
Abstract
The macronuclear (MAC) genomes of ciliates belonging to the genus Euplotes species are comprised of numerous small DNA molecules, nanochromosomes, each typically encoding a single gene. These genomes are responsible for all gene expression during vegetative cell growth. Here, we report the analysis of the MAC genome from the Antarctic psychrophile Euplotes focardii. Nanochromosomes containing bacterial sequences were not found, suggesting that phenomena of horizontal gene transfer did not occur recently, even though this ciliate species has a substantial associated bacterial consortium. As in other euplotid species, E. focardii MAC genes are characterized by a high frequency of translational frameshifting. Furthermore, in order to characterize differences that may be consequent to cold adaptation and defense to oxidative stress, the main constraints of the Antarctic marine microorganisms, we compared E. focardii MAC genome with those available from mesophilic Euplotes species. We focussed mainly on the comparison of tubulin, antioxidant enzymes and heat shock protein (HSP) 70 families, molecules which possess peculiar characteristic correlated with cold adaptation in E. focardii. We found that α-tubulin genes and those encoding SODs and CATs antioxidant enzymes are more numerous than in the mesophilic Euplotes species. Furthermore, the phylogenetic trees showed that these molecules are divergent in the Antarctic species. In contrast, there are fewer hsp70 genes in E. focardii compared to mesophilic Euplotes and these genes do not respond to thermal stress but only to oxidative stress. Our results suggest that molecular adaptation to cold and oxidative stress in the Antarctic environment may not only be due to particular amino acid substitutions but also due to duplication and divergence of paralogous genes.
Similar content being viewed by others


Introduction
Euplotes focardii is an Antarctic ciliate classified as an obligate psychrophilic stenothermal organism1,2,3,4. As all ciliates, E. focardii is characterized by the presence of cilia on its surface and by nuclear dimorphism: a micronucleus (MIC) that represents the germ line, and a macronucleus (MAC) serving as the somatic line involved in the gene expression during the vegetative stage. The MAC derives from the MIC after extensive DNA rearrangements during conjugation, including the fragmentation of chromosomes and the elimination of non-protein-coding DNA segments5. As a consequence, the MAC genome is commonly composed by nanochromosomes, i.e., tiny chromosomes capped by telomeric sequences that, in general, contain a single coding sequence (CDS)6,7. The nanochromosomes are amplified to thousands of copies (~2000)5. The copy number may oscillate since it is probably unregulated through cell replication, as it has been described for Stylonychia8 and Oxytricha9.
As a psychrophilic unicellular organism directly exposed to environmental cues, E. focardii represents an excellent model for the study of cold adaptation, offering some potential advantages over psychrophilic multicellular models10. Low temperatures exert several physicochemical constraints on cold living organisms, including the process of microtubule polymerization11. Microtubule polymers are of fundamental importance in many eukaryotic cellular processes, including cell motility, maintenance of cytoskeletal architecture, intracellular transport, and mitosis. Microtubules assemble from α- and β-tubulin heterodimers with the help of γ-tubulins, a structural component of both centrioles and basal bodies. There are other components of the tubulin superfamily, such as the δ-tubulin and the ε-tubulin, that most likely interact longitudinally with α-tubulins at the minus ends and with β-tubulins to the plus ends of microtubules, respectively12. Furthermore, δ-tubulin may be involved in the formation of the C triplet tubules in the basal bodies, while ε-tubulin may be located to the centrosome13. Generally, in organisms that live in temperate environments, the assembly of microtubules from tubulin heterodimers requires physiological temperatures and these microtubules usually disassemble at temperatures below 4 °C. In chronically cold habitats, the microtubule dynamics in psychrophilic organisms, including the Antarctic ciliate E. focardii, most likely reflect adaptive modifications of tubulin heterodimers11,14,15,16.
E. focardii is also a good model for studying adaptation to oxidative stress. Like any Antarctic marine microorganism, this ciliate is constantly exposed to a high oxygen concentration and abundant reactive oxygen species (ROS), due to the higher O2 solubility in water at low temperature and to high UV radiation due to the ozone hole. Accordingly, strengthened defenses against oxidative stress, e.g., by increasing the antioxidant enzymes system, including superoxide dismutase (SOD), catalase (CAT), peroxiredoxins (PRX), thioredoxin reductase (TRXR) and glutathione systems (glutathione synthetase GS, glutathione reductase GR, glutathione peroxidases GPx, and glutathione S-transferases GST) may be necessary. Specifically, the dismutation by SOD of the superoxide (the primary produced Reactive Oxygen Species, abbreviated as ROS), and the reduction of the produced H2O2 by CAT and GPx (often in association with thiol-containing enzymes, PRX, TRXR and glutaredoxins17) are of extreme importance for stress response in all cells18. Likewise, GSTs are important to inactivate unsaturated aldehydes, epoxides, and hydroperoxides, secondary metabolites of the reactions described above19. In this regard, it was shown that the transcription regulation of SOD, CAT, GR and GPx is strongly affected by oxidative stress in the mesophilic ciliate Tetrahymena thermophila20.
Thermal stress response could also be a further biomarker to understand the adaptation of these organisms. In mesophilic organisms, heat shock proteins (HSPs) are the main macromolecules involved in this mechanism and, acting as chaperones, provide the stabilization, the partial refolding, or the detection of proteins irreversibly damaged21,22. In particular, the Hsp70 group is represented by proteins that are quickly induced under stress conditions by the activation of the hsp70 gene expression23. However, hsp70 genes have been reported to have lost heat inducibility in several Antarctic marine organisms, including E. focardii24,25,26.
Euplotes species are characterized by an unusual and pervasive mechanism of programmed translational frameshifting27. A previous study28 reported an extensive analysis of this mechanism in the E. crassus genome and included some comparison with the E. focardii genome to gain some insights into the conservation and molecular basis of this frameshifting. Here, we report a deeper E. focardii MAC genome analysis based on reads that have been completely reassembled and annotated. Furthermore, we focused on characterizing Hsp70, tubulin and antioxidant enzymes gene families. Our results suggest that molecular adaptation to cold and oxidative stress defense in the Antarctic environment may be based on a variable number of paralogous genes.
Results and discussion
A draft E. focardii macronuclear genome assembly
The E. focardii MAC genome assembly obtained using the SPAdes algorithm showed a significant improvement (25%) of the number of 2-telomere nanochromosomes with respect to the assembly previously produced by Newbler (stored in GenBank as version MJUV01000000)28, i.e., 17,798 sequences containing telomeres at both ends (Table 1) vs 12,922 previously reported. SPAdes assembly was performed on cleaned reads, e.g., after the clean-up of all algal or bacterial contaminants (see "Materials and methods" section). A summary of the SPAdes assembly data is shown in Table 1: these data were obtained after an extra clean-up of sequences without telomeres and with a high GC content (Fig. S1). The final GC distribution shows a well-defined peak (with respect to the distribution before cleaning) centered on the 31.51% (Table 1, Fig. S2), a value consistent with those reported for other ciliates with a nanochromosomal MAC genome architecture (i.e., Stylonychia lemnae, Oxytricha trifallax, and Euplotes crassus, 31.5%, 31.2% and 36.9%, respectively)29. This homogeneous normal GC distribution is consistent with negligible bacterial contamination. Although we cannot rule out the presence of sequences belonging to yet unreported endosymbionts with low GC content, it is unlikely their sequence base composition would perfectly match that of E. focardii and produce a unimodal distribution. Nanochromosomes containing bacterial sequences were not found in the final assembly suggesting that no phenomena of horizontal gene transfer recently occurred in this ciliate even though it has a substantial associated bacterial consortium30.
The genome size is in line with expectations based on the previous studies on other ciliates5, as well as for the ciliates reported in Table 2. This number of nanochromosomes was chosen as the main parameter for the selection of the best assembly among those produced by the different versions and configurations/parameters of SPAdes algorithm described in "Materials and methods". This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession MJUV00000000. The version described in this paper is version MJUV02000000.
This assembly does not show any alternative fragmentation on the basis of the clustering analysis used in analyses of the Oxytricha MAC genome and consistent with previous observations in E. crassus7,31. Moreover, from the pairwise sequence identity analysis of the assembly (described in the "Paralog prediction" section of "Materials and methods"), E. focardii, and E. octocarinatus, do not show a peak at high (> 90%) sequence identity typical of heterozygous alleles, as instead E. crassus and E. vannus show, but only one peak of around 40% identity, likely due to the presence of a substantial number of paralogous genes (Fig. S3). The MAC genome assemblies of E. crassus, E. octocarinatus and E. vannus all have considerably more complete nanochromosomes than E. focardii (Table 1; E. crassus MAC genomes are unpublished but have in excess of 30,000 nanochromosomes). However, all three species also show an order of magnitude more paralogs around the 40% peak than E. focardii. The smaller number of complete nanochromosomes in E. focardii thus likely reflects a combination of three factors: (i) lower assembly contiguity; (ii) high genome homozygosity; (iii) fewer paralogs. In future further improvement of this genome would likely best be achieved by using long-read sequencing, such as that provided by Pacific Biosciences.
