
Abstract
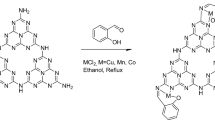
A new nano-scale Cu@salicylaldehyde-modified-chitosan (Cu@Sal-CS) was synthesized through a green, eco-friendly and cost-effective technique. The prepared catalyst was characterized using Fourier transform infrared spectroscopy (FT-IR), scanning electron microscopy (SEM), Energy-dispersive X-ray spectroscopy (EDXS), and inductively coupled plasma (ICP) analysis. The synthesized Cu@Sal-CS catalyst indicated its performance in the C–O and C–N oxidative coupling using the reaction of 1,3-dicarbonyl derivatives/2- substituted phenols with amides for the preparation of carbamates, as well as in the reaction of aldehydes and various amines in the synthesis of amides. The significant features of this work are operational simplicity of catalyst synthesis, in situ and new modification method, use of an efficient, recoverable, frequently reused and stable catalyst without any loss of catalytic activity, and high yields of the products in short times.
Similar content being viewed by others
Introduction
C–C and C–X couplings are among the most important reactions in organic synthesis1, as they can be used in the formation of new organic products2. Recently, transition-metal-catalyzed organic reactions through the C-H bond activation have attracted much attention from the atom- and step-economical points of view, and a variety of catalytic processes utilizing different modes for activating the available bond have been developed3,4. Nowadays, urethanes are considered as very important materials for the discovery and development of drugs. They are classified as one of the basic structural elements building up numerous approved human therapeutic agents5. Carbamates can be mainly classified into inorganic and organic categories. When the carbamate linkage is attached to any inorganic atoms, either metallic or nonmetallic, such compounds are referred to as inorganic carbamates6. Organic carbamates can be produced through the substitution of amino and carboxyl moieties on the unstable carbamic acid (H2N-COOH) with structurally diverse groups. They are considered as a stable class of compounds, whose characteristic is the linkage O–CO–NH-6. This group of organic compounds is widely used as agrochemicals (pesticides, herbicides, insecticides, fungicides, etc.)7, pharmaceuticals8, intermediates in organic synthesis9, protecting agent for amino groups in peptide chemistry10, and linkers in combinatorial chemistry11,12. Therefore, considerable efforts have been made in recent years to develop efficient and safe methodologies for carbamates synthesis13,14,15. Hofman16 and Curtius rearrangements17 are two methods applied for carbamates synthesis. For example, Ikegami et al. reported a method for the synthesis of carbamates through the use of sugar carboxylic acids, alcohols, and amines by using the Curtius rearrangement18. Also, gaseous carbon dioxide is another reagent used for the synthesis of carbamates through the Mitsunobu’s reaction19. The ordinary synthesis of carbamates involves intermediates such as chloroformates20 or isocyanates21, prepared by applying phosgene22 or its substitutes1. Phosgene is very harmful to the health of humans and the environment. Therefore, to avoid the use of toxic and harmful reagents, phosgene-free routes, including the oxidative carbonylation of amines using metal catalysts, have been widely reported1. Kumar and co-workers worked on the oxidative coupling of β-dicarbonyl- or 2-carbonyl-substituted phenols with N,N′-disubstituted formamides under oxidative conditions to yield carbamates by using CuBr2 as the catalyst and TBHP (t-butyl hydroperoxide) as the oxidant1. Azizi et al. reported that Fe3O4@EDTA-Cu(II) nanoparticles could catalyze the reaction of formamides with β-dicarbonyl compounds in the presence of TBHP as the oxidant to give enol carbamates in good yields3. Recently, Barve et al. reported that CuCl could catalyze the oxidative coupling of formamides with salicylaldehyde23. This team also reported the synthesis of carbamates in high yields utilizing a wide diversity of substrates including β-keto esters and 2-carbonyl-substituted phenol derivatives under oxidative conditions24. In 2017, Panahi and coworkers used nano porous metal–organic framework Cu2(BDC)2(DABCO) as a heterogeneous catalyst for the preparation of phenol carbamates25.
