
Abstract
Uveal melanoma(UM) is the most common primary intraocular malignancy in adults. However, the incidence of UM in Asia is 10 to 20 times less than in Western populations. Therefore, for the first time, we report our whole exome sequencing (WES) data analysis to discover differences in the molecular features of Asian and Western UM, and to determine the disparities between the primary tumor before brachytherapy and enucleated samples after brachytherapy. WES of 19 samples (13 primary tumors, 5 enucleation samples after brachytherapy, and 1 liver metastasis) from 13 patients diagnosed with UM and treated between 2007 and 2019 at the Yonsei University Health System (YUHS) were analyzed using bioinformatics pipelines. We identified significantly altered genes in Asian UM and changes in mutational profiles before and after brachytherapy using various algorithms. GNAQ, BAP1, GNA11, SF3B1 and CYSLTR2 were significantly mutated in Asian UM, which is similar that reported frequently in previous Western-based UM studies. There were also similar copy number alterations (M3, 1p loss, 6p gain, 8q gain) in both groups. In paired comparisons of the same patients, DICER1 and LRP1B were distinctly mutated only in tumor samples obtained after brachytherapy using rare-variant association tests (P = 0.01, 0.01, respectively). The mutational profiles of Asian UM were generally similar to the data from previous Western-based studies. DICER1 and LRP1B were newly mutated genes with statistical significance in the regrowth samples after brachytherapy compared to the primary tumors, which may be related to resistance to brachytherapy.
Similar content being viewed by others


Introduction
Uveal melanoma (UM) is the most common primary intraocular malignancy in adults. The incidence of UM varies widely between races. In Western populations, the annual incidence is 5 to 10 per million population per year1,2, whereas in Asia, it is reportedly much lower at 0.4 to 0.63,4. Various radiotherapeutic and local therapeutic options have been applied to UM. The Collaborative Ocular Melanoma Study (COMS) trial showed that UM-related mortality rates were not significantly different between enucleation and plaque brachytherapy in the patients with medium-sized UM5,6. The results justified the use of plaque radiotherapy rather than enucleation for most medium-sized UMs. Since then, this treatment has been expanded to apply to small as well as large UMs. Despite these treatment efforts, in over one-quarter to one-third of patients, metastasis develops within 10 years, usually involving the liver, and death typically occurs 1–3 years after treatment7,8. Until now, the mutation profiles of Asia UM were little known; for the first time, we performed whole exome sequencing (WES) in Asian UM, and compared the results with the Cancer Genome Atlas (TCGA)9 data, which is the largest cohort composed mainly of Western patients. In addition, we compared the mutation profiles by WES before and after brachytherapy in eyes enucleated due to post-brachytherapy tumor regrowth. Schematic diagram of workflow is written in Supplementary Fig. 1.
Results
Patient characteristics
This study included 13 UM patients that have been diagnosed and treated at the Yonsei university health system (YUHS) from August 2007 to December 2019. The study was approved by the Institutional Review Board (IRB) at YUHS. and written informed consent was obtained All study protocols adhered to the tenets of the Declaration of Helsinki.
All patients were treated with brachytherapy after local resection. Enucleation was offered as the principal treatment if the patient had a large tumor (height > 10.0 mm), however, the patients who strongly refused primary enucleation received the brachytherapy. Local resection was performed for tumor debulking or diagnostic confirmation by endoresection for choroidal tumors and exoresection for iris or ciliary body tumors. Afterwards, brachytherapy with 106 Ru plaques (Eckert & Ziegler BEBIG, Berlin, Germany) was performed, with target tumor apex radiation doses ranging 85–100 Gy. When local recurrence (tumor regrowth) was noted during follow up, enucleation was performed.
Demographical and clinical data are summarized in Table 1. The analyzed samples were composed of triple types of samples (primary, enucleation after brachytherapy, and liver metastasis). Overall, 13 samples were primary tumors and 5 were paired enucleation samples after brachytherapy. Triple -paired samples were obtained from one patient.
Somatic variant detection
Genome Analysis Tool kit (GATK)10 4.1.0.0. Mutect211 without matched normal pipeline was used to call somatic variant. The average target region sequencing depth and the average of ontarget rate of preprocessed bam were 87.21x (SD 11.28) and 96% (SD 0.5%), respectively. The sequencing coverage and quality statistics are provided in Supplementary data.
