence of osteosarcoma among men, women and children is 4.4 per million persons per year1,2,3. The latest advances in molecular genetics of osteosarcoma have changed our views on the cause of the disease and the continued treatment of patients1,2,3. Surgical removal of clinically visible tumors and systemic chemotherapy are currently popular disease management strategies1,2,3. Although the cure rate for patients with local disease is close to 70%, the 5-year overall survival rate for patients with metastatic disease is less than 25%, and most patients die from lung metastases4. Unfortunately, the treatment paradigm for OS has remained unchanged for approximately 30 years1,4. Therefore, continued efforts are urgently needed for a steady prognostic model for OS patients.
The rise of throughput sequencing technology helps clarify disease-causing genes, explore disease pathogenesis, develop biomarkers, and profoundly change our understanding of biology and human diversity5. Researchers have developed many statistical models that use genomic data to accurately predict whether the prognostic risk of cancer patients is high or low6,7,8,9,10. Many researchers have screened multiple biomarkers related to OS by mining gene expression data5. Gene signature can contain more than one single gene with a unique characteristic pattern of gene expression resulting from an altered or unaltered biological process or pathogenic medical condition11. Gene signature has a more stable ability and higher fault tolerance for prognostic prediction in cancer studies7,8,9,10,11,12.
Finding multiple molecules from the OS gene profile to construct a gene signature can better predict outcome potentially. To fill in the void and find a promising gene signature that targets OS outcomes, this work tried to identify a prognostic gene signature from the TARGET database. More importantly, the signature we found was further tested in an independent dataset for its prognostic ability and was compared to the models built in the most recent year for its superiority. In the end, the functional annotation, immune relevant signature correlation analysis, and 22 tumor-infiltrating immune cells (TICs) analysis were conducted for the full understanding of the gene signature we discovered.
Materials and methods
Database selection
The Therapeutically Applicable Research to Generate Effective Treatments (TARGET) is a dynamically updated database of the National Cancer Institute (NCI) Office of Cancer Genomics (OCG). Its mission is to advance the molecular understanding of cancer to improve patient prognosis13. The TARGET Osteosarcoma (TARGET-OS) project has elucidated a comprehensive molecular profile to identify the genetic changes that drive the occurrence and development of high-risk or difficult-to-treat childhood cancers. OS datasets are available without restrictions on their use in publications or presentations and can be obtained from the official web patrol (https://ocg.cancer.gov/programs/target/projects/osteosarcoma) or GDC Xena Hub (https://gdc.xenahubs.net). TARGET-OS was set as training cohort. Eighty-eight OS cases were included, and their gene expression profile, survival time, survival status, and clinical characteristics were obtained. Gene Expression Omnibus (GEO) is an internationally recognized and widely researched public repository for archiving and free distribution of microarray14. We searched the GEO using the keyword “Osteosarcoma” and set the filters as follows: (1) organisms: homo sapiens; (2) entry type: series; (3) study type: expression profiling by array; (4) the number of samples with expression data is greater than 50; (5) the number of samples with survival data is greater than 50. One dataset named GSE21257 (n = 53) was obtained from GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21257) and treated as validation cohorts to exam the gene signature we constructed. We strictly obeyed the guidelines of the two databases. This study was approved by the Institutional Review Board of Henan Cancer Hospital, which waived the requirement for informed consent due to the use of data obtained from the public databases. All methods were performed in accordance with relevant guidelines and regulations.
Identification of the potential prognostic genes
We implemented a univariate Cox proportional hazard model and Kaplan–Meier estimator to identify genes with potential prognostic ability. In our research, gene expression, survival status, and survival time were input into the R language. With the help of the “survival” and “survminer” R packages, Kaplan–Meier estimator could sort out genes with the ability to distinguish patients' outcomes. The Kaplan–Meier significance threshold was set to p < 0.05. Similarly, each gene’s univariate Cox model was built using the gene expression data, survival status, survival time, and the adoption of the “survival” R package. The univariate Cox model significance threshold was set to p < 0.05. The gene in both tests having a p value < 0.05 was considered the potential prognostic gene.