On the other hand, an important chemical linkage in proteins and peptides structures is the amide functional group26. The desired approach to the formation of amide bonds (as economical and available materials) is the direct amidation of aldehydes with amines by applying a suitable catalyst. Scientists take the amide functionality into account as a key unit in polymers, organic molecules, synthetic intermediates, pharmaceuticals, and natural products. As a result, the formation of amide bonds has been of great importance among all the transformations27. Furthermore, several alternative approaches, including the Schmidt, Ritter, Beckmann, Ugi, Wolff, Staudinger reactions and hydration of nitriles, have proved to be efficient to enhance the fabrication of amides28,29. Amides play a key role in materials including natural products, synthetic intermediates, and biological and synthetic polymers, and so led to an urgent need of developing novel strategies in the chemical industry for the fabrication of amides30. Recently Li et al. worked on the intermolecular dehydrogenative amidation of arenes via copper (I) bromide catalyst, in which 2-pyridyl or 1-pyrazolyl was used as the chelating group and the air was employed as the terminal oxidant at 140°C31. In 2014, Azizi et al. reported the direct oxidative amidation of alcohols using EDTA@Cu(II) functionalized superparamagnetic nanoparticle27. Due to the environmental pollution resulted from the chemical industry, enormous efforts have been made toward the development of new environmentally friendly processes using heterogeneous catalysts, because of the simple catalyst removal and recovery method. The fabrication of metal nanoparticles supported on polymeric substrates is now a hot topic in the category of nanostructured materials, finding applications in numerous fields. As a biodegradable, non-toxic substrate for metal catalysts, cellulose, which is the most abundant biopolymer, has attracted innumerable global interests32,33. Copper (Cu) and Cu-based nanoparticles, based on the earth-abundant and cost-efficient copper metal, are recently employed in a wide range of applications, especially in the field of catalysis for the preparation of efficient catalysts33,34. Among the others, the composites obtained by the immobilization of transition metals on the chitosan matrix can lead to the favorable combination of the advantages of homogeneous and heterogeneous systems. Chitosan (Scheme 1) can be used as the catalyst due to the presence of amino groups on its backbone. It is abundant, renewable, and green material and simply recoverable from the reaction mixture35.

The chemical structure of chitosan.
It has been confirmed that chitosan can adsorb transition metals via chelation36. One of the methods for chemical modification of chitosan to enhance its affinity as a complexing agent is Schiff base formation via the reaction of amine groups existing on the chitosan backbone with carbonyl groups37. Imine derivatives of chitosan have a high capacity to strongly chelate copper salts when compared with the simple chitosan33,35. Recently, modified chitosan biopolymer has attracted the researcher’s interest due to its many applications in catalyst38,39, microbial activities37, biosensors40, and etc.
In this work, modified-chitosan was obtained by treating with salicylaldehyde to enhance its copper adsorption capacity41. The catalyst was used to promote the copper-catalyzed C-O and C-N bonds formation via oxidative coupling. Under optimal conditions, various 2-carbonyl-substituted phenols and 1,3-dicarbonyl derivatives were utilized as substrates for the formation of carbamates (Electronic Supplementary Information (ESI), Figure S1).
Results and discussion
Catalyst preparation and characterization
Initially, chitosan was modified by salicylaldehyde and copper acetate through an efficient, solvent-free, and green method (ESI, Figure S2). By comparing this procedure with the previous methods reported in the literature, this protocol has some advantages such as mild and green condition, high efficiency, being straightforward, in a short time, and no need for any solvent. Here, all the steps to modify chitosan were done through mechanochemical technique, for the first time. Briefly, the Cu@modified-chitosan was synthesized in two steps. In the first step, solvent-free room temperature ball milling of the chitosan and salicylaldehyde led to the formation of an imine bond. In the second step, copper (II) acetate was added to the modified-chitosan and the mixture was grounded mechanically at room temperature. The resulting green product was characterized by using FT-IR spectroscopy, and SEM. The formation of the modified-chitosan was successfully demonstrated by the FT-IR studies33. The FT-IR data of chitosan, the modified-chitosan and Cu@Sal-CS are shown in Figure S3. (See ESI). By making a comparison between the IR spectra of chitosan and the modified-chitosan, the absence of –NH2 band and the presence of two additional bands at 1632 cm−1 and 1212 cm−1, were attributed to the formation of the imine (C=N) functional group. As it can be seen in Fig. S1, the FT-IR spectrum of the initial chitosan is identical to one reported in the literature42, investigating the IR-spectroscopic data of chitin and chitosan. It is evident that the presence of copper in the complex results in considerable changes in the shape of the broadband at 3000–3700 cm−1, attributed to various types of OH and NH vibrations in polymers. In the Cu@Sal-CS catalyst, a shoulder was observed in the spectrum of the prepared sample at 590 cm−1, assigned to the formation of bonds between NH2 groups of chitosan and Cu(I) ions42.