The results of the gene prediction performed using the AUGUSTUS software are summarized in Table 2. The number of genes (predicted in the nanochromosomes and in the contigs with no telomeres that blast with a database containing all ciliates available proteomes), the average of the CDSs’ length, of the introns’ length and of the number of introns per gene are consistent with those previously reported for Stylonychia and Oxytricha7,32, and for E. crassus28,33. Nanochromosomes encoding a single CDS represent 74.58% of the total, and nanochromosomes with more than one CDS are 8.51% of the total; these percentages are comparable to those of Stylonychia and Oxytricha (75% encoding a single CDS and higher than 7% with more than one CDS7,32,34).
To assess genome completeness, we compared the predicted proteins of the E. focardii macronuclear genome assembly with the CEG database: 93% of this database’s sequences (231) have homologs in the E. focardii assembly. This value may suggest a small amount of incompleteness of the genome assembly. However, 11 out of 17 of the sequences without matches are also absent from the Oxytricha and Stylonychia genomes (Table 3), consistently with greater evolutionary distances of ciliates from the eukaryotes included in CEG, as also proposed for Stylonychia7. Genome annotation revealed a total of 90 ribosomal proteins, of which 36 belong to the standard eukaryotic small 40S subunit and 54 to the large 60S subunit. These values strongly support the present genome analysis, considering that the total standard eukaryotic ribosomal protein set that contains 32 proteins in the 40S subunit and 48 in the 60S subunit. Rfam analysis of structural RNAs in E. focardii, in addition to confirm the annotated tRNAs described in the next section, revealed the presence in the assembly of a nanochromosome (NODE_589) encoding both 18S and 28S rRNA genes, as well as 10 additional snRNA genes, including 3 for 5S rRNA and one for 5.8S rRNA. These results agree with those previously reported for Oxytricha trifallax7 and Stylonychia lemnae32. Taken together these results support the completeness of the E. focardii MAC genome assembly.
Properties of E. focardii translation
Ciliates, including Euplotes, frequently use the standard stop codons in atypical ways compared to the usual eukaryote assignments35,36. As previously reported28,37,38, we detected abundant frameshifting motifs (AAA, AAT, ATA, AAC etc. codons followed by a stop codon, either TAA or TAG) in the E. focardii MAC genome (see "Materials and methods" section) consistent with pervasive + 1 programmed translational frameshifting in Euplotes ciliates. The frameshifting analysis on the predicted genes revealed that at least 4.2% of total genes could be affected by this phenomenon. This value could be underestimated since it excludes genes that had no BLAST hits (about 47%) to our sequence database and does not consider other possible frameshifting motifs beside those already identified in E. crassus28. Even though the comparison of the frameshifting sites between E. focardii and E. crassus revealed that these are not conserved in the same genes28, the occurrence of predicted frameshifting (just under 10%) would be in line with the observations in E. crassus by Klobutcher et al.37 and in E. octocarinatus by Wang et al.38. Considering the transcript-level quantity of these specific genes, as described in the "Materials and methods" section, the + 1 ribosomal frameshifting activity does not significantly affect the transcription in E. focardii on the basis of the statistical test used (p-value = 0.386).
61 tRNAs were predicted in the E. focardii MAC genome assembly. 55 unique tRNAs were encoded on nanochromosomes and appear to be sufficient for the translation of all codons. These tRNAs include one selenocysteine tRNA (encoded on NODE_51680), with the typical long variable arm characteristic of such tRNAs, and a putative paralogous pair of cysteine tRNA genes with TCA anticodons (tRNA-Cys(TCA)-1: NODE_55662; tRNA-Cys(TCA)-2: NODE_55665; 90% identical to each other), which resemble those previously reported for E. crassus39. The tRNA-Cys(TCA) paralogs are in turn paralogs of tRNA-Cys(GCA), as for E. crassus39.
Previously a potential stop-suppressor tRNA of UAA, suggested to play a role in the ribosomal frameshifting was reported in Euplotes octocarinatus38,40. Though we found evidence of potential translational readthrough of stop codons (see subsequent analysis), we could not detect a similar tRNA in Euplotes focardii using BLASTN searches with the putative E. octocarinatus frameshifting tRNA as a query. Furthermore, neither tRNAscan-SE nor Aragorn predicted a similar tRNA to the putative frameshifting E. octocarinatus tRNA in E. focardii. The tRNA secondary structure prediction software did however predict an unrelated tRNA with a potential stop cognate anticodon on contig NODE_32101 (Fig. 1A). NODE_32101 encodes an additional tRNA-Glu(TTC), which is 66.7% identical to the putative suppressor tRNA(CTA) (Fig. 1). A different tRNA-Glu(TTC) encoded on a separate nanochromosome (NODE_54875) is 77% identical to NODE_32101 tRNA-Glu(TTC). BLASTN searches with both of the candidate tRNAs versus the E. octocarinatus genome yield top hits to E. octocarinatus Contig33653 (for NODE_32101 tRNA(CTA)) and Contig33553 (NODE_32101 tRNA-Glu(TTC)). For both E. octocarinatus nanochromosomes tRNA-Glu(TTC) genes are predicted in the regions of the BLASTN matches. Thus it appears NODE_32101 tRNA(CTA) is a paralog of Glu(TTC) tRNAs.

(A) Secondary and tertiary structures of a potential stop-suppressor tRNA of UAG. (B) alignment of predicted tRNA-Glu genes and candidate suppressor. (C) Frequencies of amino acids aligned to TGA = Cysteine, TAA/TAG and CAA/CAG = Glutamine in tblastn matches (amino acid counts were obtained from BLAST highest scoring pairs with e-value < 1e−20, a match criterion of 21 bp amino acids centered on the codon of interest and > = 40% amino acid identity over 10 amino acids either side of subject codon – example provided below graph). “n” indicates the number of aligned codons analyzed for each bar. Query O. trifallax proteins were obtained from oxy.ciliate.org.
We observed expression of mature tRNAs for both predicted tRNAs on NODE_32101 in the YAMAT-seq reads (Supplemental Table 1), at low levels, but well within the limits of other predicted E. focardii tRNAs. It can be seen that the secondary structure predicted by RNAfold (Fig. 1A) does not place the anticodon 5’-CTA-3’ symmetrically in the anticodon loop, as is typically the case. However, this may be incorrect structure prediction, similar to the incorrect predictions by RNAfold we observed while attempting to predict the E. focardii tRNA-Cys(TCA) structures using this software. In the alignment of E. focardii and E. octocarinatus tRNAs, it can be seen that TCA aligns to the predicted TTC anticodons of the Glu-tRNAs (Fig. 1B). In future, to ascertain whether the candidate tRNA(CTA) on NODE_32101 is functional it would be necessary to search additional genomes from other Euplotes species, and observe both whether there are similar putative tRNA genes with TCA anticodons and also whether there are co-varying substitutions that support structural conservation and anticodon position.
Other than frameshifting “stop” codons, there are a few reports of potential translation of in-frame “stops”. In E. focardii, based on multiple sequence alignments, a TAG codon in a beta-tubulin gene was hypothesized to be translated as tryptophan41. Ricci et al. described the use of an in-frame TAG codon in two other Euplotes genes42. Recently, Wang et al. reported the use of TAG to encode an amino acid, likely glutamine, in the cathepsin B gene of the closely related freshwater Euplotes octocarinatus43. Given these observations and considerable plasticity in termination codon usage in ciliates44,45, we wondered if the E. focardii stop codons may occasionally be translated in other genes.
In standard genetic code organisms, which do not possess tRNAs directly cognate to “stop” codons, translational readthrough, may use near-cognate pairing of tRNAs (i.e., possessing two of the three complementary anticodon-codon pairings between bases). TAA/TAG codons are most frequently translated as glutamine and TGA as tryptophan in eukaryotic translational readthrough46. To investigate what amino acids TAA/TAG codons in E. focardii might encode, we examined the frequency of amino acids aligned to these codons in conserved alignments extracted from translated BLAST matches (Fig. 1C). We focused on matches with substantial sequence conservation up- and downstream of stops to exclude potential sites of translational frameshifting. As a baseline for comparison, we also examined the frequencies of amino acids aligned to TGA codons, which predominantly encode cysteine, and CAA/CAG codons, which encode glutamine. For both codon kinds, the most frequent aligned amino acids are the expected ones (Fig. 1C). TAA/TAG codons are most often frequently aligned to glutamine.
Whether TAA/TAG codons are translated by translational readthrough or conventionally by a tRNA such as the candidate tRNA(CTA) in E. focardii, remains to be determined. In other eukaryotes translational readthrough typically occurs at low levels, typically a small percentage of non-translational readthrough, and leads to short extensions of proteins47. Consequently, in future it will be of interest to determine the translational efficiency of in-frame stops in Euplotes, particularly in genes like the beta-tubulin paralog with an in-frame TAG occurring close to the N-terminus41, especially if it is a relatively highly translated protein like other beta-tubulins. Furthermore, it would also be of interest to determine what amino acid the candidate tRNA(CTA) may be charged with, and if there is transamidation of glutamate to glutamine, as observed in Bacillus subtilis and many other organisms48,49.