Gene signature construction and validation
Subsequently, we put the potential prognostic genes identified in the previous step into the LASSO model to detect the best lambda15,16,17,18. Specifically, we utilized the expression data of the potential prognostic genes, patients’ survival data, and the "glmnet" R package to perform the LASSO Cox regression with tenfold cross-validation. Then the R program outputted a list of prognostic genes with coefficients based on the best lambda value selected. According to the instructions and characteristics of the "glmnet" R software package, the selected genes with coefficients would be out putted. The calculation method of the risk score level of each OS is using the following formula:
In the above formula, n represents each hub gene in the gene signature; Expi represents the expression level of each gene; βi represents the coefficient of each gene.
To test our signature's ability in all studied cohorts, the Kaplan–Meier analysis was used to determine the outcome differences between high- and low-risk patients, of which the OS were classified according to the median risk score. In addition, univariate and multivariable Cox analyses further examined the predictive potential of the gene signature. The area under the curve (AUC) is a measure of the classifier's ability to distinguish classes and is used as a summary of the ROC curve19. The higher the AUC, the better the model's performance in determining between positive and negative classes.
Comparison of gene signature with previously published models
We searched PubMed (https://pubmed.ncbi.nlm.nih.gov/) using the keyword "gene signature prognosis osteosarcoma" and made the selection based on criteria we set as follows: (1) the impact factor > 4 (Journal Citation Reports Year 2020, Clarivate, https://jcr.clarivate.com/jcr/home); (2) the online publication date of the article is the most recent year (i.e. May 18, 2020, to May 18, 2021); (3) the candidate study contains specifically findings of the signature’s composition and coefficients. We extracted the gene signatures and the coefficients from the studies and applied them to the studied cohorts to calculate the risk score of each case. The most important thing was that we used the risk scores to build Kaplan–Meier analysis and Cox model to strictly assess the prognostic ability of our predecessors and ours, thus for horizontal comparison.
Function analysis of the gene signature in OS
Gene ontology (GO), including Biological Process (BP), Cellular Components (CC), and Molecular Functions (MF), and Kyoto Encyclopedia of Genes and Genomes (KEGG) were conducted to find the potential function of genes between high- and low-risk groups20,21,22. Enrich items with p value < 0.05 were considered significant.
Correlations between gene signature and immune relevant signatures
We analyzed the immune activity and tolerance of low- and high-risk groups in the training cohort. Firstly, we picked CD274, CTLA4, HAVCR2, IDO1, LAG3, and PDCD1 as immune-checkpoint-relevant signatures, and CD8A, CXCL10, CXCL9, GZMA, GZMB, IFNG, PRF1, TBX2, and TNF as immune-activity-relevant signatures. We adopted an integrated analysis including the Pearson correlation coefficient and Wilcoxon rank-sum to determine the interaction between gene signature and immune relevant signatures.
Determine the relationships between our signature and 22 TICs
We applied a comprehensive analysis based on the Pearson coefficient and Wilcoxon's rank-sum test to evaluate the relationship between 22 TICs and the signatures of this study. In the following analysis, in order to determine the prognostic ability of the 22 TICs, we combined two kinds of statistical approaches, including univariate Cox models and Kaplan–Meier analysis. Together with the evidence found in the first half of this section, we could infer the potential TICs that play crucial roles in the signature’s prognosis ability.
Statistical analysis
We adopt the "CIBERSORT" R package to estimate the abundance of 22 TICs using the gene expression data of the cohorts. LASSO regression was carried out by the "glmnet" R package. Kaplan–Meier plots were constructed by the integration of the "survival" and "survminer" R packages. Cox models, including univariate and multivariable were built via the "survival" R package. The ROC curves was made possible with the help of the “pROC” R package. R software (version 4.0.4, Windows 64-bit) carried out all the processes in this study.
Results
Cohorts’ characteristics
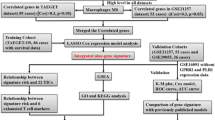
As Fig. 1 demonstrates, 88 OS cases that came from the TARGET-OS cohort were taken for model training. The dataset GSE21257, contained 53 OS cases, were selected for model validating. For patients included in the study, we have collected their clinical characteristics and shown them in Table 1 in detail.

Flow chart of the study. LASSO least absolute shrinkage and selection operator Cox regression model, ROC receiver operating characteristic, OS osteosarcoma, TICs tumor-infiltrating immune cells.
Prognostic gene signature identification
The univariate Cox regression and Kaplan–Meier analysis were conducted to test each gene’s prognostic ability. As shown in Table S1, 70 genes were identified by the Kaplan–Meier estimator, while, 80 genes were determined from the univariate Cox regression model, which has the predictive ability. We intersected them, found 57 genes suitable for our study, and included them in our next analyses (Table S2). The LASSO algorithm displayed when 17 genes existed, the model could achieve the optimized ability (Fig. 2A,B). Table 2 shows the coefficients of the 17 genes.