The morphology of the Cu@Sal-Cs was studied using scanning electron microscopy. The formation of the small particles at the nano-scale size was confirmed. The EDX analysis proved the presence of Cu, C, N, and O elements in the Cu@modified-chitosan structure. The results of SEM and EDX analysis are presented in Figure S4 (See ESI).
The amount of the bounded copper ions was analysed by ICP analysis. According to the results, 15.6 wt.% of the catalyst was copper, which was more than that previously reported for a Chitosan-Cu composite without chelating aid of salicylaldehyde36.
Study of the catalytic activity
To explore the catalytic potential of the Cu@Sal-Cs, C‒O, and C‒N coupling reactions were investigated. Initially, C‒O coupling for the synthesis of carbamates was investigated. Accordingly, the mixture of salicylaldehyde (1, 1 mmol), DMF (2, 6 mL), and TBHP as an oxidant (1.5 eq.) in the presence of a catalytic amount of Cu@Sal-Cs (20 mg) was stirred at 80 °C in an oil bath for 1 h. After completion and separation, the carbamate 3 was achieved in a yield of 70% (Entry 2, Table 1). This result encouraged us to survey the ideal condition for the reaction by changing the reaction parameters to get the highest product yield. As shown in Table 1, the presence of both oxidant and catalyst is crucial for the reaction and in the absence of any of them, no product was observed (Entries 3 and 4, Table 1). The same reaction proceeded well (85% yield after 35 min at 80 °C) when 30 mg of the catalyst has been added to the reaction mixture (Entry 5, Table 1). Moreover, the reaction does not proceed at room temperature and requires heat (Entry 1, Table 1). Screening organic and inorganic oxidants revealed that no product was found using air, benzoyl peroxide, and hydrogen peroxide even at elevated temperatures, even at high oxidant content, and under microwave irradiation (Entries 6–9 and 11–15, Table 1). By-product was produced in the presence of benzoyl peroxide and the desired carbamate was not obtained. To investigate the importance of copper in the catalyst, some reactions were carried out by using chitosan and salicylaldehyde-modified chitosan in the same model reaction. No reaction was observed after 1 h (Entries 19–20, Table 1).

The model reaction has also been studied under microwave irradiation. As it is clear from Table 1, microwave irradiation at 200 W/60 °C affords the product in a shorter reaction time (15 min) with an 81% yield (Entry 16). Increasing the amount of the catalyst to 30 mg affects the yield considerably (Compare entries 16 and 17, Table 1). Also, increasing the power and temperature resulted in higher yield and increased the reaction speed, and reduced the reaction time (Entry 18, Table 1). According to the results, we decided to use microwave irradiation for the coupling reaction. In conclusion, 30 mg of Cu@Sal-Cs catalyst and 1 mmol of the 2-carbonyl-substituted phenols with amides (6 mL) in the presence of 1.5 eq. of TBHP, under microwave irradiation was chosen as the optimized reaction condition. The model reaction has been tested in gram scale. Briefly, 300 mg of the catalyst was added to the mixture of DMF (60 mL) and salicylaldehyde (1.2 g, 10 mmol) in the presence of TBHP (15 eq.) as oxidant. The reaction mixture was subjected to microwave irradiation (80 °C /300 W, 10 min). The product (2-formylphenyl dimethylcarbamate) was synthesized in 94% yield (1.82 g).
After optimizing the reaction conditions, the generalization capability of this method was shown using the 2-carbonyl-substituted phenols and amides to construct a library of phenol and enol carbamates as shown in Table 21,3,23,25,43. We also applied this methodology in the synthesis of β-ketocarbamates using 1,3-dicarbonyls and different formamides (Entries 20–24, Table 2). The importance of this type of reaction is that the aldehyde remains intact in the presence of the oxidant. We have also investigated the effect of the carbonyl group on ortho substitutions of phenols, demonstrating that the substrates possessing no ortho substituent do not react (Entry 26, Table 2).