The tubulin super families in E. focardii genome
The protein annotation procedure allowed the identification of 15,357 proteins (Table 2), 3306 of which are enzymes. Using CD-HIT, 5222 were grouped in clusters with at least 40% of identity. We focused on the analysis of the members of tubulins, antioxidant enzymes, and Hsp70 gene families to examine whether the number of paralogs and their evolution in each superfamily may be related to the E. focardii cold-adaptation.
In vitro polymerization studies performed with E. focardii purified tubulin heterodimers demonstrated their ability to form microtubules at temperatures close to the freezing point of the Antarctic marine habitat14, as also reported for tubulin heterodimers purified from Antarctic fishes50,51. In addition, the study of the tubulin superfamily in an Antarctic psychrophilic ciliate is even more interesting with respect to other psychrophilic organisms because it contributes not only to the understanding of the molecular basis of microtubule cold adaptation but also of microtubular structure complexity. Indeed, differently from other eukaryotic microorganisms, ciliates assemble 17 different types of microtubules throughout their life cycle 52, even though all microtubule functions are carried out in a single cell.
The macronuclear genome sequencing from several ciliates7,32,53,54 allowed the characterization of up to five alpha- and beta-tubulin isotypes. Therefore, a multigenic tubulin family is a common characteristic in ciliates and these tubulin isotypes may be responsible of the formation of functionally different microtubules55 and with different dynamics properties14,16,56.
In previous papers, we reported the characterization of a single α-tubulin3, four β-tubulin1,14 and two γ-tubulin isotypes15 from E. focardii. Previously, the presence of four β-tubulin isotypes induced us to hypothesize that in E. focardii microtubules cold adaptation was based mainly on molecular modification of the β-tubulin subunit of the heterodimer rather than on the α-tubulin subunit. The comparison of these sequences with the homologs from non-cold adapted Euplotes species revealed the presence of unique amino acid substitutions in the E. focardii tubulin isotypes that may be correlated with cold adaptation14. Therefore, we further investigated this relevant class of proteins in the E. focardii genome.
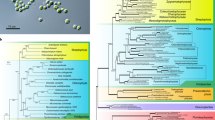
All tubulin sequences that were detected in the final assembly were checked starting from those previously characterized1,3,14,15,56. All the sequences already collected into the UniProt Knowledgebase (TBβ1—UniProt: Q9N2N6; TBβ2—UniProt: C0L7F0; TBβ3—UniProt: C0L7F1; TBα1—UniProt: Q8WRT6; TBγ1—UniProt: A3F2R1; TBγ2—UniProt: A3F2R2) were identified and confirmed as reported in Table 4, with the exception of TBβ4 (the sequence collected as C0L7F2 in UniProt is included in the phylogenetic tree of Fig. 2B). However, new isotypes were discovered after protein annotation. Specifically, a new β-tubulin isotype (named TBβ5) and six additional α-tubulin isotypes. The relationship among these isotypes and those for other Euplotes species are shown in the phylogenetic trees in Fig. 2. In the trees, we introduced homologs from Tetrahymena, Oxytricha and Stylonychia to better evidence the degree of E. focardii gene amplification and divergency. E. focardii α-tubulins branches (highlighted as bold in Fig. 2A) are scattered throughout the phylogeny of ciliate homologs: ATU1 and ATU2 isotypes cluster with the homologs from E. crassus in the same group containing the “canonical” Tetrahymena α-tubulin, whereas ATU3 to ATU7 isotypes form a separate clade with E. crassus ATU4 and ATU5, and one isotype from Oxytricha. This result suggests that the isotypes from ATU1 to ATU5 preceded the divergence of E. focardii from the other Euplotes species, and were maintained in the Antarctic ciliate, whereas ATU6 and ATU7 derived from an additional event of gene duplication in E. focardii that gave origin to new distinct isotypes. An high number of isotypes may be considered as an additional strategy for tubulin cold adaptation beside the presence of unique residues substitutions in the primary structure of tubulin heterodimers1.

Phylogenetic trees of tubulin super families: (A) α-tubulins, (B) β-tubulins and (C) γ-tubulins. Only bootstrap percentage values higher than 50%, indicated near nodes, are shown.
By contrast, E. focardii β-tubulin family appears less amplified and divergent than alpha tubulin. The branches (highlighted as bold in Fig. 2B) are less scattered and cluster with the corresponding E. crassus and E. octocarinatus homologs, even though BTU5 forms a well separated clade. BTU1 and BTU2, and BTU3 and BTU4, may be originated by recent gene duplications in E.focardii.
With the high number of different α-tubulin isotypes in the Antarctic ciliate E. focardii, we reconsidered the importance of the α-tubulin subunit in microtubule cold adaptation.
Molecular flexibility is regarded as a hallmark of cold adapted molecules, in particular enzymes, to cope with the reduction of dynamics and activity at low temperature57,58. We applied Molecular Dynamics (MD) simulation on each α-tubulin isotype of the ciliates under study. We found that only three E. focardii isotypes (2, 4 and 5; Fig. 3B) show a higher flexibility at 4 °C with respect to the E. crassus α-tubulin isotypes. The only exception in E. crassus is the isotype 2. This result can be considered as a further evidence of the E. focardii tubulin cold adaptation and suggests that not all isotypes must be flexible to function at low temperatures, as we reported also for the β-tubulin isotypes14. The similarity of E. focardii α-tubulin isotype 2 with that from E. crassus suggests a common origin of this α-isotype.

(A) trajectories data as backbone RMSDs for E. focardii α-tubulin isotype 2, for the first 10 ns, at 27 °C (red) and at 4 °C (blue). (B) the bar plot shows ΔRMSD values for each α-tubulin of E. focardii (blue), E. crassus (green), E. octocarinatus (yellow) and T. thermophila (6U0H, orange; ATU1 in Fig. 2) calculated as the difference between the RMSDmax at 27 °C and the RMSDmax at 4 °C (see "Materials and methods" section).
We also identified seven α-like and two β-like tubulins (e.g., more divergent isotypes; Table 4) and two δ and one ε isotypes. By contrast, the assembler was not able to produce two distinct complete contigs/nanochromosomes for γ-tubulin. In other words, γ-tubulin type 2 previously obtained from macronuclear DNA purified from E. focardii cells is present in the contig only in a short fragment form (see Table 4 legend). This is probably due to the high sequence similarity between the two γ-tubulin isotypes since raw reads specific for this second isotype were obtained after sequencing but difficult to align with this assembler. In the phylogenetic tree, E. focardii GTU1 and GTU2 isotypes (highlighted as bold in Fig. 2C) appear to be more related to the single γ-tubulin isotype from E. octocarinatus (that it is encoded by two similar genes) than the two distinct isotypes from E. crassus. In E. focardii, the presence of distinct γ-tubulin isotypes is associated to different roles in the nucleation of cellular microtubules (as previously reported15) more than to cold-adaptation.
The antioxidant enzymes system
A major issue for Antarctic marine organisms is oxidative stress since they experience high dissolved oxygen, more soluble in cold seawaters, typical of Antarctic marine environment59. Two key classes of enzymes involved in antioxidant defenses are the superoxide dismutases and catalases.
Superoxide dismutases (SOD, EC 1.15.1.1) are the ubiquitous metalloenzymes that catalyse the dismutation of superoxide anion into molecular oxygen and hydrogen peroxide (O2− + O2− + 2H+ → O2 + H2O2)60,61. SODs are grouped into three protein families, based on the metal cofactor they contain and on the protein fold62. Copper, zinc SODs (Cu,Zn SODs) are found in the cytoplasm of eukaryotes, in the chloroplasts of some plants and in the periplasmic space of bacteria63,64. This group of SODs is often referred to as SOD1, but if these Cu,Zn enzymes are present in the extracellular fluids of eukaryotes are referred as SOD365,66. Iron- and manganese-containing SODs (FeSOD and MnSOD, referred as SOD2) are considered the primitive forms of SODs67,68. FeSODs are found in prokaryotes and chloroplasts, while MnSODs are present both in prokaryotes and in the mitochondrial matrix of eukaryotes. Differently from MnSOD, Cu,Zn SODs were believed to be absent in protists62 until whole genomes sequencing revealed Cu,Zn SOD encoding genes in a number of different protists69,70,71,72.
In a previous paper73, two E. focardii Cu,ZnSODs and one MnSOD were biochemically characterized. All three SODs are active at 4 °C but at the same time they retain high activity upon 20 min incubation up to 55/60 °C. This feature is unusual in cold active enzymes that are often heat sensitive and undergo inactivation and unfolding even at mild temperature. Nevertheless, thermo-tolerance or even thermostability of cold adapted enzymes was previously reported4,74 suggesting that cold activity and thermo-tolerance may coexist in a molecule.
The sequences of the enzymes previously studied and present in the UniProt Knowledgebase (SOD1a—UniProt: W0FZ77; SOD1b—UniProt: W0FUJ3; SOD2a—UniProt: MG575644) were confirmed by the analysis of the genes obtained in the E. focardii genome here described (Table 5): the SOD classification was done on the base of similarity with cytoplasmic SODs (type 1) and with mitochondrial SODs (type 2) and was confirmed by the presence of Cu/Zn and Fe/Mn pattern signature, respectively. In the same genome analysis, we could predict four additional E. focardii isoforms, the SOD1d and SOD1e and two SOD3 isoforms (Fig. 4A). According to this tree, the SOD3 isoforms appear to derive from gene duplication events probably happened before the divergence of E. focardii from other Euplotes species and some isotypes were then lost in E. crassus. As general result, the E. focardii SOD encoding gene family appeared composed by a higher number of genes with respect to the mesophilic E. crassus, probably due to a repeated event of gene duplication.

