
Abstract
Southern corn rust is a destructive maize disease caused by Puccinia polysora Underw that can lead to severe yield losses. However, genomic information and microsatellite markers are currently unavailable for this disease. In this study, we generated a total of 27,295,216 high-quality cDNA sequence reads using Illumina sequencing technology. These reads were assembled into 17,496 unigenes with an average length of 1015 bp. The functional annotation indicated that 8113 (46.37%), 1933 (11.04%) and 5516 (31.52%) unigenes showed significant similarity to known proteins in the NCBI Nr, Nt and Swiss-Prot databases, respectively. In addition, 2921 (16.70%) unigenes were assigned to KEGG database categories; 4218 (24.11%), to KOG database categories; and 6,603 (37.74%), to GO database categories. Furthermore, we identified 8,798 potential SSRs among 6653 unigenes. A total of 9 polymorphic SSR markers were developed to evaluate the genetic diversity and population structure of 96 isolates collected from Guangdong Province in China. Clonal reproduction of P. polysora in Guangdong was dominant. The YJ (Yangjiang) population had the highest genotypic diversity and the greatest number of the multilocus genotypes, followed by the HY (Heyuan), HZ (Huizhou) and XY (Xinyi) populations. These results provide valuable information for the molecular genetic analysis of P. polysora and related species.
Similar content being viewed by others


Introduction
Southern corn rust caused by Puccinia polysora is one of most devastating fungal diseases of corn; it was first described and named by Underw in 18971. SCR is distributed in tropical, subtropical and temperate cultivation regions, including Asia2, the United States3,4, Australia5 and Africa1,6. An increase in disease occurrence has been reported in recent years in China. The lack of varieties resistant to SCR and long-distance migration of the disease are the main reasons for severe disease epidemics3,7,8.
SCR appears to have great potential to damage plants. Symptoms initially appear on leaves and expand through an entire plant, including the leaves, stalks, leaf sheaths and husks, in late-season planting, which can result in leaf necrosis and complete destruction of photosynthetic areas, followed by death of the plant9,10,11,12. The risk of the pandemics of this disease is a big threat11. Although several physiologic races of P. polysora have been described13,14,15,16,17 and resistance has been reported3,7, the majority of commercial hybrids grown in China and the United Stated are rated as susceptible to P. polysora18,19,20. In the past, the disease has caused several serious yield losses in various countries, including West Africa (50%6), the Philippines (80–84%21), Brazil (40%22), the United States (30–50%3) and China (42–53%23).
The life cycle and mating system of P. polysora remain unknown. The alternate host of P. polysora has never been found. Urediniospores serve as both primary and secondary inoculum sources of mini cycle of the disease2. The aecidial and pycnidial stages of P. polysora are absent9. Teliospores are rarely or not produced2. All experiments attempting to induce the germination of teliospores have been unsuccessful2,9. The function of teliospores in the life cycle remains mystery, although they could be of significance in assuring extended survival of the pathogen. The highly successful uredo stage and the ease of continuity of this stage may have resulted in the suppression of the sexual phase in the life cycle9. Due to the lack of evidence, Cammack9 provisionally classified P. polysora as a microcyclic and autoecious hemiform.
Although the disease is a main problem of corn production, few nucleotide sequences of P. polysora have been deposited in GenBank. The lack of whole-genome sequences means that little molecular information is available for the pathogen, which limits the research on this pathogen. Illumina sequencing of the transcriptomes of model and nonmodel organisms previously confirmed that short reads can be effectively assembled24,25,26. For nonmodel organisms with limited genomic information, transcriptome sequencing, which focuses on the sequencing of functional and protein-coding RNAs, is a cost-effective method27,28. Transcriptome data are invaluable in the discovery of gene functions, metabolic pathways and molecular markers25.
Polymorphic simple sequence repeats (SSRs) have been proven to be important for assessing genetic diversity and population structure29,30, however, no SSR markers for P. polysora have been developed and reported to date. Without effective molecular markers, the genetic diversity of P. polysora has rarely been reported. The population diversity of P. polysora was investigated using restriction fragment length polymorphisms (RFLPs) in Japan31 and inter-simple sequence repeats (ISSRs) in Thailand32 and China33. The traditional and standard methods for developing SSR markers are time-consuming and expensive, whereas transcriptome sequencing provides a high-throughput source for investigating SSRs34,35.