Possible mechanism
A plausible mechanism has been proposed for the reaction catalyzed by Cu@Sal-CS (ESI, Figure S5). In the proposed mechanism, at first, a complex is formed between the substrate (1) and the catalyst (I). Microwave irradiation decomposes TBHP (2) to form hydroxyl and tert-butoxyl (3) radicals, which in turn transform formamide to the radical 6. The complex III will be formed between the radical 6 and complex II. Finally, this complex affords the desired carbamate. Similar to the 2-carbonyl-substituted phenol complex, a coordinated complex is suggested for the enol tautomer of the dicarbonyl moiety. This complex can be transformed to different carbamates. To demonstrate that this reaction absolutely promotes via a radical mechanism, we used the radical scavengers 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and hydroquinone, and no reaction was observed in these cases (Entries 27, 28, Table 2).
C–N coupling of benzaldehyde derivatives with amines to amides
The successful C-N coupling of benzaldehyde derivatives with amines was found to be facilitated in high yields in the presence of the Cu@Sal-Cs catalyst with TBHP and resulted in the desired amide products (3) (ESI, Figure S6). At first, to select the best and optimum condition, an amount of 20 mg catalyst was tested with 1 mmol p-methoxybenzaldehyde and 6 mL of pyrrolidine at room temperature. After 60 min no reaction occurred (Entry 1, Table 3). In the next step, 1.5 eq. TBHP as oxidant was added, but no amide was obtained (Entry 2, Table 3). Increasing the temperature to 80 °C resulted in 70% yield (Entry 3, Table 3). The use of 30 mg catalyst in the presence of 1.5 eq. TBHP led to an increase of yield in lesser reaction time (Entry 4, Table 3). Additionally, the catalytic activity of 30 mg catalyst in the presence of ethanol and acetonitrile, as the solvent, and without the oxidant was tested, which did not result in the formation of the desired product (Entries 5–6, Table 3). By the addition of TBHP, this reaction represented good yields. The reaction has also been studied under microwave irradiation. It was found that using a catalyst (30 mg), oxidant TBHP (1.5 eq), in the absence of solvent, and at the power of 300 W, led to the formation of the amide in good yield (Entry 9, Table 3).

With the optimized conditions in hand, we further investigated the scope of the reaction using different substituted benzaldehydes and amines as shown in Table 426,44,45,46,47. A variety of aromatic aldehydes having electron-donating or electron-withdrawing substituents, regardless of their positions, participated well in the reaction, indicating no obvious electronic impact. Substituents such as OMe, F, Cl groups on the phenyl ring (Entries 1, 4, 9, 14, Table 4)26,45,46 were also found to be compatible under standard reaction conditions and did not hamper the reaction processes. Aldehydes bearing several aliphatic groups such as –CH3 afforded the corresponding product in good yields, as well (Entries 7,8, Table 4)26,45. It is noteworthy that not only secondary amines but primary amines reacted well.
On the other hand, to demonstrate the advantages of our catalyst over other catalysts reported in literature for the synthesis of the carbamates and amides, we compared the yield and condition of the reaction with other catalysts, based on the amount of the catalyst, the amount of oxidant and reaction time (Table 5). The present catalyst has the advantages of being green, recoverable, and affords the products in short reaction times and with high yields (Entry 4, Table 5).
The result of comparison between catalytic activity of Cu@modified-chitosan and other catalysts in literature in the reaction between aldehyde 1d and amide 2c forming 3n is reported in Table 626,46,49. It can be clearly seen that our catalyst is superior to other catalysts in terms of time, higher yield, being green, and availability (Entries 1–3, Table 6).
Reusability of the catalyst
The reusability of Cu@modified-chitosan in the synthesis of carbamates and amides was also studied. After each reaction, the catalyst was easily filtered and washed with ethanol to remove any organic impurities. It was then dried at 80 °C and used for the next cycle. As shown in Figure S7 (See ESI file), the catalyst could be reused in five successive reactions without any significant loss of its catalytic activity. The SEM image and FT-IR spectra of the recovered catalyst after the fifth run demonstrated that its structure remained intact (ESI, Figure S8).
In order to evaluate the chelation strength, ICP analysis for the recycled catalyst from the last run of the model reaction was compared to the fresh catalyst. According to the results, copper content of the recycled catalyst after the fifth run was measured as 15.2%, which was slightly less than in the fresh catalyst (15.6%). Taking into account that only 0.4% of copper ions was lost, this can suggest an acceptable enough strong chelation of copper ions by the substrate.