Phylogenetic trees of SODs (A) and CATs (B). Only bootstrap percentage values higher than 50%, indicated near nodes, are shown. Copper/zinc SODs are highlighted in green, iron/manganese SODs in blue, and nickel SODs in red.
A similar situation is evident for the catalase (CAT) genes (Fig. 4B). CAT (EC 1.11.1.6), that inhibits the DNA damage by decomposing the H2O2 into oxygen and water induced by nitrofurazone, was previously considered a good biomarker for detecting oxidative stress and, consequently, ecotoxicity in aquatic ecosystems75. The number of E. focardii CATs genes is higher compared to the mesophilic E. crassus, probably due to a repeated event of gene duplication. As shown in Fig. 4B, CAT branches are scattered throughout the phylogeny of ciliate homologs. The low bootstrap values at these branches suggest that in E. focardii the CAT gene family underwent several events of recent gene duplications with potential adaptive outcomes that imply high divergence and consequently less supported phylogeny.
In general, the antioxidant enzymes system appears amplified in E. focardii (Table 6) suggesting that gene amplification may have contributed to combating the effects of increased oxygen concentration in the Antarctic seawaters. On the other hand, E. focardii possesses few gene encoding Thioredoxin NADPH Reductase (TrxR) and Glutathione Reductase (GR) isoforms. With the exception of E. octocarinatus, the genes for these enzymes are present in small number in the ciliate genomes known so far (see Table 6): Tetrahymena, as an exception in ciliates, has no TRXRs or GRs genes but 6 isotypes of thioredoxin–glutathione reductase (TGR) genes, that are composed by a fusion of the sequences of TRXR and glutaredoxin domains and are capable of transporting electrons from NADPH to both Trx and GSH systems76. E. octocarinatus has only 3 isotypes of TGR. In conclusion, the E. focardii antioxidant system appears to be based mainly on numerous SODs and CATs enzymes.
The E. focardii heat-shock protein 70 gene family
The E. focardii macronuclear genome possesses seven distinct nanochromosomes that encode Hsp70 isoforms. According to their predicted C-terminal domain sequences77, we identified the respective Hsp70 subfamilies defined by the putative subcellular localization (Fig. 5 and Table SII). Figure 5 shows the phylogenetic relationships of the Euplotes focardii Hsp70 isoforms to orthologs from the mesophilic Euplotes species E. crassus and E. octocarinatus and from O. trifallax, T. thermophila, and S. lemnae. The topology of the tree supported sister relationships of all E. focardii Hsp70s with the orthologs from E. crassus, that represents the closest mesophilic Euplotes. Furthermore, our tree showed that all the E. focardii isoforms are of equal or even lower number than the corresponding orthologous from the other ciliates, indicating that in E. focardii genome there is no amplification of the hsp70 encoding genes.

Phylogenetic tree of heat-shock protein Hsp70 super families. Only bootstrap percentage values higher than 50%, indicated near nodes, are shown.
La Terza et al.26 demonstrated that the canonical cytoplasmic E. focardii hsp70 gene (GenBank acc. no. AAP51165.1, Protein_06381 in this paper) is not inducible by acute thermal stress, in contrast to the orthologous gene in E. nobili also found in Antarctica. Unlike in the Antarctic fish Trematomus bernacchii25, the hsp70 gene does respond to several oxidative stressors, such as hydrogen peroxide in E. focardii78. In order to understand whether the Euplotes Hsp70 isoforms newly described here respond to thermal and/or oxidative stressors, we tested the inducibility of these genes in cultures subjected to heat shock (18 °C) or oxidative stress (H2O2). Figure 6 shows that thermal stress did not induce transcription of the any of the seven Hsp70 genes relative to the control temperature (4 °C). In contrast, six of the seven genes (excluding isoform named Protein_05367) were induced by oxidative stress, strikingly so in the case of Protein_07400 isoform. We suggest that E. focardii, like several other Antarctic organisms, maintained a constitutive synthesis of Hsp70 isoforms to preserve protein function in the cold environment and evolved an oxidative stress response involving inducible Hsp70 synthesis. Moreover, this Antarctic ciliate has clearly lost the capability to induce the classical heat shock response when confronted with elevated temperatures.

Hsp70 transcript abundance in stressed E. focardii cells normalized to the abundance in control cells. mRNA levels were determined by qPCR of the Hsp70 isoforms (reported as protein assembly code) using the comparative threshold method. Mean ± SD (n = 4). Total RNA was purified from cells grown at their standard temperature of 4° C (i.e., not shocked), from cells which were exposed to 18 °C (orange) via a heat ramp (see "Materials and methods" section), or from cells exposed to 100 µM of hydrogen peroxide (blue); control and experimental treatments were for 30 min. Data are reported as the mean of three experiments.
Conclusions
The Antarctic ciliate E. focardii represents an optimal model for studying genome adaptation to cold environments. In this paper, we reported the analysis of the E. focardii MAC genome after a significant improvement of the assembly that were also cleaned-up of algal or bacterial contaminants. In particular, we obtained 17,798 sequences containing telomeres at both ends vs 12,922 previously reported28. Even though some assembly parameters remain lower in comparison to the other ciliates reported, complete nanochromosomes are now closer, as a percentage of the total assembly, to those from other Euplotes species38,79 . This new report on the E. focardii MAC genome will provide additional information to investigate translation mechanisms in organisms with alternative genetic codes associated with the evolution of novel tRNA variants, including a putative suppressor tRNA, and to investigate how cold adaptation may have evolved.
The analysis of this improved E. focardii genome assembly allowed a better characterization of gene families, in particular that of the tubulins, that were previously only partially identified by single gene cloning approaches1,3. We identified a new β-tubulin isotype (TBβ5) and six additional divergent α-tubulin isotypes. In combination with the β-tubulin diversity, the role of the high number of different α-tubulins in microtubules cold adaptation should be reconsidered. Furthermore, we found that SODs and CATs families are composed by a higher number of genes with respect to the mesophilic Euplotes. The opposite trend was observed for the hsp70 genes: in this case isoform’s diversification appears reduced with respect to homologs from other Euplotes species. Furthermore, expression of these genes was not induced by heat stress (18 °C for 30 min vs. a physiological temperature of 4 °C). On the other hand, hsp70 expression was raised during oxidative stress. We can conclude from these results that as for other Antarctic organisms, it is more important for E. focardii to cope with cold denaturation of proteins and oxidative stress than to respond to thermal stress. Consequently, the Hsp70 gene family did not expand like SODs and CATs families, that are involved in the antioxidant responses.
All these results suggest potential roles for paralogy in environmental adaptation, warranting future experimental investigation. Genomic expansions of specific protein gene families contributing to physiological fitness in freezing polar conditions have previously been reported for Antarctic notothenioids80. Gene diversification has been proved to produce a differential gene expression in specific adaptive conditions, as reported for the cold acclimation of the tea plant Camellia sinensis81 and also for the E. focardii βT3-tubulin during cilia regeneration14 and the E. focardii SOD 1b during cold stress73. In future, RNA-seq analyses of E. focardii transcriptome in different environmental conditions coupled to detailed molecular analyses will provide deeper insights into the role of duplicated genes.
In conclusion, we propose that the molecular basis of cold adaptation that enabled E. focardii to thrive in the Antarctic Ocean may not be solely due to particular amino acid substitutions that enable these molecules to function at low temperatures but may have also arisen via gene duplications that increased protein functional diversity.
Materials and methods
SPAdes assembly
The Illumina HiSeq 2000 PE (paired-end, 100 bp, with BioProject ID SRX1959352) reads obtained after sequencing of Euplotes focardii macronuclear genome, previously trimmed using the Trimmomatic software (version 0.36)82 and checked using the FastQC software (version 0.11.5)83, were assembled using the SPAdes algorithm (version 3.10.1)84 with the “careful” option and the BayesHammer error correction algorithm85. Other parameters were set to default values. Additionally, SPAdes version 3.9, version 3.11.1 and a different set of k-mer lengths were also used to check and identify the best version and configuration of the assembler for these reads.
Possible redundant “chaff” contigs were removed from the assembly, as previously reported32, by mapping contigs shorter than 500 bp that had matches to the other contigs with greater than 80% coverage and 90% sequence identity.
Assembly clean-up and properties
To perform a quality assessment of the obtained assembly from SPAdes, avoiding bacterial contamination, the assembly was further analyzed checking the GC content by the QUAST software (version 4.5)86. The contigs having GC content higher than 45%, and a coverage lower than 10, were removed from the assembly using a custom algorithm written in Perl. This GC content percentage threshold was also set on the base of the minimum GC content of the most abundant bacteria in the consortium associated to E. focardii (data unpublished). Further bacterial contaminations were analyzed by using BLAST of the assembly versus the Genbank nr database setting bacteria as taxonomy filter, with 80% of hit coverage and 95% sequence identity of the matches. To check the goodness of the assembly, the SPAdes procedure was further repeated after this decontamination step. Currently, Genbank nr database also includes data of the last reported Euplotes endosymbiotic bacteria87,88.