The objectives of this study were to (1) sequence the transcriptomes of urediniospores of P. polysora using an RNA-seq sequencing platform, Illumina HiSeq2000, (2) assemble and annotate the unigenes of P. polysora, (3) develop polymorphic SSR markers, and (4) genotype and characterize the population genetics of P. polysora isolates collected from the cities of Xinyi, Huizhou, Yangjiang and Heyuan in Guangdong Province. The transcriptome sequences provide a valuable genomic resource for the molecular study of P. polysora, and the findings provide the first perspective into the P. polysora population structure in Guangdong Province.
Results
Sequence assembly
We sequenced the transcriptomes of urediniospores of P. polysora. A total of 28,252,282 raw reads (4.09 Gbp) were obtained. The percentages of Q20 and Q30 were 96.62% and 91.76%, respectively. The GC content was approximately 49.49%. After quality control, the remaining high-quality reads were assembled into 10,539 transcripts with an average length of 1,229 bp and an N50 of 2,083 bp (Table 1). We obtained 17,496 unigenes varying from 201 bp to 15,021 bp with an average of 1,015 bp and an N50 of 1,922 bp (Table 1). The length distribution of the unigenes showed that 10,539 unigenes were 201–500 bp, 4,795 unigenes were 500–1,000 bp, 6,292 unigenes were 1,000–2,000 bp and 5,623 unigenes were longer than 2,000 bp (Fig. 1).

The transcript and unigene length distribution of P. polysora.
Functional annotation
The functional annotation of all the unigenes was conducted by searching against published databases. Based on the sequence similarity, a total of 9550 (54.58%) unigenes were annotated. Because of the lack of genome information for P. polysora, 7946 unigenes could not be matched to known genes. The overall functional annotation across each database is described in Table 2.
There were 6,603 unigenes categorized into three main GO categories: biological process (17,725), cellular component (7,824) and molecular function (12,942) (Fig. 2, Table S1). Within the three categories, ‘cellular process’ (4,040), ‘binding’ (3,495), ‘cell part’ (2,685) and ‘cell’ (2,685) were the most prevalent. A high percentage of genes as classified under the ‘single-organism process’ (2,958), ‘catalytic activity’ (2,964) and ‘metabolic process’ (3,772) terms. The categories ‘metallochaperone activity’ (3), ‘cell aggregation’ (1) and ‘cell killing’ (2) represented the smallest groups.

Gene ontology (GO) classification of P.polysora unigenes. A total of 6603 unigenes with significant similarity in Nr database categorized into three main GO categories.
A total of 4,218 unigenes were clustered into 25 orthologous groups (KOG/COG) (Fig. S1, Table S2). Among these categories, the cluster ‘General function prediction only’ (567) was the largest group, followed by the cluster ‘Translation, ribosomal structure and biogenesis’ (533) and ‘Posttranslational modification, protein turnover, chaperones’ (477), while ‘Cell motility’ (3) and ‘Extracellular structures’ (2) represented the smallest groups.
To further analyse the transcriptome of P. polysora, all the unigenes were analysed in the KEGG pathway database. A total of 2,921 unigenes with significant matches were assigned to 32 KEGG pathways in five main categories (Fig. 3, Table S3). Among these five main categories, ‘Translation’ (480) constituted the largest category, followed by ‘Signal transduction’ (249), ‘Sorting and degradation’ (237) and ‘Amino acid metabolism’ (203).

Pathway assignment based on the Kyoto Encyclopedia of Genes and Genomes (KEGG). (A) Cellular process. (B) Environmental information processing. (C) Genetic information processing. (D) Metabolism. (E) Organismal systems.
SSR marker detection
After a bibliographic research, we have not found any SSR markers for P. polysora; therefore, we detected novel SSRs. In this study, all the assembled unigenes were used to mine potential SSRs. We identified a total of 8,798 potential SSRs in 6,653 unigenes (Table S4, Table S5). There were 2,157 unigenes containing more than one SSR. The largest fraction of identified SSR markers consisted of mononucleotides, which accounted for 52.4% (4,609), followed by those with trinucleotide repeats (2,461), dinucleotide repeats (1,351) and tetranucleotide repeats (290) (Table 3). In the mononucleotide repeats, the dominant repeat motif was A/T (3,631), followed by C/G (978). For the dinucleotide repeats, the AG/CT (898) motif was much more common than AC/GT (252), AT/AT (198) and CG/CG (3). For the trinucleotide repeats, the most common motif was ATC/ATG (826), followed by AGG/CCT (375), ACC/GGT (312) and AAG/CTT (278) (Table 3).