Conclusions
To draw the conclusion, we efficiently synthesized Cu@Sal-Cs catalyst by using the solvent-free ball milling technique. This catalyst was utilized in the C-O and C-N bond formation for the synthesis of carbamates and amides with excellent yields and selectivity. The advantages of this study are the introduction of a green, selective, efficient, and easily recoverable catalyst in the oxidative coupling reactions. This catalyst was easily recovered without loss of its catalytic activity and selectivity. The advantages of our method are the easy separation and reusability of the fabricated catalyst. Moreover, during the reaction, the oxidation-sensitive functional groups (as formyl groups) remain intact. The use of microwave irradiation in the reaction process facilitates the reaction.
Experimental section
Materials and instrumentation
All the reagents and starting materials used in this research were of analytical grade, obtained commercially from Merck or Sigma–Aldrich and used without further purification. The chitosan used was with high molecular weight 600,000 to 800,000 Da. A MM400 Retsch, ball-milling apparatus was used for the solvent-free synthesis reactions. Two stainless steel balls with a diameter of 7 mm were used in a 10 mL stainless steel container at a frequency of 28 Hz. An industrial microwave Milestone MicroSYNTH was used for accelerating of reactions. The progress of the reactions was monitored by TLC (Thin Layer Chromatograghy) or GC (Gas Chromatography) and the actual loading of copper was determined by Inductively Coupled Plasma (ICP) analysis on sequential plasma spectrometer, Shimadzu (ICPS-7000 ver. 2). FT-IR spectra of products were recorded with a Shimadzu 8400s FT-IR spectrometer using potassium bromide pellets. 1H NMR and 13C NMR spectra were recorded with a Bruker DRX-500 Avance spectrometers with Me4Si as the internal standard.
Step by step modification of chitosan biopolymer with salicylaldehyde and copper (II) acetate:
Synthesis of salicylaldehyde-chitosan (modified-chitosan)
The catalyst was synthesized by a similar procedure reported in literature33. Using the ball mill technique and solvent-free process without heating in a short time is the innovation in this study. A mixture of chitosan (2 g) and salicylaldehyde (0.87 mL) was ball milled at 28 Hz, under the solvent-free condition at room temperature. The process was monitored by TLC and completed within 40 min. Then obtained crude was washed with hot ethanol (3*10 mL) and dried at 60 °C to yield modified-chitosan as a yellow powder (Sal-Cs).
Synthesis of Cu@Sal-CS
1 g of the modified-chitosan obtained from the previous step and Cu(OAC)2.H2O (1.5 g) were mixed by using the ball mill for 80 min at room temperature. After the completion of the reaction, the mixture was filtered, washed several times with hot ethanol, and then dried at ambient temperature, to produce the green powder of Cu@Sal-CS (ESI, Figure S9).
One-pot modification of chitosan biopolymer by using salicylaldehyde and copper (II) acetate
In a 10 mL stainless steel ball mill container equipped with two 7 mm stainless steel balls, 2 g of chitosan with 0.87 mL of salicylaldehyde was placed and mixed at a frequency of 28 Hz for 10 min. After that, 1.5 g of Cu(OAC)2 was added to this mixture and milled at the same frequency for 1 h at room temperature to complete. The resulting solid powder was washed several times with hot ethanol, then dried at ambient temperature to yield Cu@Sal-CS as a green powder.
General procedure for synthesis of carbamates through the C–O coupling reaction
A 20-mL round-bottomed flask was charged with 2-substituted phenols or 1–3 dicarbonyl derivative (1 mmol), formamide derivatives (6 mL), Cu@Sal-CS (30 mg), and t-butyl hydroperoxide (TBHP, 70 wt.% in water, 1.5 eq.) as oxidant. The reaction mixture was irradiated under microwave condition for the desired time at 80 °C with the power of 300 Watt. The reaction progress was monitored by TLC or GC techniques. After the completion of the reaction, the catalyst was separated by filtration and the obtained solution was extracted with ethyl acetate by decanter funnel, and washed with water (3*5 mL). The solvent was removed in vacuum and the crude product was purified by plate chromatography or column chromatography on silica gel (Ethyl acetate/n-hexane: 1/9) to afford the pure product.
All of the products were identified by comparison of their melting point and/or spectral data with those reported in previous literature.