Moreover, to remove possible contaminant algal sequences, the Dunaliella salina genome (used to feed E. focardii) was compared to the E. focardii macronuclear genome assembly, using blastn and cd-hit-2d software with a threshold of 0.95 (see "Paralog prediction" section for details).
After all the steps of assembling and cleaning, the assembly was evaluated using another custom Perl script providing information about the size, the number of contigs, the number of telomeres, the number of 2, 1, and 0 telomere contigs and the mean length of contigs.
Gene prediction and frameshifting analysis
The gene prediction procedure on the E. focardii macronuclear genome assembly was performed using the AUGUSTUS software (version 3.3.3)89 previously trained and tested on a manually curated data set, with no cases of translation frameshifting, from Euplotes crassus (data unpublished). The software was run using the following parameters: “–species = euplotes_crassus –UTR = on –alternatives-from-evidence = true –genemodel = complete –codingseq = on”. The results of this prediction were assessed/processed with another Perl script.
Genes predicted were additionally analyzed checking 12 potential frameshifting sites (5’-AAATAA-3’, 5’-AATTAA-3’, 5’-ATTTAA-3’, 5’-TTATAA-3’, 5’-AACTAA-3’, 5’-ATATAA-3’, 5’-GAGTAA-3’, 5’-AAATAG-3’, 5’-GTATAA-3’, 5’-TTATAG-3’, 5’-ATATAG-3’ and 5’-TCCTAA-3’, ranked by abundance), previously detected in E. crassus28, located at the end of these sequences and comparing them with the E. focardii proteome obtained after the Protein Annotation step. Sequences having these potential sites (7023) were checked and selected on the base of sequence length (lower than the related blast top hit length) and BLAST coverage (higher than 80%) versus the best hit obtained from nr reference database. Moreover, the transcript-level quantification of these genes, in comparison with all the others, was estimated using pair-end Illumina transcriptome reads of E. focardii and RSEM software package90, which allocates the multi-mapping reads on the base of an expectation maximization approach. A two-sample independent t-test was used to perform this comparison (p-value < 0.05 were considered to be significant).
Protein annotation
The proteins predicted from the E. focardii macronuclear genome assembly were searched and annotated using the OmicsBox software (version 1.4.11)91,92. The parameter settings used for the procedure were: blastp as blast program, nr as blastdb, 1.0e−3 as E-value and 20 as number of BLAST hits. Other settings for the annotation were set to the default values.
Assessment of genome completeness
The assessment of genome completeness was firstly conducted analyzing the percentage of conserved core eukaryotic genes (CEGs)93 searching the number of protein sequences contained in the CEG database (composed by 248 proteins) that were likely homologs with those of the E. focardii macronuclear genome assembly (i.e., with blastp E-values lower than 1e-10 and a match coverage higher than 70% of the length of the CEG proteins).
The tRNAs were initially predicted with the Aragorn algorithm (version 1.2.41)94. Ribosomal proteins were counted after the protein annotation, as previously described, and structural RNAs were identified by BLAST searches of the assembly against the Rfam database. Secondary and tertiary structures of a potential stop-suppressor tRNA were determined using the RNAfold web server95 and the RNAComposer automated RNA structure modeling server96, respectively.
Paralog prediction
The first step of this analysis was to cluster the E. focardii macronuclear proteome using the cd-hit software (version 4.7)97,98 with a sequence identity threshold of 0.95 to merge alleles (26,680 clusters). Therefore, the clustering was performed using a threshold of 0.4 to identify the largest and most represented protein families in the proteome (21,850 clusters of which 2312 with at least two elements). In this work, we have focused our interest on the Hsp70, tubulins and antioxidant enzymes family. Moreover, CD-HIT (and CD-HIT-EST) software was also used with a threshold of 0.95 to identify possible alternative fragmentation in the whole genome.
Pairwise sequence identity searches were performed on the E. focardii MAC genome assembly, in comparison with the E. crassus, E. octocarinatus and E. vannus assemblies, to estimate the distribution of alleles and paralogous sequences. By a custom Perl script, an alignment of all contigs against each other was performed into the assembly invoking the blastn algorithm and extracting the best non-self BLAST hits; then, MAFFT algorithm (–clustalout –maxiterate 1000 –globalpair / –localpair) was invoked to align the two sequences of each pair obtained; finally, its sequence identity was calculated.
tRNA sequencing, mapping and quantification
Total RNA was extracted using TRI Reagent (Sigma) according to the manufacturer’s protocol, deacylated at 37 °C for 40 min in 20 mM Tris–HCl (pH 9.0) and precipitated with 5 M ammonium acetate in 75% ethanol. This RNA was used to produce a library of mature tRNAs by the YAMAT-seq method99. The resulting cDNA library was multiplexed and sequenced on an Illumina MiSeq sequencer in paired-end mode (75 bp reads).
Paired-end YAMAT-seq reads were merged with BBMerge (default parameters), from the BBTools package100. Forward and reverse adapters were trimmed off the merged reads using cutadapt 3.2 (default parameters; -m 20)101. This procedure yielded 623,000 reads, which were mapped to E. focardii tRNAs predicted by tRNAscan-SE 2.0 as a part of the tRAX pipeline102 (default parameters) which was also used to obtain read counts (available as Supplementary Table 1).
Hsp70, tubulins and antioxidant enzymes system classification
The classes of Hsp70, tubulins and antioxidant enzymes in E. focardii previously detected and analyzed1,69 were confirmed and extended after the protein annotation and ""Paralog prediction stages. Clustal Omega algorithm103 was used to produce each multi-alignment, and MEGA version X104 was used to infer each phylogeny using the Maximum Likelihood statistical method, the JTT substitution model with a single substitution rate category (for SODs, it was used with four substitution rate categories), and generate 100 bootstrap pseudo-replicates. MEGA version X104 was also used to plot each phylogenetic tree.
Homology modeling and molecular dynamics of alpha tubulins
Three dimensional structures of α-tubulins of Euplotes species, not already available by X-ray crystallographic or NMR analysis, were obtained by homology modeling using the Modeller software (version 9.20)105 and the Swiss-Model server106. Three dimensional structures (reported as pdb ID) detected as best template to model the α-tubulins of Euplotes, on the base of the Global Model Quality Estimation (GMQE) score, the coverage and the sequence identity in the Swiss-Model server, were: 6U0H10, used for TBα1 (with 96.20% of identity), TBα2 (94.63%), and TBα3 (83.87%) of E. focardii, for TBα1 (93.93%), TBα2 (92.19%), and TBα4 (90.02%) of E. crassus, and for TBα1 (95.74%) of E. octocarinatus; 6E88108, used for TBα4 (52.53%), and TBα7 (49.31%) of E. focardii; 1IA0109, used for TBα5 (56.06%) of E. focardii; 5W3F110, used for TBα6 (52.58%) of E. focardii; 6U42111, used for TBα3 (52.58%) of E. crassus; 6U0T107, used for TBα5 (52.58%) of E. crassus. Final homology models were assessed using the zDOPE score and the estimated RMSD/Overlap in Modeller and the Q-MEAN score in the Swiss-Model server.
Molecular dynamics (MD) was performed using GROMACS (version 2019.3)112 and the all-atom OPLS force field. After the preliminary topology generation, macromolecules were soaked in a SPC water molecules cubic box in the presence of Na+/Cl− ions. The entire system was minimized, until the variation of potential energy was smaller than 100 kJ mol−1 nm−1, using the steepest descent algorithm and equilibrated under a constant Number of particles, Volume, and Temperature (NVT) and a constant Number of particles, Pressure and Temperature (NPT) ensemble. Then, the MD simulation was run for 50 ns. Temperatures in the equilibration and MD simulation steps were set either at 4 °C (277 K) or at 27 °C (300 K). The backbone root mean square deviation (RMSD) and the protein root mean square fluctuation (RMSF) were determined using the GROMACS tools “gmx rms” and “gmx rmsf”, respectively, while the maximum RMSD value (RMSDmax) for each dynamic was calculated fitting the trajectory with the equation RMSD = t * RMSDmax / (tn + const) (Fig. 3A).
Hsp70 gene transcription by E. focardii under stress
We evaluated the inducibility of E. focardii Hsp70 genes in cultures subjected to thermal or oxidative stress. Cells entering stationary phase after ~ 1 week of vegetative proliferation in the presence of food (the green alga Dunaliella tertiolecta) were pelleted by low-speed centrifugation (500×g, 3 min), and pellets were resuspended in seawater to a density of ~ 5 × 103 cells/mL. Heat-shock was performed by warming cells from 4 to 18 °C over 30 min. Control cells were incubated at 4 °C for 30 min. Oxidative stress was produced by incubating cells at 4 °C in the presence of 100 µM H2O2 for 30 min. Total RNA from control or stressed cells was extracted with Trizol reagent (GIBCO BRL), and cDNA was synthesized from each template using the StrataScript Reverse Transcriptase (Stratagene).