Identification of novel polymorphic SSR markers
Based on their flanking sequences, a total of 10,440 primer pairs for 3,480 out of 8,798 potential SSRs were successfully designed. We selected 400 high-quality SSR primer pairs for amplification and polymorphism detection via PCR with 96 isolates of P. polysora sampled in Guangdong Province. Of the primers we designed, 9 primers produced polymorphic fragments with the 96 isolates tested (Table 4). For the polymorphic loci, the number of alleles per locus ranged from 2 to 4, with an average of 3.0 alleles per locus. Nei’s gene diversity (Hexp), which estimates gene diversity, ranged from 0.021 to 0.50236. Simpson’s index λ (the genotypic diversity index) ranged from 0.021 to 0.499, and evenness ranged from 0.353 to 0.998 (Table 4)37. Locus Ppoly8 had the highest Simpson diversity (0.499) and the most evenly distributed alleles (0.998). A genotype accumulation curve showed that these 9 loci are informative for population genetic analysis (Fig. S2).
Population genetic diversity
The population genetic diversity of four populations (HY, HZ, XY and YJ) of P. polysora was analysed (Table 5). A total of 32 multilocus genotypes (MLGs) were detected in the 96 isolates based on the 9 novel SSR markers. The number of MLGs observed for each population ranged from 6 to 14. To reduce the influence of sample size, a more appropriate comparison, eMLG, was performed. eMLG is a measure of the number of genotypes that would be expected at the largest, shared sample size based on rarefaction. The YJ population had the highest genotypic diversity and the most eMLGs, followed by HZ, HY and XY. In addition, YJ was also the most even population, followed by XY, HZ and HY. Except for the XY population, the index of association \(\overline{r}_{d}\) and the standardized index of association IA showed that P. polysora reproduced clonally.
Population structure
Discriminant analysis of principal components (DAPC) was performed to analyse the genetic structure of 96 P. polysora isolates. The optimal number of PCs (30) was assessed using the find.clusters function. The cluster membership probabilities of each isolate based on the discriminant functions of DAPC were conducted with cluster numbers (K) from 2 to 8 (Fig. 4). With the increase in cluster number, population subdivision was gradually generated among the populations. All the populations in Guangdong Province exhibited admixture with other populations, and YJ showed the most admixture.

Discriminant analysis of principal components (DAPC) of the 96 P. polysora isolates. Posterior membership probabilities of each isolate based on the discriminant functions of DAPC. The group K was set from 2 to 8. Each isolate is represented by a vertical bar.
A neighbour-joining (NJ) phylogenetic tree and minimum spanning network (MSN) were conducted to further assess the population structure of P. polysora. The structure of P. polysora was dominated by the presence of two clonal linages (Fig. 5B). There were 44 and 17 isolates from all the populations clustered into these two clonal linages, respectively. Most isolates with high genetic distance from the YJ population were clustered into groups with cryptic diversity. The MSN results showed that two dominant MLGs were observed in all the populations (Fig. 5A). Populations HY and HZ contributed substantially to the major groups of MLGs, and HY had more unique MLGs than the other populations. The MSN results were consistent with the NJ tree analysis.

Minimum spanning network (MSN) and neighbour-joining (NJ) tree of 96 P. polysora isolates. Samples are colored according to the cities they were collected from. (A) MSN is constructed using the genetic distance of Bruvo. All the mutilocus genotypes are shown as a circle. The circle scale shows the number of isolates sharing the same mutilocus genotype. The ratio of colors in each circle is proportional to the ratio of isolates collected in different cities with the same mutilocus genotype. (B) Neighbour-joining (NJ) tree of 96 P. polysora isolates. Only values greater than 75 are shown based on 999 bootstraps performed.
Discussion
Recently, southern corn rust has become research focus because of its wide distribution, great damage and high incidence in China. Although southern corn rust is a devastating disease worldwide, the complete genome sequence and transcriptomic data for P. polysora remain unknown. Transcriptome analysis is an attractive alternative to examine the properties of a transcriptome as a proxy for the whole genome38. Transcriptome sequencing is a powerful and cost-effective method for generating large-scale transcriptomes and is used to annotate novel genes for model and nonmodel organisms and to develop molecular markers24,25,26. To our knowledge, the present study is the first attempt to perform de novo assembly of the transcriptome and to develop SSR markers of P. polysora.