General procedure for the synthesis of amides through the C–N coupling reaction
Similar to the oxidative coupling reaction of C-O for the synthesis of carbamates, 30 mg of the catalyst was added to the mixture of amines (1.3 mmol) and aldehyde derivatives (1 mmol) in the presence of TBHP (70 wt.% in water, 1.5 eq.) as oxidant agent. The reaction mixture was subjected to microwave irradiation (80 °C /300 W). The progress of the reaction was monitored by TLC or GC analysis. After the reaction was complete, followed by extraction and purification to yield desired amides. The catalyst was separated by filtration and the reaction mixture was extracted from aqueous mixture with ethyl acetate by decanter funnel, and washed with water (3*5 mL). The solvent was removed in vacuum and the crude product was purified by plate chromatography or column chromatography on silica gel (Ethyl acetate/n-hexane: 1:5) to afford the pure amide. In the case of solid products, recrystallization from ethanol was chosen.
References
Kumar, G. S., Maheswari, C. U., Kumar, R. A., Kantam, M. L. & Reddy, K. R. Copper-catalyzed oxidative C–O coupling by direct C–H bond activation of formamides: synthesis of enol carbamates and 2-carbonyl-substituted phenol carbamates. Angew. Chem. Int. Ed. 50, 11748–11751 (2011).
Dhakshinamoorthy, A., Asiri, A. M. & Garcia, H. Metal–organic frameworks catalyzed C–C and C–heteroatom coupling reactions. Chem. Soc. Rev. 44, 1922–1947 (2015).
Azizi, K., Karimi, M. & Heydari, A. Oxidative coupling of formamides with β-dicarbonyl compounds and the synthesis of 2-aminobenzothiazole using Cu (II)-functionalized Fe3O4 nanoparticles. Tetrahedron Lett. 56, 812–816 (2015).
Verma, S., Baig, R. N., Nadagouda, M. N. & Varma, R. S. Oxidative CH activation of amines using protuberant lychee-like goethite. Sci. Rep. 8, 1–7 (2018).
Vessally, E., Mohammadi, R., Hosseinian, A., Edjlali, L. & Babazadeh, M. Three component coupling of amines, alkyl halides and carbon dioxide: an environmentally benign access to carbamate esters (urethanes). J. CO2 Util. 24, 361–368 (2018).
Chaturvedi, D. Perspectives on the synthesis of organic carbamates. Tetrahedron 1, 15–45 (2012).
Goto, T. et al. The high throughput analysis of N-methyl carbamate pesticides in fruits and vegetables by liquid chromatography electrospray ionization tandem mass spectrometry using a short column. Anal. Chim. Acta 555, 225–232 (2006).
Ray, S. & Chaturvedi, D. Application of organic carbamates in drug design. Part 1: Anti-cancer agents-recent reports. Drugs Fut 29, 343–357 (2004).
Dangerfield, E. M., Timmer, M. S. & Stocker, B. L. Total synthesis without protecting groups: pyrrolidines and cyclic carbamates. Org. Lett. 11, 535–538 (2008).
Greene, T. & Wuts, P. (Wiley, Hoboken, 2002).
Mayer, J. P., Lewis, G. S., Curtis, M. J. & Zhang, J. Solid phase synthesis of quinazolinones. Tetrahedron Lett. 38, 8445–8448 (1997).
Buchstaller, H.-P. Solid phase synthesis of oxazolidinones via a novel cyclisation/cleavage reaction. Tetrahedron 54, 3465–3470 (1998).
Phan, N. T., Nguyen, T. T. & Vu, P. H. A copper metal-organic framework as an efficient and recyclable catalyst for the oxidative cross-dehydrogenative coupling of phenols and formamides. ChemCatChem 5, 3068–3077 (2013).
Prasad, K. R., Suresh, P., Ravikumar, B., Reddy, N. V. & Reddy, K. R. Synthesis of functionalized carbamates and quinones via sequential oxidation of salicylaldehydes using TBHP as the oxidant. Tetrahedron Lett. 55, 6307–6310 (2014).
Ali, W. et al. Copper-catalyzed cross dehydrogenative coupling of N, N-disubstituted formamides and phenols: A direct access to carbamates. Adv. Synth. Catal. 357, 515–522 (2015).
Yoshimura, A., Luedtke, M. W. & Zhdankin, V. V. (Tosylimino) phenyl-λ3-iodane as a reagent for the synthesis of methyl carbamates via hofmann rearrangement of aromatic and aliphatic carboxamides. J. Org. Chem. 77, 2087–2091 (2012).