LASSO regression analysis for the construction of prognostic gene signature. (A) Cross-validation for tuning parameter screening upon LASSO regression analysis. (B) Screening of optimal parameter (lambda) at which the vertical lines were drawn. LASSO the least absolute shrinkage and selection operator Cox regression model.
Confirmation of the prognostic capacity of the seventeen-gene signature
In the risk plot in Fig. 3, we displayed the survival time, survival status, and relative expression of the hub genes for each sample, so as to show the distinguishing ability of the signature in a macroscopic view. In the training cohort, UBE2L3, PLD3, SLC45A4, CLTC, CTNNBIP1, FBXL5, MKL2, SELPLG, C3orf14, WDR53, and ZFP90 have protective abilities for OS patients, while UHRF2, ARX, CORT, DDX26B, MYC, and SLC16A3 display unfavored for the OS prognosis (Figure S1A).

The overall distributions of the risk score (upper), survival status (middle), and gene expression profiles (bottom) of the seventeen-gene signature in the training (A) and validation (B) cohorts.
After drawing the risk plot, we first chose the Kaplan–Meier estimator to estimate the ability of the model we built. As shown in Fig. 4, the survival probability of the high-risk group in the training cohort is lower than that of the low-risk group (p value = 1.764E−08), the same is happening in the validation cohort (p value = 8.915E−03), which demonstrated significant survival differences occurred in the signature distinguished patients.

Kaplan–Meier estimator that evaluating the prognosis capacity of the seventeen-gene signature in the training (A) and validation (B) cohorts. The bottom part indicates the number of patients at risk. The two-sided log-rank test measured the differences between the high- and low-risk groups with a p value < 0.05.
The univariate and multivariable Cox regression were established to exam the signature’s prognostic capacity (Fig. 5). Analysis in the training cohort showed that the risk score affected the OS patients’ outcomes (p value ≤ 5.42E−06). Consistently, the results in the validation cohort proved that risk score was the best one affecting prognosis in either univariate or multivariable examination, furtherly confirmed the powerful predictive capacity of the gene signature (p value ≤ 4.53E−05).

Univariate and multivariate Cox proportional-hazards models that built for testing the predicting ability of the seventeen-gene signature in two cohorts. HR hazard ratio, CI confidence interval.
As shown in Fig. 6A, ROC analysis indicated that the area under the curve (AUC) for our seventeen-gene signature risk score reached 0.891 (95% CI 0.780–0.995, best cutoff = − 5.633), which was the best among other clinical factors. In the GSE21257 cohort, the AUC as well arrived at 0.777 (95% CI 0.780–0.995, best cutoff = − 9.553), topping other characteristics (Fig. 6B).

ROC curves that constructed for examining the predictive ability of the seventeen-gene signature in the training (A) and validation (B) cohorts. ROC receiver operating characteristic, AUC area under the ROC curve, 95% CI 95% confidence interval.
Our gene signature is superior to previous ones
Based on the screening criteria set, we found nine studies that suit for our comparison (Table 3). We applied these discovered signatures and their risk score equations to our training and validation cohorts to calculate the risk score of each OS patient. Then, the Kaplan–Meier estimators were built against our signature and previous models (Fig. 7), demonstrating Yang et al.'s and ours have the ability predicting the outcomes of OS. However, our gene signature (p value ≤ 8.915E−03) seemed to be stronger than Yang et al.'s (p value ≤ 3.602E−02) in terms of their p values.

Comparisons between the seventeen-gene signature and previous studies conducted in the training and validation cohorts using Kaplan–Meier estimator. The two-sided log-rank test measured the differences between the high- and low-risk groups. The bold p value indicates that < 0.05, which considers significantly.
Additionally, Cox univariate and multivariable regression were constructed using these selected prognosis models (Fig. 8). The results in the training cohort demonstrated that only our gene signature (p value ≤ 1.33E−06) having the prognosis capabilities in both the univariate and multivariable analyses. The Cox analysis of the validation cohort determined that only our gene signature passed all the univariate and multivariable tests (p value ≤ 4.31E−06).