Since we have called variant of FFPE samples without matched blood samples, the optimized filtration pipeline for tumor only sequencing12 was applied applied on somatic variant candidates from MuTect2. After filtration to distinguish somatic alterations from germline and sequencing artifacts, the average number of somatic alterations of primary samples (n = 13) was 21.46 (SD 12.38). This was not statistically different to the number of somatic mutations in TCGA UM (Wilcoxon Rank Sum test, P = 0.203, Supplementary Fig. 2).
Significant mutated gene analysis
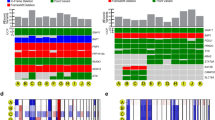
In MutSigCV, a total of 10 genes were found to be significant in the YUHS data (Fig. 1A, Supplementary Fig. 3). GNAQ, BAP1, GNA11, SF3B1, EIF1AX, PTPRD and CYSLTR2 were similarly found to be significant genes in the TCGA data using the same algorithm, whereas the other 3 genes (SLFN11, KTN1 and FANCL) were not. Using the OncodriveCLUST algorithm, 6 genes (GNA11, BAP1, GNAQ, SF3B1, CYSLTR2 and SLFN11) were noted to be significant in the YUHS data (Fig. 1A, Supplementary Fig. 4); and except for BAP1, CYSLTR2 and SLFN11, these were also significant in the TCGA data. Altogether, GNAQ, BAP1, SF3B1, GNA11, and CYSLTR2 were significant in both algorithms, and have been reported as frequently mutated genes in a previous UM study13. We could find common recurrent alterations (p.Q209P/L of GNAQ, p.Q209L of GNA11, p.L129Q of CYSLTR2 and p.R625H/C of SF3B1) in both datasets (Supplementary Figs. 5–10).

(a) Summary of Enrolled Uveal Melanoma Exome Analysis. The nonsynonymous somatic altered genes are color coded in waterfall plot. Left color coded bar indicates genes that significant in each tests such as MutSigCV, OncodriveCLUST and SKAT-O. (b) Comparison of somatic alterations in primary, enucleation and metastasis from YUHS 12 patient.
Copy number alterations
In previous UM studies13,14, recurrent copy number alterations (CNAs) of chromosome 1p, 3, 6p, and 8q have been reported. Those CNAs were also observed in YUHS UMs (Supplementary Fig. 11). There were 3 YUHS samples (23%) and 22 TCGA samples (27.5%) with chromosome 8q gain (Supplementary Fig. 12). The loss of chromosome 1p were found in 2 YUHS UMs (15.4%) and 21 (26.2%) TCGA UMs. The proportion of the two CNAs in YUHS UMs was similar with TCGA UM (Fisher’s exact test; 8q gain, P = 1.00; 1p loss, P = 0.51). On the other hand, there were 6 YUHS UMs (46.1%) and 17 TCGA UMs (21%) with chromosome 6p gain (P = 0.02). While two samples (15.4%) with monosomy chromosome 3 (M3) were found in YUHS UM samples, 42 samples (52.5%) had M3 in the TCGA UMs cohort (P = 0.03).
Molecular changes between before and after brachytherapy
In paired comparisons using the SKAT-O test, DICER1 and LRP1B were newly mutated genes with statistical significance in enucleation samples when compared to the primary tumor of the same patients (P = 0.01 and 0.01, respectively; Fig. 1A). The 4 types of somatic alterations of DICER1 and 5 somatic alterations of LRP1B were exclusively present in the enucleation samples (Table 2).
In a triple comparison of one patient (YUHS 12), comprising of the primary tumor, an enucleated sample after brachytherapy, and a liver metastasis sample, when the latter samples were compared to the primary tumor, one gene (METRK) mutations in the enucleation sample and six gene (FANCA, KMT2D, PCLO, PGR, TET3, and ZBTB7A) mutations in the metastasis were found to be newly occurred, and were each exclusive without common mutations (Fig. 1B).
Mutational signature analysis
Mutational signature analysis with primary variants data indicated mainly 5 signatures (Cosine similarity: 0.919), which were single base substitution (SBS) 5 (34.44%), SBS32 (23.1%), SBS1 (17.46%), SBS7b (17.28%), and SBS7a (7.76%) in primary samples (Fig. 2). SBS7b and SBS7a have been found in skin cancers from sun exposed areas and thus likely to be due to exposure to ultraviolet (UV) light. However, none of the signatures from TCGA UM was related to exposure to UV (Supplementary Fig. 13). Additionally, SBS5 was also found in TCGA UM signatures and SBS1 has been known to be related to various malignancies and aging. Therefore, we found both known signatures (SBS1, SBS5) from previous Western-based UM studies. Similar to a previous study showing ultraviolet radiation signatures were observed in iris tumors14, SBS7b and SBS7a were predominantly (≥ 50%) observed in YUHS iris melanoma samples (YUHS7, YUHS9) (Supplementary Fig. 14).