Lebel, H. & Leogane, O. Curtius rearrangement of aromatic carboxylic acids to access protected anilines and aromatic ureas. Org. Lett. 8, 5717–5720 (2006).
Sawada, D., Sasayama, S., Takahashi, H. & Ikegami, S. A new and facile synthesis of carbamate-and urea-linked glycoconjugate using modified Curtius rearrangement. Tetrahedron Lett. 47, 7219–7223 (2006).
Chaturvedi, D., Kumar, A. & Ray, S. A high yielding one-pot, novel synthesis of carbamate esters from alcohols using Mitsunobu’s reagent. Tetrahedron Lett. 44, 7637–7639 (2003).
Satchell, D. & Satchell, R. Acylation by ketens and isocyanates. A mechanistic comparison. Chem. Soc. Rev. 4, 231–250 (1975).
Sauriat-Dorizon, H. & Guibé, F. Enantioconservative synthesis and ring closing metathesis of disubstituted dialkenic amides. Tetrahedron Lett. 39, 6711–6714 (1998).
Nowick, J. S., Powell, N. A., Nguyen, T. M. & Noronha, G. An improved method for the synthesis of enantiomerically pure amino acid ester isocyanates. J. Org. Chem. 57, 7364–7366 (1992).
Barve, B. D. et al. Copper-catalyzed oxidative coupling of formamides with salicylaldehydes: Synthesis of carbamates in the presence of a sensitive aldehyde group. J. Org. Chem. 79, 3206–3214 (2014).
Barve, B. D. et al. Synthesis of carbamates by direct C–H bond activation of formamides. Eur. J. Org. Chem. 2012, 6760–6766 (2012).
Panahi, L., Naimi-Jamal, M. R., Mokhtari, J. & Morsali, A. Mechanochemically synthesized nanoporous metal-organic framework Cu2 (BDC) 2 (DABCO): An efficient heterogeneous catalyst for preparation of carbamates. Microporous Mesoporous Mater. 244, 208–217 (2017).
Leow, D. Phenazinium salt-catalyzed aerobic oxidative amidation of aromatic aldehydes. Org. Lett. 16, 5812–5815 (2014).
Azizi, K., Karimi, M., Nikbakht, F. & Heydari, A. Direct oxidative amidation of benzyl alcohols using EDTA@ Cu (II) functionalized superparamagnetic nanoparticles. Appl. Catal. A 482, 336–343 (2014).
Bahsis, L. et al. Cellulose-copper as bio-supported recyclable catalyst for the clickable azide-alkyne [3+ 2] cycloaddition reaction in water. Int. J. Biol. Macromol. 119, 849–856 (2018).
Drageset, A. & Bjørsvik, H. R. Synthesis of amides from alcohols and amines through a domino oxidative amidation and telescoped transamidation process. Eur. J. Org. Chem. 2018, 4436–4445 (2018).
Li, G., Ji, C.-L., Hong, X. & Szostak, M. Highly chemoselective, transition-metal-free transamidation of unactivated amides and direct amidation of alkyl esters by N–C/O–C Cleavage. J. Am. Chem. Soc. (2019).
Li, G. et al. Copper (I)-catalyzed dehydrogenative amidation of arenes using air as the oxidant. Adv. Synth. Catal. 357, 1311–1315 (2015).
Goswami, M. & Das, A. M. Synthesis of cellulose impregnated copper nanoparticles as an efficient heterogeneous catalyst for CN coupling reactions under mild conditions. Carbohydr. Polym. 195, 189–198 (2018).
Kumari, S. & Pathak, D. D. Synthesis and development of Chitosan anchored copper (II) Schiff base complexes as heterogeneous catalysts for N-arylation of amines. Tetrahedron Lett. 56, 4135–4142 (2015).
Gawande, M. B. et al. Cu and Cu-based nanoparticles: synthesis and applications in catalysis. Chem. Rev. 116, 3722–3811 (2016).
Chtchigrovsky, M. et al. Functionalized chitosan as a green, recyclable, biopolymer-supported catalyst for the [3+ 2] Huisgen cycloaddition. Angew. Chem. Int. Ed. 48, 5916–5920 (2009).
Kucherov, A., Kramareva, N., Finashina, E., Koklin, A. & Kustov, L. Heterogenized redox catalysts on the basis of the chitosan matrix: 1. Copper complexes. J. Mol. Catal. A Chemical 198, 377–389 (2003).