Comparisons between the seventeen-gene signature and previous studies conducted in the training and validation cohorts using Cox models. HR hazard ratio, CI confidence interval. *The seventeen-gene signature that identified in this study; the bold p value indicates that < 0.05, which considers significantly.
GO and KEGG enrichment analysis with the seventeen-gene signature
According to the risk score for each case in the TARGET-OS cohort, we conducted GO and KEGG enrichment analysis between high-risk and low-risk groups. The GO enrichment result showed the differences between the two groups mainly focus on extracellular matrix organization, extracellular structure organization, collagen—containing extracellular matrix, endoplasmic reticulum lumen, and extracellular matrix structural constituent (Figure S2A). KEGG analysis was showed that the enriched items were mainly related to protein digestion and absorption, complement and coagulation cascades, and Wnt signaling pathway (Figure S2B).
Relationships between the seventeen-gene signature and immune relevant signatures
We observed that 8/15 of the immune relevant signatures in the high-risk group were significantly under expressed, as demonstrated by the Wilcoxon test (Fig. 9A). The Pearson coefficient test discovered 7/15 of the immune relevant signatures correlated with the seventeen-gene signature (Fig. 9B, Table S3). Incorporating the above findings, six genes, including PRF1, CD8A, HAVCR2, LAG3, CD274, and GZMA were identified associating with the seventeen-gene signature.

Identification of the relationships between the seventeen-gene signature and immune relevant signatures. (A) Wilcoxon rank-sum was adopted to differentiate immune relevant signatures between the high- and low-risk groups. (B) The Pearson coefficient was applied for the correlation test between the immune relevant signatures and seventeen-gene signature. Only correlations with p value < 0.05 were plotted. ns: p value > 0.05; *p value < 0.05; **p value < 0.01; ***p value < 0.001; p value < 0.05 was considered statistically significant.
The seventeen-gene signature and 22 TICs
The GO and KEGG analysis suggested that the difference between the two groups was related to the immune response, so we further conducted 22 TICs analysis to better study how the seventeen-gene signature interact with the immune microenvironment. CIBERSORT algorithm was used to determine the proportion of the tumor-infiltrating immune subpopulations. We visual outputted the 22 TICs distribution and inner correlation in Figure S3.
Combining the findings from difference analysis (Fig. 10A) and correlation analysis (Fig. 10B, Table S4), three TICs (Fig. 10C), including T cells CD8, Mast cells activated, and T cells CD4 memory activated were identified associating with the seventeen-gene signature. Among them, Mast cells activated were found positively correlated with the gene signature, while the others negatively.

Integrating analysis for the relationship between TICs and the seventeen-gene signature. (A) Wilcoxon rank-sum was adopted to differentiate each of 22 TICs between the high- and low-risk groups. (B) The Pearson coefficient was applied for the correlation test between the TICs and the seventeen-gene signature. Only correlations with p value < 0.05 were plotted. (C) The Venn diagram that integrating the results from (A) and (B). TIC: tumor-infiltrating immune cell; *p value < 0.05; p value < 0.05 was considered statistically significant.
We further tested the 22 TICs prognostic abilities by consulting the Kaplan–Meier estimator and univariate Cox proportional-hazard model. As displayed in Fig. 11, the univariate Cox proportional-hazard model (Fig. 11A) indicated that T cells CD8, T cells CD4 memory activated, T cells CD4 naïve, Dendritic cells resting, and Mast cells activated impacted prognosis. Additionally, Kaplan–Meier estimator (Fig. 11B; Table S5) highlighted that T cells CD8, T cells CD4 naïve, and Mast cells activated can predict the survival rate of OS. From the above survival analysis, it can be determined that T cells CD8, T cells CD4 naïve, and Mast cells activated have potential prognostic ability in OS.

Univariate Cox proportional-hazards model (A) and Kaplan–Meier estimator (B) that built for evaluating the prognostic ability of 22 TICs. Only graphs with a p value < 0.05 in the log-rank test were plotted in (B). The bold p value indicates that < 0.05, which considers significant. TIC tumor-infiltrating immune cell.
The results of this part found that T cells CD8 and Mast cells activated were significantly related to our signature and closely related to the OS prognosis, potentially implying that T cells CD8 and Mast cells activated infiltrations play vital roles in the discovered signature in OS patients.