Transcript levels corresponding to the seven E. focardii Hsp70 genes were measured in control and stressed DNA samples by comparative-threshold qPCR using the SYBR green DNA-binding method113 and the primer pairs given in Supplemental Tables 2 and 3; the Euplotes SSrDNA gene (GenBank ID: EF094961) was used for normalization. To 100 ng of E. focardii cDNA were added 12.5 μl of 2 × SYBR Green JumpStart Taq ReadyMix (Sigma-Aldrich, Milan), 5 pg each of gene-specific forward and reverse primers, and water to a final volume of 25 μl. Amplification reactions were performed in triplicate in a Multicolor qPCR MX3000P thermocycler (Stratagene, Milan, Italy), with an initial denaturation step (95 °C for 2 min) to activate the polymerase followed by 45 cycles of denaturation at 95 °C for 30 s, and annealing and extension at 60 °C for 15 s. During annealing/extension, the increase in fluorescence at 495 nm was monitored, and the threshold value was set at 30 units. To verify that the primer pairs gave specific PCR products without non-specific amplification, the DNA samples were subjected to melting curve analysis by ramping the thermocycler temperature from 50 to 95 °C at 0.05 °C/sec.
The relative expression of the Hsp70 genes was calculated by the method of Pfaffl114:
where Ct is the PCR cycle number at which the fluorescent signal is above the set threshold, ∆Ct is the Ct difference (control minus sample) of the target or reference gene, and E is the real-time PCR efficiency of the target or reference gene (E = 10–1/slope, calculated from plots of Ct vs. cDNA input). The relative expression ratios of transcripts under investigation were tested for statistical significance by a pairwise, fixed reallocation randomization test implemented in REST MCS version 2 software115.
References
Pucciarelli, S. et al. Molecular cold-adaptation of protein function and gene regulation: the case for comparative genomic analyses in marine ciliated protozoa. Mar Genomics 2, 57–66. https://doi.org/10.1016/j.margen.2009.03.008 (2009).
Pucciarelli, S., Marziale, F., Di Giuseppe, G., Barchetta, S. & Miceli, C. Ribosomal cold-adaptation: characterization of the genes encoding the acidic ribosomal P0 and P2 proteins from the Antarctic ciliate Euplotes focardii. Gene 360, 103–110. https://doi.org/10.1016/j.gene.2005.06.007 (2005).
Pucciarelli, S. & Miceli, C. Characterization of the cold-adapted alpha-tubulin from the psychrophilic ciliate Euplotes focardii. Extremophiles 6, 385–389. https://doi.org/10.1007/s00792-002-0268-5 (2002).
Yang, G. et al. Characterization and comparative analysis of psychrophilic and mesophilic alpha-amylases from Euplotes species: a contribution to the understanding of enzyme thermal adaptation. Biochem Biophys Res Commun 438, 715–720. https://doi.org/10.1016/j.bbrc.2013.07.113 (2013).
Prescott, D. M. The DNA of ciliated protozoa. Microbiol Rev 58, 233–267 (1994).
Mollenbeck, M. & Klobutcher, L. A. De novo telomere addition to spacer sequences prior to their developmental degradation in Euplotes crassus. Nucleic Acids Res 30, 523–531 (2002).
Swart, E. C. et al. The Oxytricha trifallax macronuclear genome: a complex eukaryotic genome with 16,000 tiny chromosomes. PLoS Biol 11, e1001473. https://doi.org/10.1371/journal.pbio.1001473 (2013).
Heyse, G., Jonsson, F., Chang, W. J. & Lipps, H. J. RNA-dependent control of gene amplification. Proc Natl Acad Sci U S A 107, 22134–22139. https://doi.org/10.1073/pnas.1009284107 (2010).
Nowacki, M., Haye, J. E., Fang, W., Vijayan, V. & Landweber, L. F. RNA-mediated epigenetic regulation of DNA copy number. Proc Natl Acad Sci U S A 107, 22140–22144. https://doi.org/10.1073/pnas.1012236107 (2010).
Dayeh, V. R. et al. Comparing a ciliate and a fish cell line for their sensitivity to several classes of toxicants by the novel application of multiwell filter plates to Tetrahymena. Res Microbiol 156, 93–103. https://doi.org/10.1016/j.resmic.2004.08.005 (2005).
Detrich, H. W., 3rd, Parker, S. K., Williams, R. C., Jr., Nogales, E. & Downing, K. H. Cold adaptation of microtubule assembly and dynamics. Structural interpretation of primary sequence changes present in the alpha- and beta-tubulins of Antarctic fishes. J Biol Chem 275, 37038–37047. https://doi.org/10.1074/jbc.M005699200 (2000).
Manka, S. W. & Moores, C. A. Microtubule structure by cryo-EM: snapshots of dynamic instability. Essays Biochem 62, 737–751. https://doi.org/10.1042/EBC20180031 (2018).
Inclan, Y. F. & Nogales, E. Structural models for the self-assembly and microtubule interactions of gamma-, delta- and epsilon-tubulin. J Cell Sci 114, 413–422 (2001).
Chiappori, F. et al. Structural thermal adaptation of beta-tubulins from the Antarctic psychrophilic protozoan Euplotes focardii. Proteins 80, 1154–1166. https://doi.org/10.1002/prot.24016 (2012).
Marziale, F. et al. Different roles of two gamma-tubulin isotypes in the cytoskeleton of the Antarctic ciliate Euplotes focardii: remodelling of interaction surfaces may enhance microtubule nucleation at low temperature. FEBS J 275, 5367–5382. https://doi.org/10.1111/j.1742-4658.2008.06666.x (2008).
Pucciarelli, S., Miceli, C. & Melki, R. Heterologous expression and folding analysis of a beta-tubulin isotype from the Antarctic ciliate Euplotes focardii. Eur J Biochem 269, 6271–6277 (2002).
Gromer, S., Urig, S. & Becker, K. The thioredoxin system–from science to clinic. Med Res Rev 24, 40–89. https://doi.org/10.1002/med.10051 (2004).
Birben, E., Sahiner, U. M., Sackesen, C., Erzurum, S. & Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J 5, 9–19. https://doi.org/10.1097/WOX.0b013e3182439613 (2012).
Alin, P., Danielson, U. H. & Mannervik, B. 4-Hydroxyalk-2-enals are substrates for glutathione transferase. FEBS Lett 179, 267–270 (1985).
Juganson, K. et al. Mechanisms of toxic action of silver nanoparticles in the protozoan Tetrahymena thermophila: From gene expression to phenotypic events. Environ Pollut 225, 481–489. https://doi.org/10.1016/j.envpol.2017.03.013 (2017).
Clark, M. S., Fraser, K. P. & Peck, L. S. Antarctic marine molluscs do have an HSP70 heat shock response. Cell Stress Chaperones 13, 39–49. https://doi.org/10.1007/s12192-008-0014-8 (2008).
Tomanek, L. The heat-shock response: its variation, regulation and ecological importance in intertidal gastropods (genus Tegula). Integr Comp Biol 42, 797–807. https://doi.org/10.1093/icb/42.4.797 (2002).
Morimoto, R. I., Kline, M. P., Bimston, D. N. & Cotto, J. J. The heat-shock response: regulation and function of heat-shock proteins and molecular chaperones. Essays Biochem 32, 17–29 (1997).
Gonzalez-Aravena, M. et al. HSP70 from the Antarctic sea urchin Sterechinus neumayeri: molecular characterization and expression in response to heat stress. Biol Res 51, 8. https://doi.org/10.1186/s40659-018-0156-9 (2018).
Hofmann, G. E., Buckley, B. A., Airaksinen, S., Keen, J. E. & Somero, G. N. Heat-shock protein expression is absent in the antarctic fish Trematomus bernacchii (family Nototheniidae). J Exp Biol 203, 2331–2339 (2000).
La Terza, A., Papa, G., Miceli, C. & Luporini, P. Divergence between two Antarctic species of the ciliate Euplotes, E. focardii and E. nobilii, in the expression of heat-shock protein 70 genes. Mol Ecol 10, 1061–1067. https://doi.org/10.1046/j.1365-294x.2001.01242.x (2001).
Klobutcher, L. A. & Farabaugh, P. J. Shifty ciliates: frequent programmed translational frameshifting in euplotids. Cell 111, 763–766 (2002).
Lobanov, A. V. et al. Position-dependent termination and widespread obligatory frameshifting in Euplotes translation. Nat Struct Mol Biol 24, 61–68. https://doi.org/10.1038/nsmb.3330 (2017).
Coordinators, N. R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 45, D12–D17. https://doi.org/10.1093/nar/gkw1071 (2017).
Pucciarelli, S. et al. Microbial consortium associated with the antarctic marine ciliate Euplotes focardii: an investigation from genomic sequences. Microb Ecol 70, 484–497. https://doi.org/10.1007/s00248-015-0568-9 (2015).
Klobutcher, L. A. et al. Conserved DNA sequences adjacent to chromosome fragmentation and telomere addition sites in Euplotes crassus. Nucleic Acids Res 26, 4230–4240. https://doi.org/10.1093/nar/26.18.4230 (1998).
Aeschlimann, S. H. et al. The draft assembly of the radically organized Stylonychia lemnae macronuclear genome. Genome Biol Evol 6, 1707–1723. https://doi.org/10.1093/gbe/evu139 (2014).