Hassan, M. A., Omer, A. M., Abbas, E., Baset, W. M. & Tamer, T. M. Preparation, physicochemical characterization and antimicrobial activities of novel two phenolic chitosan Schiff base derivatives. Sci. Rep. 8, 1–14 (2018).
Al-Azmi, A. & Keshipour, S. Cross-linked chitosan aerogel modified with Pd (II)/phthalocyanine: Synthesis, characterization, and catalytic application. Sci. Rep. 9, 1–10 (2019).
Alirezvani, Z., Dekamin, M. G. & Valiey, E. Cu (II) and magnetite nanoparticles decorated melamine-functionalized chitosan: a synergistic multifunctional catalyst for sustainable cascade oxidation of benzyl alcohols/Knoevenagel condensation. Sci. Rep. 9, 1–12 (2019).
Vahid, N. F., Marvi, M. R., Naimi-Jamal, M. R., Naghib, S. M. & Ghaffarinejad, A. X-Fe2O4-buckypaper-chitosan nanocomposites for nonenzymatic electrochemical glucose biosensing. Anal. Bioanal. Electrochem 11, 930–942 (2019).
Koyama, Y. & Taniguchi, A. Studies on chitin X. Homogeneous cross-linking of chitosan for enhanced cupric ion adsorption. J. Appl. Polym. Sci. 31, 1951–1954 (1986).
Kramareva, N., Finashina, E., Kucherov, A. & Kustov, L. Copper complexes stabilized by chitosans: Peculiarities of the structure, redox, and catalytic properties. Kinet. Catal. 44, 793–800 (2003).
Movahed, S. K., Piraman, Z. & Dabiri, M. A nitrogen-doped porous carbon derived from copper phthalocyanines on/in ZIF-8 as an efficient photocatalyst for the degradation of dyes and the CH activation of formamides. J. Photochem. Photobiol., A 351, 208–224 (2018).
Pandey, G., Koley, S., Talukdar, R. & Sahani, P. K. Cross-dehydrogenating coupling of aldehydes with amines/R-OTBS ethers by visible-light photoredox catalysis: Synthesis of amides, esters, and ureas. Org. Lett. 20, 5861–5865 (2018).
Yang, S. et al. Copper-catalyzed dehydrogenative reaction: Synthesis of amide from aldehydes and aminopyridine. Tetrahedron 69, 6431–6435 (2013).
Wang, J., Li, J., Xu, F. & Shen, Q. Anionic bridged bis (amidinate) lithium lanthanide complexes: efficient bimetallic catalysts for mild amidation of aldehydes with amines. Adv. Synth. Catal. 351, 1363–1370 (2009).
Yazdani, E. et al. A magnetically recoverable copper–salen complex as a nano-catalytic system for amine protection via acetylation using thioacetic acid. Res. Chem. Intermed. 45, 1775–1793 (2019).
Sharma, R., Dutta, S. & Sharma, S. Quinoline-2-carboimine copper complex immobilized on amine functionalized silica coated magnetite nanoparticles: a novel and magnetically retrievable catalyst for the synthesis of carbamates via C-H activation of formamides. Dalton Trans. 44, 1303–1316 (2015).
Inagawa, H., Uchida, S., Yamaguchi, E. & Itoh, A. Metal‐free oxidative amidation of aromatic aldehydes using an anthraquinone‐based organophotocatalyst. Asian J. Org. Chem. (2019).
Acknowledgements
We acknowledge Iran University of Science and Technology and Iran National Science Foundation for partial financial support of this work.
Author information
Authors and Affiliations
Contributions
The work is a part of the M.Sc. thesis of M. A. She made the experiments, collected the data, and wrote the main manuscript text. M. R. N.-J. was her supervisor and made corrections to the text and the scientific discussion. L. P., as a Ph.D. candidate, has made a substantial contribution in the design of the work, interpretation of data, and preparation of the draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Asadi, M., Naimi-Jamal, M.R. & Panahi, L. Green synthesis of carbamates and amides via Cu@Sal-Cs catalyzed C–O and C–N oxidative coupling accelerated by microwave irradiation. Sci Rep 11, 18105 (2021). https://doi.org/10.1038/s41598-021-97554-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-97554-3
This article is cited by
-
Metal–Organic Frameworks (MOFs): The Next Generation of Materials for Catalysis, Gas Storage, and Separation
Journal of Inorganic and Organometallic Polymers and Materials (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