Discussion
In this study, we innovatively discovered a robust seventeen-gene prognostic signature for the OS prognosis by mining TARGET and GEO databases. Specifically, our novelty lay in using univariate Cox model, Kaplan–Meier estimator, and LASSO regression in the model training phase. The adoption of an independent cohort, Kaplan–Meier analysis, Cox regression, ROC curve in the validation process, moreover, highlighted our innovativeness. Most importantly, we compared our signature with published research to prove ours' superiority. At the end of the study, we discovered important mechanisms related to gene signature through function annotations, immune gene correlation analysis, and immune infiltration analysis and speculated that the T cells CD8 and Mast cells activated might potentially help the predictive ability of the signature. This study we worked on designed to shed light on the development of future OS research.
Our signature consists of seventeen genes (Table 2), which were UBE2L3, PLD3, SLC45A4, CLTC, CTNNBIP1, FBXL5, MKL2, SELPLG, C3orf14, WDR53, ZFP90, UHRF2, ARX, CORT, DDX26B, MYC, and SLC16A3, respectively. After tested in the two cohorts (Figure S1), UBE2L3, PLD3, SLC45A4, CTNNBIP1, FBXL5, SELPLG, WDR53, and ZFP90, showed solid protective impacts on OS, while UHRF2, ARX, CORT, DDX26B, MYC, and SLC16A3 witnessed effects on OS prognosis unfavorably. Our findings suggest that CTNNBIP1 is a suppressor of cancer migration, thus making it a potential prognostic predictor for OS. Rothzerg et al. also demonstrated that high expression of CTNNB1 was associated with a good OS prognosis, which is consistent with our findings23. In the UHRF family, UHRF1 and UHRF2 have a multidomain architecture and have similarities in sequence and domain organization24. UHRF1 is a well-known epigenetic regulator. Significant UHRF1 overexpression has been shown in many kinds of tumors25. Liu et al. reported that UHRF1 promotes the proliferation of human OS cells and increases the invasiveness of human OS cells by down-regulating the E-cadherin expression and increasing EMT in an Rb1-dependent manner25. CORT is an endogenous cyclic neuropeptide, which can regulate the growth and metastasis of lung cancer and thyroid cancer, and regulate inflammation by inhibiting immune infiltration26. Wu et al. found that the expression of CORT was higher in high-risk OS populations, confirmed that the high expression of CORT was related to the poor prognosis of OS27. According to previous studies, MYC is widely involved in many cancers, and its expression is estimated to be elevated or dysregulated in up to 70% of human cancers28. MYC mediated transcriptional amplification through super enhancers is an important hallmark of cancer29. The dysregulated expression of the oncogene MYC is usually associated with the oncogenesis and progression of OS30. MYC proto-oncogene boosts the oncogenic transcription amplification process in cancer and is a crucial target for cancer therapy30. It is reported that the MYC gene is amplified in OS, and its expression is often up regulated in patients with OS30. MYC overexpression, coupled with the loss of Ink4a/Arf, can further the transformation of bone marrow stromal cells into OS30. Above all, high MYC levels are related to low apoptosis and poor outcomes in patients with OS30. Chen's team recently demonstrated that MYC-driven super-enhancer signaling is essential for OS tumorigenesis, and the MYC/super-enhancer axis targeting therapeutic strategy to be a promising perspective for OS patients30. The genes ARX31,32, DDX26B33,34, and SLC16A335,36 have been reported to be involved in the occurrence and development of certain cancers, but whether they play an important role in OS has not yet been revealed, implied more efforts are needed.
Freshly, with the widespread application of bioinformatics, potential gene signatures associated with OS prognosis were generated from the publicly databases, which witnessed by more and more involved research. To judge the pros or cons of our signature, we found nine studies published in the most recent past year and compared them horizontally37,38,39,40,41,42,43,44,45. The comparison results once again confirmed our discovery is superior in predicting the prognosis of OS.
KEGG analysis was showed that the enriched items were mainly related to protein digestion and absorption, complement and coagulation cascades, and Wnt signaling pathway (Figure S3B). Wnt signaling is one of the key cascades regulating development and stemness, and it is also closely related to cancer46. The role of Wnt signaling in carcinogenesis has been most prominently described in colorectal cancer, but abnormal Wnt signaling has been observed in more cancer entities46. Constitutive Wnt signal activation is common in human OS, while gene mutations that activate components of the Wnt pathway are rare in OS1. Wnt signaling may play a key role in OS proliferation, metastasis and OS cancer stem cell maintenance1.