Swart, E. C. (personal communication).
Cavalcanti, A. R., Stover, N. A., Orecchia, L., Doak, T. G. & Landweber, L. F. Coding properties of Oxytricha trifallax (Sterkiella histriomuscorum) macronuclear chromosomes: analysis of a pilot genome project. Chromosoma 113, 69–76. https://doi.org/10.1007/s00412-004-0295-3 (2004).
Lozupone, C. A., Knight, R. D. & Landweber, L. F. The molecular basis of nuclear genetic code change in ciliates. Curr Biol 11, 65–74. https://doi.org/10.1016/s0960-9822(01)00028-8 (2001).
Salas-Marco, J. et al. Distinct paths to stop codon reassignment by the variant-code organisms Tetrahymena and Euplotes. Mol Cell Biol 26, 438–447. https://doi.org/10.1128/MCB.26.2.438-447.2006 (2006).
Klobutcher, L. A. Sequencing of random Euplotes crassus macronuclear genes supports a high frequency of +1 translational frameshifting. Eukaryot Cell 4, 2098–2105. https://doi.org/10.1128/EC.4.12.2098-2105.2005 (2005).
Wang, R., Xiong, J., Wang, W., Miao, W. & Liang, A. High frequency of +1 programmed ribosomal frameshifting in Euplotes octocarinatus. Sci Rep 6, 21139. https://doi.org/10.1038/srep21139 (2016).
Turanov, A. A. et al. Genetic code supports targeted insertion of two amino acids by one codon. Science 323, 259–261. https://doi.org/10.1126/science.1164748 (2009).
Maehigashi, T., Dunkle, J. A., Miles, S. J. & Dunham, C. M. Structural insights into +1 frameshifting promoted by expanded or modification-deficient anticodon stem loops. Proc Natl Acad Sci U S A 111, 12740–12745. https://doi.org/10.1073/pnas.1409436111 (2014).
Miceli, C., Ballarini, P., Di Giuseppe, G., Valbonesi, A. & Luporini, P. Identification of the tubulin gene family and sequence determination of one beta-tubulin gene in a cold-poikilotherm protozoan, the antarctic ciliate Euplotes focardii. J Eukaryot Microbiol 41, 420–427. https://doi.org/10.1111/j.1550-7408.1994.tb06100.x (1994).
Ricci, F. et al. The sub-chromosomic macronuclear pheromone genes of the ciliate Euplotes raikovi: comparative structural analysis and insights into the mechanism of expression. J Eukaryot Microbiol 66, 376–384. https://doi.org/10.1111/jeu.12677 (2019).
Wang, R., Liu, J., Di Giuseppe, G. & Liang, A. UAA and UAG may Encode Amino Acid in Cathepsin B Gene of Euplotes octocarinatus. J Eukaryot Microbiol 67, 144–149. https://doi.org/10.1111/jeu.12755 (2020).
Heaphy, S. M., Mariotti, M., Gladyshev, V. N., Atkins, J. F. & Baranov, P. V. Novel ciliate genetic code variants including the reassignment of all three stop codons to sense codons in condylostoma magnum. Mol Biol Evol 33, 2885–2889. https://doi.org/10.1093/molbev/msw166 (2016).
Swart, E. C., Serra, V., Petroni, G. & Nowacki, M. Genetic codes with no dedicated stop codon: context-dependent translation termination. Cell 166, 691–702. https://doi.org/10.1016/j.cell.2016.06.020 (2016).
Roy, B., Leszyk, J. D., Mangus, D. A. & Jacobson, A. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc Natl Acad Sci U S A 112, 3038–3043. https://doi.org/10.1073/pnas.1424127112 (2015).
Dunn, J. G., Foo, C. K., Belletier, N. G., Gavis, E. R. & Weissman, J. S. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. Elife 2, e01179. https://doi.org/10.7554/eLife.01179 (2013).
Frechin, M., Duchene, A. M. & Becker, H. D. Translating organellar glutamine codons: a case by case scenario?. RNA Biol 6, 31–34. https://doi.org/10.4161/rna.6.1.7564 (2009).
Wilcox, M. & Nirenberg, M. Transfer RNA as a cofactor coupling amino acid synthesis with that of protein. Proc Natl Acad Sci U S A 61, 229–236. https://doi.org/10.1073/pnas.61.1.229 (1968).
Detrich, H. W. 3rd., Fitzgerald, T. J., Dinsmore, J. H. & Marchese-Ragona, S. P. Brain and egg tubulins from antarctic fishes are functionally and structurally distinct. J Biol Chem 267, 18766–18775 (1992).
Detrich, H. W. 3rd., Johnson, K. A. & Marchese-Ragona, S. P. Polymerization of Antarctic fish tubulins at low temperatures: energetic aspects. Biochemistry 28, 10085–10093 (1989).
Wloga, D. et al. Glutamylation on alpha-tubulin is not essential but affects the assembly and functions of a subset of microtubules in Tetrahymena thermophila. Eukaryot Cell 7, 1362–1372. https://doi.org/10.1128/EC.00084-08 (2008).
Eisen, J. A. et al. Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote. PLoS Biol 4, e286. https://doi.org/10.1371/journal.pbio.0040286 (2006).
Aury, J. M. et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444, 171–178. https://doi.org/10.1038/nature05230 (2006).
Pucciarelli, S. et al. Distinct functional roles of beta-tubulin isotypes in microtubule arrays of Tetrahymena thermophila, a model single-celled organism. PLoS ONE 7, e39694. https://doi.org/10.1371/journal.pone.0039694 (2012).
Pucciarelli, S. et al. Tubulin folding: the special case of a beta-tubulin isotype from the Antarctic psychrophilic ciliate Euplotes focardii. Polar Biol 36, 1833–1838. https://doi.org/10.1007/s00300-013-1390-9 (2013).
Pucci, F. & Rooman, M. Physical and molecular bases of protein thermal stability and cold adaptation. Curr Opin Struct Biol 42, 117–128. https://doi.org/10.1016/j.sbi.2016.12.007 (2017).
Aqvist, J., Isaksen, G. V. & Brandsdal, B. O. Computation of enzyme cold adaptation. Nat Rev Chem 1, 0051. https://doi.org/10.1038/s41570-017-0051 (2017).
Lesser, M. P. Oxidative stress in marine environments: biochemistry and physiological ecology. Annu Rev Physiol 68, 253–278. https://doi.org/10.1146/annurev.physiol.68.040104.110001 (2006).
McCord, J. M. & Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 244, 6049–6055 (1969).
McCord, J. M. & Fridovich, I. Superoxide dismutase: the first twenty years (1968–1988). Free Radic Biol Med 5, 363–369 (1988).
Miller, A. F. Superoxide dismutases: ancient enzymes and new insights. FEBS Lett 586, 585–595. https://doi.org/10.1016/j.febslet.2011.10.048 (2012).
Benov, L. T. & Fridovich, I. Escherichia coli expresses a copper- and zinc-containing superoxide dismutase. J Biol Chem 269, 25310–25314 (1994).
Steinman, H. M. & Ely, B. Copper-zinc superoxide dismutase of Caulobacter crescentus: cloning, sequencing, and mapping of the gene and periplasmic location of the enzyme. J Bacteriol 172, 2901–2910. https://doi.org/10.1128/jb.172.6.2901-2910.1990 (1990).
Antonyuk, S. V., Strange, R. W., Marklund, S. L. & Hasnain, S. S. The structure of human extracellular copper-zinc superoxide dismutase at 1.7 A resolution: insights into heparin and collagen binding. J Mol Biol 388, 310–326. https://doi.org/10.1016/j.jmb.2009.03.026 (2009).
Marklund, S. L. Extracellular superoxide dismutase and other superoxide dismutase isoenzymes in tissues from nine mammalian species. Biochem J 222, 649–655. https://doi.org/10.1042/bj2220649 (1984).
Bannister, J. V., Bannister, W. H. & Rotilio, G. Aspects of the structure, function, and applications of superoxide dismutase. CRC Crit Rev Biochem 22, 111–180 (1987).
James, E. R. Superoxide dismutase. Parasitol Today 10, 481–484. https://doi.org/10.1016/0169-4758(94)90161-9 (1994).
Ferro, D. et al. Cu, Zn superoxide dismutases from Tetrahymena thermophila: molecular evolution and gene expression of the first line of antioxidant defenses. Protist 166, 131–145. https://doi.org/10.1016/j.protis.2014.12.003 (2015).
Arnaiz, O. & Sperling, L. ParameciumDB in 2011: new tools and new data for functional and comparative genomics of the model ciliate Paramecium tetraurelia. Nucleic Acids Res 39, D632-636. https://doi.org/10.1093/nar/gkq918 (2011).
Fink, R. C. & Scandalios, J. G. Molecular evolution and structure–function relationships of the superoxide dismutase gene families in angiosperms and their relationship to other eukaryotic and prokaryotic superoxide dismutases. Arch Biochem Biophys 399, 19–36. https://doi.org/10.1006/abbi.2001.2739 (2002).
Lee, Y. M., Friedman, D. J. & Ayala, F. J. Superoxide dismutase: an evolutionary puzzle. Proc Natl Acad Sci U S A 82, 824–828. https://doi.org/10.1073/pnas.82.3.824 (1985).