Decomposition plots of mutational signature analysis with primary samples (n = 13). Plots of Original and reconstructed data (left) and decomposed 5 mutational signature plots (right).
Discussion
We analyzed Asian UM and confirmed that significantly mutated genes were similar to those of TCGA, the largest western cohort.
In primary sample analysis, we found similar mutational profiles with western data. The 5 genes (GNAQ, BAP1, SF3B1, GNA11 and CYSTLR2) that have been reported as recurrent mutated or driver genes in other UM study were significant in two algorithms (MutsigCV and OncodriveCLUST). In addition, SLFN11 that never have been reported as UM related gene was also significant in both of them. This could be novel UM related cancer gene. The previously reported recurrent CNAs of UM were similarly observed in Asian UMs. Although the proportion of some CNAs (6p gain, M3) was different between the two cohorts, it is difficult to determine whether the results are from a difference between Asian and Western populations due to the small sample size of our cohort.
In the cases where the tumor regrows post-brachytherapy, enucleation should be considered. We performed paired comparison of WES data before and after brachytherapy and confirmed that two novel genes, DICER1 and LRP1B, were significantly mutated in the regrowth samples compared to the primary tumors. These two genes have already been associated with malignancy in non-ocular tumors15. It is not clear whether these newly developed mutations are the result of cancer evolution or the effect of brachytherapy; however, they could be related to resistance to brachytherapy.
In a triple comparison of one patient (Fig. 1B), the enucleated regrowth sample after brachytherapy and liver metastasis sample did not share any newly developed mutations. This may indicate that the liver metastasis event occurred earlier and independently of the tumor regrowth after brachytherapy.
Conclusion
This report, albeit a small sample size, is the first WES analysis in Asian UM, and through paired comparisons, novel and insightful results can be drawn.
Methods
Sample preparation and data generation
DNA was extracted from 19 formalin fixed paraffin embedded (FFPE) tissue from 13 UM patients that have been diagnosed and treated at the Yonsei university health system (YUHS) from August 2007 to December 2019 and captured using SureSelectHuman All Exon V6 for whole exome sequencing. After library QC of DNA, the libraries were sequenced on the Illumina HiSeq X ten platform. This study was approved by the Institutional Review Board (IRB) at YUHS and written informed consent was obtained. All study protocols adhered to the tenets of the Declaration of Helsinki.
Sequencing data processing and somatic variant analysis
The paired end reads of FASTA files from sequencing were aligned using BWA-mem16 on human genome (hg) 19. After duplicated reads of the aligned bam files were marked and removed using Picard, the base quality of reads in bam files was recalibrated using Genome Analysis Tool kit (GATK)10 4.1.0.0. Mutect211 without matched normal mode was used to call somatic variant candidates with gNomad17 as germline resources.
Variant filtration for only tumor sequencing and Annotation
To remove sequencing artifact, the variants with "bad_haplotype", "chimeric_original_alignment","base_quality","duplicate_evidence","fragment_length","low_avg_alt_quality","mapping_quality","multiallelic","n_ratio","read_orientation_artifact", "read_position", "str_contraction","strand_artifact" and "strict_strand_bias" were filtered out. We selected genes that have been reported as cancer genes in both OncoKB18, cBioportal19, for uveal melanoma. We also added MAPKAPK5 which was not listed in OncoKB but it has been reported to be a frequently mutated gene in uveal melanoma data from TCGA. If the population allele frequency of the variants is more than 1% in any subpopulation of 1000 genomes project20, ExAC21, KOVA22, gNomad and Korean Genome Project23 data, the variants were excluded as germline mutations. The second exclusion criteria is that the variant were present in the Korean 1,000 depression exome data24, which is a panel of normal. It removes not only germline mutation but also potential platform specific artifacts. Then we reviewed the all variants using Integrative Genomics Viewer25. If there is a missed variant that is found in another tissue from a same patient, we used blastn26 to align all sequence on neighboring region(± 50 bp) of the variant. We consider the variant exist if more than each 2 paired aligned reads that encoding same nucleotide of variants. The variants which passed the above criteria were selected as analysis ready somatic variants. All passed variants were annotated with SIFT27, Polylphen228 and CADD29 score using ANNOVAR30.