In this study, we obtained a total of 27,295,216 high-quality reads with a Q20 of 96.62% using Illumina paired-end sequencing. A total of 17,496 unigenes were assembled, which is similar to the gene numbers predicted for other rust fungi, such as Melampsora larici-populina (16,39939), M. lini (16,27140) and P. striiformis (20–25,00041,42). The average length of the unigenes with an N50 of 1,922 bp was 1,015 bp. The GC content was 49.49%. These results indicated that the transcripts of P. polysora were of high quality and provided a large amount of valuable transcriptome information for gene annotation, novel gene discovery, and the investigation of molecular evolutionary mechanisms in P. polysora24,25,26.
To annotate the biological function of unigenes at the transcriptome level, sequence similarity searching was conducted against the Nr, Nt, Swiss-Prot, GO, Pfam, KOG/COG and KEGG databases. More than half of the unigenes (9,550; 54.58%) were matched with known proteins in public databases. Due to the absence of a reference genome for P. polysora, almost half of the unigenes might represent novel genes whose function is unknown. In the Nr database, most unigenes (77.00%) were annotated to Puccinia graminis f. sp. tritici, which causes stem rust of wheat. This result confirmed that P. polysora is closely related to P. graminis which has been completely sequenced43. In addition, we categorized 6,603, 4,218 and 2,921 unigenes into GO categories, orthologous groups (KOG/COG) and KEGG pathways, respectively, indicating that one-third of the unigenes could be well annotated with potential functions. The annotated unigenes might be useful in the investigation of gene function and valuable for further research in the future. These results again demonstrated that genes can be investigated by transcriptome analysis for nonmodel plant species.
In the present research, a total of 8,798 potential SSRs were identified in 6,653 unigenes. We selected 400 high-quality SSR primer pairs for amplification and polymorphism detection via PCR with 96 isolates of P. polysora. Only 9 primer pairs with the expected size were polymorphic among the 96 isolates of P. polysora. The primer pairs resulting in PCR products larger or smaller than expected may be due to the presence of introns, insertions or repeats, a lack of specificity, or assembly errors44. The low percentage of polymorphic loci may be due to the close geographic origin of isolates or the clonal reproduction of the pathogen45.
Previous genetic diversity studies of P. polysora populations fully support our results32,33. Using ISSR markers, Unartngam et al.32 found that P. polysora isolates from different provinces in Thailand have similar genetic characteristics and are present in the same groups, likely resulting from spore migration. The same result was found in Chinese populations using ISSR markers33. The urediniospores of P. polysora can be dispersed by air currents up to altitudes greater than 15,000 ft high and spread long distances by wind1. The present study again confirmed that the isolates from different localities presented similar genetic characteristics, which might be due to spore migration.
All the populations in Guangdong Province exhibited admixture with other populations and were clustered into two clonal groups, while the YJ population showed the highest genotypic diversity and the most eMLGs. More isolates with unique MLGs were found in the YJ population. The pathogen can survive year-round in the most south city Yangjiang belonging to the tropical zone, which might increase the possibility of new genotypes arising.
Our results confirm the previous study showing that P. polysora is a microcyclic and autoecious hemiform pathogen9,32. The index of association \(\overline{r}_{d}\) and the standardized index of association IA rejected the null hypothesis of linkage equilibrium in three out of four populations, suggesting high rates of selfing and clonal reproduction. Population XY was the exception and was characterized by only 6 MLGs, of which 5 MLGs were shared with other populations. Eight out of 13 isolates clustered into the clonal groups, and 5 isolates showed slight genetic differentiation. The differences remaining in population XY could lead to not reject the null hypothesis of linkage equilibrium, despite the pathogen truly reproducing clonally. Failure to reject the null hypothesis of linkage equilibrium does not mean the confirmation of sexual reproduction. More isolates in population XY are needed for further confirmation. The majority of isolates were clustered into two dominant clonal groups, which could be additional support for clonality and selfing.