Immunotherapy is a type of therapy that helps the individual's immune system eliminate or control cancer47. Recently, immunotherapy has begun to show good prospects in various adult cancers, but whether this method is effective in OS is still rarely reported. Several biological characteristics of OS suggest that the regulation of the immune response may bring benefits, and the various immune approaches available now make immunotherapy potential for OS48. One of the main challenges of immunotherapy is identifying biomarkers that predict response so that treatments can be tailored to maximize benefits48. In the present study, six genes, including PRF1, CD8A, HAVCR2, LAG3, CD274 (PD-L1), and GZMA, were identified as closely related to our seventeen-gene signature and might guide future OS immunotherapy.
Combining the findings of immune infiltration analysis and the 22 TICs survival analysis, we speculated that the extensive infiltration of T cells CD8 and Mast cells activated in tumors may help our signature to achieve stable predictive ability. There is evidence that PD-1 is involved in the progression of OS disease, and the percentage of PD-1 in peripheral blood CD4 + and CD8 + T lymphocytes in OS patients is significantly up-regulated49. More importantly, in vivo and in vitro experiments conducted by researchers have confirmed that PD-L1 in OS is significantly expressed49. Therefore, inhibition of PD-1/PD-L1 is an interesting therapeutic target that can restore the function of the immune system to OS cells49. Mast cells are immune cells that accumulate in tumors and their microenvironment during disease progression50. They play a multi-faceted role in the tumor microenvironment by regulating various events in tumor biology, such as angiogenesis, cell proliferation, and survival50. Invasion and transfer. Mast cells are recruited in the early stages of tumor development and play a key role in angiogenesis and tissue remodeling and promote tumor occurrence and growth50. As tumor growth progresses, mast cells recruit immune cells or suppress anti-tumor responses50. We know from previous studies that mast cells affect the homeostasis of OS and affect tumor progression, but we have not yet understood its underlying mechanism50,51,52. Interestingly, our research showed that T cells CD8 and Mast cells activated can potentially target the gene signature in OS treatment. Thus, further research should consider closely to the roles that the T cells CD8 and Mast cells activated play in the remodeling of the tumor microenvironment.
In the end, we must clarify the limitations of this research. The seventeen-gene signature we derived was from retrospective data. We believe that more prospective data can make our results more effective and rigorous. In addition, although it has absolute superiority compared with previous studies, its proof results are derived from the analysis results of three public databases. There is still no wet laboratory data to explain and support the prognostic ability of these 17 genes and their role in immune infiltration. Therefore, ongoing research is needed to reveal more evidence to for the seventeen -gene signature’s promising future.
Conclusion
The present work identified a novel and robust seventeen-gene signature for the OS prognosis by mining TARGET and GEO databases. In addition, we determined the reliability and applicability of the signature by applying it to an independent cohort. Through comparison, we confirm that our signature is superior to previous research. We identified our signature’s potential immunotherapy targets and the important role of T cells CD8 and Mast cells activated in the seventeen-gene signature prognostic capacity. The real-world influence of the seventeen-gene signature and the underlying mechanisms between it and tumor immunity in OS remained a lack of research and warranted further investigation.
Data availability
Publicly available datasets were used in this study. Data from TARGET-OS (https://ocg.cancer.gov/programs/target/projects/osteosarcoma) and data from GSE21257 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21257) were downloaded and analyzed in this work.
References
Fang, F. et al. Targeting the Wnt/beta-catenin pathway in human osteosarcoma cells. Oncotarget 9, 36780–36792 (2018).
Harrison, D. J., Geller, D. S., Gill, J. D., Lewis, V. O. & Gorlick, R. Current and future therapeutic approaches for osteosarcoma. Expert Rev. Anticancer Ther. 18, 39–50 (2018).
Isakoff, M. S., Bielack, S. S., Meltzer, P. & Gorlick, R. Osteosarcoma: Current treatment and a collaborative pathway to success. J. Clin. Oncol. 33, 3029–3035 (2015).
Whelan, J. S. & Davis, L. E. Osteosarcoma, chondrosarcoma, and chordoma. J. Clin. Oncol. 36, 188–193 (2018).
Jia, Y., Liu, Y., Han, Z. & Tian, R. Identification of potential gene signatures associated with osteosarcoma by integrated bioinformatics analysis. PeerJ 9, e11496 (2021).