Significant mutated genes
The filtered non-synonymous variants were analyzed for significantly mutated genes using two algorithms, MutSigCV31 and OncodriveCLUST32. We defined the genes that have P less than 0.05 as significant in MutSicCV. on Genepattern33. with default parameter. OncodriveCLUST is used to identify genes with a significant bias of mutation clustering within the protein sequence. If Q value is less than 0.05 in OncodriveCLUST, the gene is significant. Maftools34 was used to run OncodriveCLUST. We also ran the same algorithms on uveal melanoma somatic variants data from TCGA. The somatic variant data were produced from tumor bam files from GDC data portal with same calling pipeline for YUHS.
Identifying copy number alterations
CNVkit35 was used to identify copy number alterations in UMs. Because construction of a normal reference from pooled normal samples is necessary for the somatic copy number calling pipeline, 1,000 WES of normal blood from the Korean 1000 database were used. Fused Lasso (flasso) algorithm was used as a segmentation method. The thresholds of log2 transformed relative ratio to reference ploidy for copy number 0,1,2,3 were -1.1, -0.4, 0.3, 0.7, respectively. The same pipeline was also utilized on TCGA tumor-normal paired data.
Rare variant association test
Since single variant association test has little power for testing of rare variants in relatively small samples, an alternative approach is needed. Sequence kernel association optimal test (SKAT-O)36 is an optimal unified approach for rare variant association testing in case control sequencing studies. The test was applied on non-synonymous variants of genes between primary and matched enucleation samples. R library SKAT was used to run the statistical test.
Mutational signature analysis
The 96 different contexts of single base substitutions from filtered variants were analyzed for generating mutational signatures. The signatures are identified as causes of mutational process, due to their unique mutational pattern and specific activity on the genome. The python packages, SigProfilerMatrixGenerator and SigProfilerExtractor, were used for this analysis. The iteration time was set 100 times to be performed to extract each signature.
Data availability
All data generated or analyzed during this study are included in this article. Raw sequence data have been deposited in Sequence Read Archive under PRJNA701837https://dataview.ncbi.nlm.nih.gov/object/PRJNA701837?reviewer=u0rm0asqvhrug0vv24usm3eijf.
References
Singh, A. D., Turell, M. E. & Topham, A. K. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology 118, 1881–1885. https://doi.org/10.1016/j.ophtha.2011.01.040 (2011).
Baily, C. et al. Uveal melanoma in Ireland. Ocul. Oncol. Pathol. 5, 195–204. https://doi.org/10.1159/000492391 (2019).
Park, S. J. et al. Nationwide incidence of ocular melanoma in South Korea by using the national cancer registry database (1999–2011). Invest. Ophthalmol. Vis. Sci. 56, 4719–4724. https://doi.org/10.1167/iovs.15-16532 (2015).
Tomizuka, T., Namikawa, K. & Higashi, T. Characteristics of melanoma in Japan: a nationwide registry analysis 2011–2013. Melanoma Res. 27, 492–497. https://doi.org/10.1097/CMR.0000000000000375 (2017).
Group & TCOMS,. The COMS randomized trial of Iodine 125 brachytherapy for choroidal melanoma, III: initial mortality findings: COMS report No. 18. Archiv. Ophthalmol. 119, 969–982. https://doi.org/10.1001/archopht.119.7.969 (2001).
The COMS Randomized Trial of Iodine 125 Brachytherapy for Choroidal Melanoma: V. Twelve-Year Mortality Rates and Prognostic Factors: COMS Report No. 28. Archiv. Ophthalmol. 124, 1684–1693, https://doi.org/10.1001/archopht.124.12.1684 (2006).
Shields, C. L. et al. Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch. Ophthalmol. 127, 989–998. https://doi.org/10.1001/archophthalmol.2009.208 (2009).
Zimmerman, L. E., McLean, I. W. & Foster, W. D. Does enucleation of the eye containing a malignant melanoma prevent or accelerate the dissemination of tumour cells. Br. J. Ophthalmol. 62, 420–425. https://doi.org/10.1136/bjo.62.6.420 (1978).
Cancer Genome Atlas Research, N. et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120. https://doi.org/10.1038/ng.2764 (2013).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. https://doi.org/10.1101/gr.107524.110 (2010).