The observed cryptic diversity appeared to be sampling bias. Members of the diverse groups were from HY, HZ and XY; however, most isolates of these populations were clustered into the dominant clonal groups. Furthermore, the index of association \(\overline{r}_{d}\) and the standardized index of association IA showed that the populations HY and HZ are highly clonal or selfing. Therefore, we propose that the observed cryptic diversity is because of sampling bias. Isolates of the cryptically diversity groups likely belong to their own clonal group or groups, that were not sampled deeply enough to obtain their membership.
This work provides novel insights into the transcriptome, annotated unigenes, polymorphic SSRs and population genetics of P. polysora. More work is needed to investigate the gene function and population structure of P. polysora.
Methods
Pathogen materials
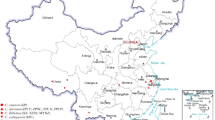
There is no research report on the physiological races and pathogenic types of P. polysora in China. To investigate the population genetics of P. polysora in Guangdong Province, we collected diseased leaves from the cities of Xinyi, Huizhou, Yangjiang and Heyuan in Guangdong Province. In each city, two to ten sampling sites with distances of at least 500 m apart from each other were identified, and 10 diseased leaves were randomly collected in each sampling site (Table 6). The plant materials used in this study were obtained from the wild and permission was obtained to collect samples. The collection of plant materials also complied with institutional, national, or international guidelines and legislation. The voucher specimens were preserved in the public herbarium of the Key Lab of Pest Monitoring and Green Management, Ministry of Agriculture and Rural Affairs of China.
To obtain enough pure urediniospores for RNA extraction, reproduction of P. polysora was conducted in a greenhouse. P. polysora is an obligate parasite. Three-leaf maize seedlings of susceptible ‘Zhengdan 958’ grown in a pot were rubbed to remove wax and sprayed with water mist. The urediniospores collected from diseased leaves were inoculated on the leaf surface. The inoculated plants were moisturized at 25 to 30 °C for 24 h in a sealed box to promote infection. The inoculated plants were transferred to a greenhouse at 25 to 30 °C. After 10 to 15 days, urediniospores were harvested and used for RNA extraction.
Populations were grouped based on cities and assigned as follows: XY (Xinyi), HZ (Huizhou), YJ (Yangjiang) and HY (Heyuan). A single pustule was collected from the sampled leaf to obtain each isolate on the assumption that each pustule was caused by a single urediniospore. To obtain enough urediniospores for DNA extraction, reproduction of isolates was conducted using the same method described above. A total of 98 isolates were obtained (Tables 5, 6).
RNA extraction
Total RNA was extracted using a TRIzol Kit (Promega, Beijing, China) following the manufacturer’s instructions. Residual DNA was removed using RNase-free DNase I (Takara Biotech Incorporation, Otsu, Japan) for 30 min at 37 °C. The purified RNA quality and quantity were verified using a spectrophotometer (Thermo Scientific, Waltham, MA, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
cDNA library construction and Illumina sequencing
Poly(A) mRNA was enriched by oligo (dT) magnetic beads. All mRNA sequences were cut into short fragments by using fragmentation buffer. Taking the short fragments as templates, random hexamer primers were used to synthesize first-strand cDNA. Then, buffer, dNTPs, polymerase I and RNase H were added to synthesize second-strand cDNA. The cDNA fragments were purified using the AMPure XP system (Beckman Coulter, Beverly, MA, USA), resolved with EB buffer for end repair and A tailing, and connected with sequencing adapters. PCR was performed using Phusion High-Fidelity DNA polymerase, universal PCR primers and an index primer. PCR products were purified using the AMPure XP system and assessed on DNA high-sensitivity chips using the Agilent Bioanalyzer 2100 system to construct the final cDNA library. The cDNA library was sequenced by Illumina HiSeq2000 sequencing performed by Novogene Bioinformatics Technology Co., Ltd., Beijing, China.
Sequence assembly and gene annotation
The high-quality clean reads for assembly were separated from adapters. Reads with more than 50% Q20 bases and those with more than 10% ambiguous “N” bases were removed. De novo transcriptome assembly was conducted using Trinity software with an optimized k-mer length of 2546. Unigenes of the transcriptome were annotated using the NCBI nonredundant protein (Nr) database, NCBI nonredundant nucleotide sequence (Nt) database, Kyoto Encyclopedia of Genes and Genomes (KEGG) database, a manually annotated and reviewed protein sequence (Swiss-Prot) database, Cluster of Orthologous Groups of proteins (KOG/COG) database, Gene Ontology (GO) database and protein family (Pfam) database. The statistical enrichment of the unigenes in KEGG pathways was tested by KOBAS software as shown previously47,48. The annotation with the Nr, Nt, Swiss-Prot and KOG databases used NCBI Blast version 2.2.28 + with an e-value of 1e−5. GO analysis was performed using Blast2GO version 2.549. Pfam protein domain prediction was conducted using HMMER version 3.050.