Zhang, A., Yang, J., Ma, C., Li, F. & Luo, H. Development and validation of a robust ferroptosis-related prognostic signature in lung adenocarcinoma. Front. Cell Dev. Biol. 9, 616271 (2021).
Wang, Y. et al. Prognostic implications of immune-related eight-gene signature in pediatric brain tumors. Braz. J. Med. Biol. Res. 54, e10612 (2021).
Luo, H. & Ma, C. A novel ferroptosis-associated gene signature to predict prognosis in patients with uveal melanoma. Diagnostics (Basel) 11, 20 (2021).
Luo, H., Ma, C., Shao, J. & Cao, J. Prognostic implications of novel ten-gene signature in uveal melanoma. Front. Oncol. 10, 567512 (2020).
Ma, C. et al. Identification of a novel tumor microenvironment-associated eight-gene signature for prognosis prediction in lung adenocarcinoma. Front. Mol. Biosci. 7, 571641 (2020).
Cantini, L. et al. Classification of gene signatures for their information value and functional redundancy. NPJ Syst. Biol. Appl. 4, 2 (2018).
Yu, S., Shao, F., Liu, H. & Liu, Q. A five metastasis-related long noncoding RNA risk signature for osteosarcoma survival prediction. BMC Med. Genom. 14, 124 (2021).
Mao, R. et al. Prognostic nomogram for childhood acute lymphoblastic leukemia: A comprehensive analysis of 673 patients. Front. Oncol. 10, 1673 (2020).
Clough, E. & Barrett, T. The gene expression omnibus database. Methods Mol. Biol. 1418, 93–110 (2016).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Goeman, J. J. L1 penalized estimation in the Cox proportional hazards model. Biom. J. 52, 70–84 (2010).
Sauerbrei, W., Royston, P. & Binder, H. Selection of important variables and determination of functional form for continuous predictors in multivariable model building. Stat. Med. 26, 5512–5528 (2007).
Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 16, 385–395 (1997).
Cao, R. & Lopez-de-Ullibarri, I. ROC curves for the statistical analysis of microarray data. Methods Mol. Biol. 1986, 245–253 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551 (2021).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Rothzerg, E., Xu, J., Wood, D. & Koks, S. 12 Survival-related differentially expressed genes based on the TARGET-osteosarcoma database. Exp. Biol. Med. (Maywood) 246, 2072–2081 (2021).
Iguchi, T. et al. Identification of UHRF2 as a negative regulator of epithelial-mesenchymal transition and its clinical significance in esophageal squamous cell carcinoma. Oncology 95, 179–187 (2018).
Liu, W. et al. UHRF1 promotes human osteosarcoma cell invasion by downregulating the expression of Ecadherin in an Rb1dependent manner. Mol. Med. Rep. 13, 315–320 (2016).
Delgado-Maroto, V. et al. The neuropeptide cortistatin attenuates experimental autoimmune myocarditis via inhibition of cardiomyogenic T cell-driven inflammatory responses. Br. J. Pharmacol. 174, 267–280 (2017).
Wu, Z. L. et al. Development of a novel immune-related genes prognostic signature for osteosarcoma. Sci. Rep. 10, 18402 (2020).
Dang, C. V. MYC on the path to cancer. Cell 149, 22–35 (2012).
Kress, T. R., Sabo, A. & Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 15, 593–607 (2015).
Chen, D. et al. Super enhancer inhibitors suppress MYC driven transcriptional amplification and tumor progression in osteosarcoma. Bone Res. 6, 11 (2018).
Hackeng, W. M. et al. Assessment of ARX expression, a novel biomarker for metastatic risk in pancreatic neuroendocrine tumors, in endoscopic ultrasound fine-needle aspiration. Diagn. Cytopathol. 48, 308–315 (2020).
Liu, W. B. et al. Epigenetic silencing of Aristaless-like homeobox-4, a potential tumor suppressor gene associated with lung cancer. Int. J. Cancer 134, 1311–1322 (2014).
Chen, H. et al. Small RNA-induced INTS6 gene up-regulation suppresses castration-resistant prostate cancer cells by regulating beta-catenin signaling. Cell Cycle 17, 1602–1613 (2018).
Lui, K. Y. et al. Integrator complex subunit 6 (INTS6) inhibits hepatocellular carcinoma growth by Wnt pathway and serve as a prognostic marker. BMC Cancer 17, 644 (2017).
Yu, S. et al. Comprehensive analysis of the SLC16A gene family in pancreatic cancer via integrated bioinformatics. Sci. Rep. 10, 7315 (2020).