Benjamin, D. et al. Calling Somatic SNVs and Indels with Mutect2. bioRxiv, 861054. https://doi.org/10.1101/861054 (2019).
Sukhai, M. A. et al. Somatic tumor variant filtration strategies to optimize tumor-only molecular profiling using targeted next-generation sequencing panels. J. Mol. Diagn. 21, 261–273. https://doi.org/10.1016/j.jmoldx.2018.09.008 (2019).
Robertson, A. G. et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 32, 204-220.e215. https://doi.org/10.1016/j.ccell.2017.07.003 (2017).
Johansson, P. A. et al. Whole genome landscapes of uveal melanoma show an ultraviolet radiation signature in iris tumours. Nat. Commun. 11, 2408. https://doi.org/10.1038/s41467-020-16276-8 (2020).
Dobrijević, Z. et al. Association between genetic variants in DICER1 and cancer risk: an updated meta-analysis. Gene 766, 145132. https://doi.org/10.1016/j.gene.2020.145132 (2021).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 (2013).
Wang, Q. et al. Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes. Nat. Commun. 11, 2539. https://doi.org/10.1038/s41467-019-12438-5 (2020).
Chakravarty, D. et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017. https://doi.org/10.1200/PO.17.00011 (2017).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1–pl1. https://doi.org/10.1126/scisignal.2004088 (2013).
Siva, N. 1000 Genomes project. Nat. Biotechnol. 26, 256–256. https://doi.org/10.1038/nbt0308-256b (2008).
Karczewski, K. J. et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 45, D840–D845. https://doi.org/10.1093/nar/gkw971 (2016).
Lee, S. et al. Korean variant archive (KOVA): a reference database of genetic variations in the Korean population. Sci. Rep. 7, 4287. https://doi.org/10.1038/s41598-017-04642-4 (2017).
Jeon, S. et al. Korean Genome Project: 1094 Korean personal genomes with clinical information. Science Advances 6, eaaz7835. https://doi.org/10.1126/sciadv.aaz7835 (2020).
Kang, H.-J. et al. Genetic markers for later remission in response to early improvement of antidepressants. Int. J. Mol. Sci. 21, 4884 (2020).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. https://doi.org/10.1038/nbt.1754 (2011).
Ye, J., McGinnis, S. & Madden, T. L. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 34, W6–W9. https://doi.org/10.1093/nar/gkl164 (2006).
Ng, P. C. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. https://doi.org/10.1093/nar/gkg509 (2003).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 76, 7.20.21–27.20.41. https://doi.org/10.1002/0471142905.hg0720s76 (2013).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. https://doi.org/10.1093/nar/gky1016 (2018).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164. https://doi.org/10.1093/nar/gkq603 (2010).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. https://doi.org/10.1038/nature12213 (2013).
Tamborero, D., Gonzalez-Perez, A. & Lopez-Bigas, N. OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 29, 2238–2244. https://doi.org/10.1093/bioinformatics/btt395 (2013).
Reich, M. et al. GenePattern 2.0. Nature genetics 38, 500–501. https://doi.org/10.1038/ng0506-500 (2006).
Mayakonda, A., Lin, D.-C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756. https://doi.org/10.1101/gr.239244.118 (2018).
Talevich, E., Shain, A. H., Botton, T. & Bastian, B. C. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12. https://doi.org/10.1371/journal.pcbi.1004873 (2016).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237. https://doi.org/10.1016/j.ajhg.2012.06.007 (2012).
Acknowledgements
The funding organization had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Funding
This research was supported by the Healthcare Bigdata Showcase Project by Korea Disease Control and Prevention Agency in the Republic of Korea (no.4800-4848-501); the Education and Research Encouragement Fund of Seoul National University Hospital.
Author information
Authors and Affiliations
Contributions
Drs J.L. and J.H.K. had full access to all of the data in the study, and takes responsibility for the integrity of the data and the accuracy of the data analysis. Conception and design: J.L. and J.H.K. Analysis and interpretation: W.S.L., J.L., J.J.C., S.C.L., and J.H.K. Data collection: W.S.L. and J.L., J. Manuscript preparation: W.S.L. and J.L., and H.G.K. Obtaining funding: J.H.K. Overall responsibility: J.L. and J.H.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, W.S., Lee, J., Choi, J.J. et al. Paired comparisons of mutational profiles before and after brachytherapy in asian uveal melanoma patients. Sci Rep 11, 18594 (2021). https://doi.org/10.1038/s41598-021-98084-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-98084-8
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