Polymorphic SSR validation and assessment
Microsatellites were identified using MISA software51. The parameters were adjusted to identify mono-, di-, tri-, tetra-, penta- and hexanucleotide motifs with a minimum of ten, six, five, five, five and five repeats, respectively. Primer 3 version 2.2.3 was used to design the primer pairs with default settings. Forward primers were labelled with fluorescent tags (FAM, HEX or TAMRA) to improve the separation of overlapping markers during multiplexing. Ninety-six isolates sampled from Guangdong Province were used for PCR amplification and polymorphic identification. Amplifications were performed with a thermal cycler (Eppendorf AG) under the following conditions: initial denaturation at 94 °C for 5 min, followed by 35 cycles of 94 °C for 30 s, annealing temperature (Tm) (Table 1) for 30 s, and 72 °C for 1 min, and a final extension at 72 °C for 10 min. Electrophoresis and visualization of alleles were performed on an ABI 3730 DNA analyser (Applied Biosystems, Carlsbad, CA, USA) by Beijing Tsingke Biotech Co., Ltd. Allele sizes were estimated using GeneMarker version 2.2.0 software and compared to a GS500 (35–500 bp) internal standard.
Population genetics data analysis
We used various R packages to perform population genetic analysis52. The R package poppr53 was used to calculate the number of MLGs and eMLGs, genotypic diversity (the Shannon–Wiener index H)54, Stoddard and Taylor’s index G55, and Simpson’s index λ37, evenness E556, Nei’s unbiased gene diversity Hexp36, and index of association \(\overline{r}_{d}\) and standardized index of association IA57. eMLG estimates the number of expected MLGs that share the same sample size based on rarefaction. H and G both measure richness and evenness, increasing as genotypic richness and evenness increase. G weights abundant MLGs more heavily than less abundant MLGs. λ is an estimation of the probability that two randomly selected genotypes are different. E5 is a measure of the distribution of genotype abundances. E5 is equal to one when a population has equally abundant genotypes and closer to zero when it is dominated by a single genotype. Hexp estimates the average heterozygosity. \(\overline{r}_{d}\) and IA are measures of linkage disequilibrium. Significant disequilibrium is expected due to linkage among loci when a population is clonal, and linkage among loci is not expected in sexually reproducing populations.
The package ape58 was used to draw a neighbour-joining dendrogram with Prevosti’s distance59. Minimum spanning networks (MSNs) were constructed with the genetic distance of Bruvo60 using the package poppr. Discriminant analysis of principal components (DAPC) was plotted using the packages adegenet61 and ade462.
Data deposit
The transcriptome has been submitted to the National Center for Biotechnology Information (NCBI). BioSample accession: SAMN18713839.
References
Orian, G. Occurrence of Puccinia polysora underwood in the Indian ocean area. Nature 173, 505 (1954).
Brewbaker, J. L., Kim, S. K., So, Y. S. & Logrono M. General resistance in maize to southern rust (Puccinia polysora Underw). Crop Sci. 51, 1393–1409 (2011).
Futrell, M. C., Hookerm, A. L. & Scott, G. E. Resistance in maize to corn rust, controlled by a single dominant gene. Crop Sci. 15, 597–599 (1975).
Sim, T. IV. Southern rust of corn recognized in Kansas. Plant Dis. 64, 500 (1980).
Barker, S. J. Testing of maize hybrids resistant to Puccinia polysora on the Atherton Tableland, Queensland. J. Agric. Anim Sci. 26, 319–327 (1969).
Rhind, D., Waterston, J. M. & Deighton, F. C. Occurrence of Puccinia polysora Underw. in West Africa. Nature 169, 631 (1952).
Scott, G. E., King, S. B. & Armour, J. W. Jr. Inheritance of resistance to southern corn rust in maize populations. Crop Sci. 24, 268–267 (1984).
Crouch, J. A. & Szabo, L. J. Real-time PCR detection and discrimination of the southern and common corn rust pathogens Puccinia polysora and Puccinia sorghi. Plant Dis. 95, 624–632 (2011).