Pischedda, A. et al. Antarctic marine ciliates under stress: superoxide dismutases from the psychrophilic Euplotes focardii are cold-active yet heat tolerant enzymes. Sci Rep 8, 14721. https://doi.org/10.1038/s41598-018-33127-1 (2018).
Yang, G. et al. Characterization of the first eukaryotic cold-adapted patatin-like phospholipase from the psychrophilic Euplotes focardii: Identification of putative determinants of thermal-adaptation by comparison with the homologous protein from the mesophilic Euplotes crassus. Biochimie 95, 1795–1806. https://doi.org/10.1016/j.biochi.2013.06.008 (2013).
Li, J., Zhou, L., Lin, X., Yi, Z. & Al-Rasheid, K. A. Characterizing dose-responses of catalase to nitrofurazone exposure in model ciliated protozoan Euplotes vannus for ecotoxicity assessment: enzyme activity and mRNA expression. Ecotoxicol Environ Saf 100, 294–302. https://doi.org/10.1016/j.ecoenv.2013.08.021 (2014).
Prast-Nielsen, S., Huang, H. H. & Williams, D. L. Thioredoxin glutathione reductase: its role in redox biology and potential as a target for drugs against neglected diseases. Biochim Biophys Acta 1262–1271, 2011. https://doi.org/10.1016/j.bbagen.2011.06.024 (1810).
Kabani, M. & Martineau, C. N. Multiple hsp70 isoforms in the eukaryotic cytosol: mere redundancy or functional specificity?. Curr Genomics 9, 338–248. https://doi.org/10.2174/138920208785133280 (2008).
La Terza, A., Miceli, C. & Luporini, P. The gene for the heat-shock protein 70 of Euplotes focardii, an Antarctic psychrophilic ciliate. Antarct. Sci. 16, 23–28. https://doi.org/10.1017/S0954102004001774 (2004).
Chen, X. et al. Genome analyses of the new model protist Euplotes vannus focusing on genome rearrangement and resistance to environmental stressors. Mol Ecol Resour 19, 1292–1308. https://doi.org/10.1111/1755-0998.13023 (2019).
Chen, Z. et al. Transcriptomic and genomic evolution under constant cold in Antarctic notothenioid fish. Proc Natl Acad Sci U S A 105, 12944–12949. https://doi.org/10.1073/pnas.0802432105 (2008).
Li, Y. et al. Comparative transcriptomic analysis reveals gene expression associated with cold adaptation in the tea plant Camellia sinensis. BMC Genomics 20, 624. https://doi.org/10.1186/s12864-019-5988-3 (2019).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Andrews, S. (2010).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Nikolenko, S. I., Korobeynikov, A. I. & Alekseyev, M. A. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genomics 14 Suppl 1, S7. https://doi.org/10.1186/1471-2164-14-S1-S7 (2013).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. https://doi.org/10.1093/bioinformatics/btt086 (2013).
Boscaro, V., Husnik, F., Vannini, C. & Keeling, P. J. Symbionts of the ciliate Euplotes: diversity, patterns and potential as models for bacteria-eukaryote endosymbioses. Proc Biol Sci 286, 20190693. https://doi.org/10.1098/rspb.2019.0693 (2019).
Serra, V. et al. Morphology, ultrastructure, genomics, and phylogeny of Euplotes vanleeuwenhoeki sp. nov. and its ultra-reduced endosymbiont "Candidatus Pinguicoccus supinus" sp. nov. Sci Rep 10, 20311. https://doi.org/10.1038/s41598-020-76348-z (2020).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435-439. https://doi.org/10.1093/nar/gkl200 (2006).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. https://doi.org/10.1186/1471-2105-12-323 (2011).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. https://doi.org/10.1093/bioinformatics/bti610 (2005).
Gotz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36, 3420–3435. https://doi.org/10.1093/nar/gkn176 (2008).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. https://doi.org/10.1093/bioinformatics/btm071 (2007).
Laslett, D. & Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 32, 11–16. https://doi.org/10.1093/nar/gkh152 (2004).
Gruber, A. R., Lorenz, R., Bernhart, S. H., Neubock, R. & Hofacker, I. L. The Vienna RNA websuite. Nucleic Acids Res 36, W70-74. https://doi.org/10.1093/nar/gkn188 (2008).
Popenda, M. et al. Automated 3D structure composition for large RNAs. Nucleic Acids Res 40, e112. https://doi.org/10.1093/nar/gks339 (2012).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. https://doi.org/10.1093/bioinformatics/bts565 (2012).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. https://doi.org/10.1093/bioinformatics/btl158 (2006).
Shigematsu, M. et al. YAMAT-seq: an efficient method for high-throughput sequencing of mature transfer RNAs. Nucleic Acids Res 45, e70. https://doi.org/10.1093/nar/gkx005 (2017).
Bushnell, B., Rood, J. & Singer, E. BBMerge: accurate paired shotgun read merging via overlap. PLoS ONE 12, e0185056. https://doi.org/10.1371/journal.pone.0185056 (2017).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3. https://doi.org/10.14806/ej.17.1.200 (2011).
Holmes, A. D., Howard, J. M., Chan, P. P. & Lowe, T. M. tRNA Analysis of eXpression (tRAX): A tool for integrating analysis of tRNAs, tRNA-derived small RNAs, and tRNA modifications. (Submitted) (2020).
Sievers, F. & Higgins, D. G. Clustal omega. Curr Protoc Bioinformatics 48, 3 13 11–16. https://doi.org/10.1002/0471250953.bi0313s48 (2014).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35, 1547–1549. https://doi.org/10.1093/molbev/msy096 (2018).
Webb, B. & Sali, A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinform. 54, 5 6 1–5 6 37. https://doi.org/10.1002/cpbi.3 (2016).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46, W296–W303. https://doi.org/10.1093/nar/gky427 (2018).
Ichikawa, M. et al. Tubulin lattice in cilia is in a stressed form regulated by microtubule inner proteins. Proc Natl Acad Sci U S A 116, 19930–19938. https://doi.org/10.1073/pnas.1911119116 (2019).
Chaaban, S. et al. The Structure and Dynamics of C. elegans Tubulin Reveals the Mechanistic Basis of Microtubule Growth. Dev Cell 47, 191–204 e198. https://doi.org/10.1016/j.devcel.2018.08.023 (2018).
Kikkawa, M. et al. Switch-based mechanism of kinesin motors. Nature 411, 439–445. https://doi.org/10.1038/35078000 (2001).
Howes, S. C. et al. Structural differences between yeast and mammalian microtubules revealed by cryo-EM. J Cell Biol 216, 2669–2677. https://doi.org/10.1083/jcb.201612195 (2017).
Ma, M. et al. Structure of the Decorated Ciliary Doublet Microtubule. Cell 179, 909–922 e912. https://doi.org/10.1016/j.cell.2019.09.030 (2019).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. https://doi.org/10.1016/j.softx.2015.06.001 (2015).
Morrison, T. B., Weis, J. J. & Wittwer, C. T. Quantification of low-copy transcripts by continuous SYBR Green I monitoring during amplification. Biotechniques 24, 954–958, 960, 962 (1998).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45. https://doi.org/10.1093/nar/29.9.e45 (2001).
Pfaffl, M. W., Horgan, G. W. & Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30, e36. https://doi.org/10.1093/nar/30.9.e36 (2002).
Acknowledgements
We would like to thank Dr. Alexey V. Lobanov and Prof. Vadim N. Gladyshev of the Harvard Medical School of Boston, and Prof. Mariusz Nowacki of the Institute of Cell Biology of University of Bern for the providing some of the E. focardii and E. crassus data, respectively. We also thank the Gordon and Betty Moore Foundation for the grant to C.M. in the framework of the Marine Microbiology Initiative "Genetic manipulation of Ciliates" (Grant Number 4961) and the University of Camerino for grants FAR-JAU BVI000082 to M.M. and PNRA18_00133 to C.M.
Author information
Authors and Affiliations
Contributions
M.M., S.P., E.C.S. and C.M. wrote the main manuscript text and performed the main analyses. A.P. and C.E. performed the tRNA analysis. G.M. participated to the assembling and the cleaning of the assembly. P.B. maintained the cell cultures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mozzicafreddo, M., Pucciarelli, S., Swart, E.C. et al. The macronuclear genome of the Antarctic psychrophilic marine ciliate Euplotes focardii reveals new insights on molecular cold adaptation. Sci Rep 11, 18782 (2021). https://doi.org/10.1038/s41598-021-98168-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-98168-5
This article is cited by
-
From germline genome to highly fragmented somatic genome: genome-wide DNA rearrangement during the sexual process in ciliated protists
Marine Life Science & Technology (2024)
-
Extreme environments offer an unprecedented opportunity to understand microbial eukaryotic ecology, evolution, and genome biology
Nature Communications (2023)
-
Comparative genome analysis of three euplotid protists provides insights into the evolution of nanochromosomes in unicellular eukaryotic organisms
Marine Life Science & Technology (2023)
-
Taxonomy and SSU rRNA gene-based phylogeny of two new Euplotes species from China: E. chongmingensis n. sp. and E. paramieti n. sp. (Protista, Ciliophora)
BMC Microbiology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