Javaeed, A. & Ghauri, S. K. MCT4 has a potential to be used as a prognostic biomarker—a systematic review and meta-analysis. Oncol. Rev. 13, 403 (2019).
Yang, M. et al. Identification of a novel glycolysis-related gene signature for predicting the prognosis of osteosarcoma patients. Aging (Albany NY) 13, 12896–12918 (2021).
Fu, Y. et al. Development and validation of a hypoxia-associated prognostic signature related to osteosarcoma metastasis and immune infiltration. Front. Cell Dev. Biol. 9, 633607 (2021).
Cao, M. et al. Identification and development of a novel 4-gene immune-related signature to predict osteosarcoma prognosis. Front. Mol. Biosci. 7, 608368 (2020).
Xiao, B. et al. Identification and verification of immune-related gene prognostic signature based on ssGSEA for osteosarcoma. Front. Oncol. 10, 607622 (2020).
Chen, Z., Huang, H., Wang, Y., Zhan, F. & Quan, Z. Identification of immune-related genes MSR1 and TLR7 in relation to macrophage and type-2 T-helper cells in osteosarcoma tumor micro-environments as anti-metastasis signatures. Front. Mol. Biosci. 7, 576298 (2020).
Wen, C. et al. A three-gene signature based on tumour microenvironment predicts overall survival of osteosarcoma in adolescents and young adults. Aging (Albany NY) 13, 619–645 (2020).
Song, Y. J. et al. Immune landscape of the tumor microenvironment identifies prognostic gene signature CD4/CD68/CSF1R in osteosarcoma. Front. Oncol. 10, 1198 (2020).
Yu, Y. et al. Development of a prognostic gene signature based on an immunogenomic infiltration analysis of osteosarcoma. J. Cell Mol. Med. 24, 11230–11242 (2020).
Zhu, N. et al. Co-expression network analysis identifies a gene signature as a predictive biomarker for energy metabolism in osteosarcoma. Cancer Cell Int. 20, 259 (2020).
Zhan, T., Rindtorff, N. & Boutros, M. Wnt signaling in cancer. Oncogene 36, 1461–1473 (2017).
Viale, P. H. The American Cancer Society’s Facts & Figures: 2020 Edition. J. Adv. Pract. Oncol. 11, 135–136 (2020).
Wedekind, M. F., Wagner, L. M. & Cripe, T. P. Immunotherapy for osteosarcoma: Where do we go from here?. Pediatr. Blood Cancer 65, e27227 (2018).
Wang, Z., Li, B., Ren, Y. & Ye, Z. T-cell-based immunotherapy for osteosarcoma: Challenges and opportunities. Front. Immunol. 7, 353 (2016).
Inagaki, Y. et al. Dendritic and mast cell involvement in the inflammatory response to primary malignant bone tumours. Clin. Sarcoma Res. 6, 13 (2016).
Maciel, T. T., Moura, I. C. & Hermine, O. The role of mast cells in cancers. F1000Prime Rep. 7, 09 (2015).
Campillo-Navarro, M. et al. Mast cells in lung homeostasis: Beyond type I hypersensitivity. Curr. Respir. Med. Rev. 10, 115–123 (2014).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
J.Y., A.Z., and C.M. drafted the original manuscript. H.L. plotted all figures for data visualization. H.L. and C.M. did the final proofread of the manuscript. All authors reviewed the current form of the manuscript and approved its publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, J., Zhang, A., Luo, H. et al. Construction and validation of a novel gene signature for predicting the prognosis of osteosarcoma. Sci Rep 12, 1279 (2022). https://doi.org/10.1038/s41598-022-05341-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-05341-5
This article is cited by
-
Identification of an early survival prognostic gene signature for localized osteosarcoma patients
Scientific Reports (2024)
-
Ferroptosis-related lncRNAs guiding osteosarcoma prognosis and immune microenvironment
Journal of Orthopaedic Surgery and Research (2023)
-
Single-Cell RNA Sequencing Pro-angiogenic Macrophage Profiles Reveal Novel Prognostic Biomarkers and Therapeutic Targets for Osteosarcoma
Biochemical Genetics (2023)
-
Transcriptomic characterization and construction of M2 macrophage-related prognostic and immunotherapeutic signature in ovarian metastasis of gastric cancer
Cancer Immunology, Immunotherapy (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