Cammack, R. H. Studies on Puccinia polysora underw: II. A consideration of the method of introduction of P. Polysora into Africa. Trans. Br. Mycol. Soc. 42, 27–32 (1959).
Futrell, M. C. Puccinia polysora on maize associated with cropping practice and genetic homogeneity. Phytopathology 65, 1041–1042 (1975).
Rodriguez-Ardon, R., Scott, G. E. & King, S. B. Maize yield losses caused by southern corn rust. Crop Sci. 20, 812–814 (1980).
Scott, G. E. & Futrell, M. C. Southern corn rust. Crops Soils Mag. April-May, 16–18 (1976).
Casela, C. R. & Ferreira, A. S. Variability in isolates of Puccinia polysora in Brazil. Fitopatol. Bras. 4, 414–416 (2002).
Robert, A. L. Host range and races of the corn rusts. Phytopathology 52, 1010–1015 (1962).
Ryland, A. K. & Storey, H. H. Physiological races of Puccina polysora Underw. Nature 176, 655–656 (1955).
Storey, H. H. & Howland, K. A. Resistance in maize to a third east African race of Puccinia polysora Underw. Ann. Appl. Biol. 60, 297 (1967).
Ullstrup, A. J. Inheritance and linkage of a gene determining resistance in maize American race of Puccinia polysora. Phytopathology 55, 425–430 (1965).
Chen, C. X. et al. Molecular tagging and genetic mapping of the disease resistance gene RppQ to southern corn rust. Theor. Appl. Genet. 108, 945–950 (2004).
Ru-Hong, Z., King, S. B. & Scott, G. E. A study of slow rusting of southern rust of corn: preliminary report. USDA Bull, 925 (1983).
Wu, X. J. et al. Geographic and genetic identification of RppS, a novel locus conferring broad resistance to southern corn rust disease in China. Euphytica 205, 17–23 (2015).
Reyes, G. M. An epidemic outbreak of the maize rust in eastern and central Visayas, Philippines. Philipp. J. Agric. 18, 115–128 (1953).
Von Bülow, J. F. W. As ferrugens (Puccinia sorghi, P. polysora, Physopella zeae) do milho (Zea mays). I. Revisão Bibliográfica. Pesq. Agropec. Bras. 1, 249–262 (1966).
Liu, Y. Y. & Wang, J. Southern corn rust occurred in Hebei province in 1998. Plant Prot. 25, 53–58 (1999).
Hegedűs, Z. et al. Deep sequencing of the zebrafish transcriptome response to mycobacterium infection. Mol. Immunol. 46, 2918–2930 (2009).
Liu, J. J. et al. Comparative transcriptomics and RNA-Seq-based bulked segregant analysis reveals genomic basis underlying Cronartium ribicola vcr2 Virulence. Front. Microbiol. 12, 1–17 (2021).
Rosenkranz, R., Borodina, T., Lehrach, H. & Himmelbauer, H. Characterizing the mouse ES cell transcriptome with Illumina sequencing. Genomics 92, 187–194 (2008).
Geng, Y. et al. A De novo transcriptome assembly of Ceratopteris richardii provides insights into the evolutionary dynamics of complex gene families in land plants. Genome Biol. Evol. 13, 1–14 (2021).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Cowger, C., Parks, R. & Kosman, E. Structure and migration in US Blumeria graminis f. sp tritici populations. Phytopathology 106, 295–304 (2016).
Wang, C. C. et al. Genetic diversity and population structure of Puccinia striiformis f. sp. tritici reveal its migration from central to eastern China. Crop Prot. 128(104974), 0261–2194 (2020).
Hirayae, K. et al. Genetic variation of southern rust fungus of corn in Japan. Kyushu Pl. Prot. Res. 44, 12–14 (1998).
Unartngam, J., Janruang, P. & To-anan, C. Genetic diversity of Puccinia polysora in Thailand based on inter simple sequence repeat (ISSR) markers analysis. Int. J. Agric. Technol. 7, 1125–1137 (2011).
Guo, Y. Y. et al. Genetic diversity of Puccinia polysora Underw. in China. Sci. Agric. Sin. 46, 4523–4533 (2013).
Cardoso, S. D. et al. Efficient isolation of polymorphic microsatellites from high throughput sequence data based on number of repeats. Mar. Genom. 11, 11–16 (2013).
Chen, W., Liu, Y. X. & Jiang, G. F. De novo assembly and characterization of the testis transcriptome and development of EST-SSR markers in the cockroach Periplaneta americana. Sci. Rep. 5, 11144. https://doi.org/10.1038/srep11144 (2015).
Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590 (1978).
Simpson, E. H. Measurement of diversity. Nature 163, 688. https://doi.org/10.1038/163688a0 (1949).
Onda, Y. et al. Transcriptome analysis of thermogenic Arum concinnatum reveals the molecular components of floral scent production. Sci. Rep. 5, 8753. https://doi.org/10.1038/srep08753 (2015).
Duplessis, S. et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. 108, 9166–9171 (2011).
Nemri, A. et al. The genome sequence and effector complement of the flax rust pathogen Melampsora lini. Front. Plant Sci. 5, 98. https://doi.org/10.3389/fpls.2014.00098 (2014).
Cantu, D. et al. Genome analyses of the wheat yellow(stripe) rust pathogen Puccinia striiformis f. sp. tritici reveal polymorphic and haustorial expressed secreted proteins as candidate effectors. BMC Genom. 14, 270. https://doi.org/10.1186/1471-2164-14-270 (2013).
Zheng, W. et al. High genome heterozygosity and endemic genetic recombination in the wheat stripe rust fungus. Nat. Commun. 4, 2673 (2013).
Upadhyaya, N. M. et al. Comparative genomics of Australian isolates of the wheat stem rust pathogen Puccinia graminis f. sp. tritici reveals extensive polymorphism in candidate effector genes. Front. Plant Sci. 5, 759. https://doi.org/10.3389/fpls.2014.00759 (2015).
Wei, W. L. et al. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genom. 12, 451 (2011).
Wu, J., Cai, C. F., Cheng, F. Y., Cui, H. L. & Zhou, H. Characterisation and development of EST-SSR markers in tree peony using transcriptome sequences. Mol. Breed. 34, 1853–1866 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Mao, X., Tao, C. J. G. O. & Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793 (2005).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435 (2008).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 39, 29–37 (2011).
Thiel, T., Michalek, W., Varshney, R. K. & Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422 (2013).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (2017).
Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281. https://doi.org/10.7717/peerj.281 (2014).
Shannon, C. E. The mathematical theory of communication. 1963. MD Comput. 14, 306–317 (1997).
Stoddart, J. A. & Taylor, J. F. Genotypic diversity: Estimation and prediction in samples. Genetics 118, 705–711 (1988).
Pielou, E. C. Ecological Diversity. Wiley, New York. ISBN-10: 0471689254 (1975).
Grünwald, N. J., Goodwin, S. B., Milgroom, M. G. & Fry, W. E. Analysis of genotypic diversity data for populations of microorganisms. Phytopathology 93, 738–746 (2003).
Paradis, E. & Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Prevosti, A., Ocaña, J. & Alonso, G. Distances between populations of Drosophila subobscura, based on chromosome arrangement frequencies. Theor. Appl. Genet. 45, 231–241 (1975).
Bruvo, R., Michiels, N. K., D’Sousa, T. G. & Schulenberg, H. A simple method for calculation of microsatellite genotypes irrespective of ploidy level. Mol. Ecol. 13, 2101–2106 (2004).
Jombart, T. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Dray, S. & Dufour, A. B. The ade4 Package: Implementing the duality diagram for ecologists. J. Stat. Softw. https://doi.org/10.18637/jss.v022.i04 (2007).
Acknowledgements
We thank National Agro-tech Extension and Service Center, and plant protective stations for collecting disease samples. We thank Brian J. Knaus and Shankar K. Shakya for excellent technical support. This study was supported by the Natural Science Foundation of China (grant 31772101, 31972211).
Author information
Authors and Affiliations
Contributions
Q.Y.S., Y.L., and Z.H.M. conceived and designed the experiments. Q.Y.S. performed the experiments, analysed the data, and wrote the paper. J.L. and C.H. participated in collection of samples of SCR. K.Y.Z., L.F.L. and J. Y. D. helped in propagating pathogens.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, Q., Liu, J., Zhang, K. et al. De novo transcriptome assembly, polymorphic SSR markers development and population genetics analyses for southern corn rust (Puccinia polysora). Sci Rep 11, 18029 (2021). https://doi.org/10.1038/s41598-021-97556-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-97556-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
