
Abstract
In this research, an efficient heterogeneous catalyst based on graphitic carbon nitride nanosheets (CN) has been reported. The CN was functionalized by 1,3-dibromopropane as a linker (CN–Pr–Br) and subsequently modified with metformin (CN–Pr–Met). Furthermore, the copper(II) was coordinated on modified CN (CN–Pr–Met–Cu(II)) and during this process, 7.94% copper(II) was loaded into the catalyst structure. The synthesized catalyst was evaluated by various techniques including fourier-transform infrared spectroscopy (FT-IR), energy dispersive X-ray spectroscopy (EDS), field emission scanning electron microscopy (FE-SEM), thermogravimetric analysis (TGA), X-ray diffraction (XRD), and inductively coupled plasma atomic emission spectroscopy (ICP-OES). CN–Pr–Met–Cu(II) was used as a catalyst in the synthesis of amides via the oxidation of benzyl alcohols. The conditions of this reaction were optimized in terms of temperature, time, amount of catalyst, type of base, oxidant, and solvent. Moreover, a variety of amides with an efficiency of 75–95% were synthesized. The reaction was carried out in the presence of benzyl alcohols, amine hydrochloride salts, tert-butyl hydroperoxide (TBHP), CaCO3, and CN–Pr–Met–Cu(II) at 80 °C of acetonitrile solvent. The synthesized catalyst can be easily separated from the reaction medium and reused for 7 consecutive runs without a significant reduction in reaction efficiency.
Similar content being viewed by others


Introduction
Carbon-based nanomaterials including carbon nanotubes, graphene, and graphitic carbon nitride (g-C3N4) have become important topics in a variety of scientific fields in recent years1,2. Among these materials, the g-C3N4 with features such as high chemical and thermal stability, non-toxicity, cost-effectiveness, and easy synthesis has been considered in organic synthesis and catalyst1, supercapacitor3, biosensor4, environment5, energy6, and medical applications6. The tri-s-triazine units in the structure of the g-C3N4 are one of the reasons for catalytic applications of g-C3N47. In this regard, various forms of g-C3N4 including bulk8, nanosheets9, mesoporous10, quantum dot11, and nanotubes have been reported12. Recently, g-C3N4 in the form of nanosheets has been functionalized covalently or non-covalently by various molecules to improve its properties and applications13. Functionalization of g-C3N4 is usually accomplished by oxidation/carboxylation, amidation, sulfonation, phosphorylation, polymer grafting, electrostatic interaction, π–π interaction, and hydrogen bonding13. Also, various molecules including L-arginine9, ferrocene14, thiamine15, 1,4-butane sultone16, 5-bromovaleryl chloride17, 1, 3-dibromopropane have been used to functionalized g-C3N415. Metformin (dimethyl biguanide) as a polydentate ligand can be also useful. Metformin is an effective drug for type 2 diabetics that lowers blood glucose levels18. This molecule has been used as auxiliary medicine for cancer19, aging20, and covid-1921. Metformin can form chelates with transition metals such as copper, nickel, palladium, and cobalt for catalytic applications22,23,24,25,26,27. In a study performed in 2020 by Hamed and Ali, Cu(II)–metformin was immobilized on graphene oxide as an efficient catalyst for the Beckmann rearrangement23. In another study performed in 2016 by Kojoori, palladium(II) was coordinated on metformin-SBA-15 and used as a catalyst in the partial hydrogenation of alkynes26.
Amides are an important functional group found extensively in pharmacology such as acetaminophen, cephalexin, lidocaine, diazepam, and diperodon. Also, they were used in the synthesis of polymeric materials, such as nylon, hydrogels, artificial silks, and supported catalysts. Various methods, include Staudinger reaction28, Beckmann rearrangement29, Schmidt reactions30, acylation of amines31, hydroamination of alkynes32, transamidation33, have been used to produce amides. Synthesis of amide by the mentioned methods has disadvantages such as high cost, producing a lot of wastes, low efficiency, and the use of toxic reagents. Therefore, alternative methods such as oxidative amidation of aldehydes or alcohols have been recommended34. Tandem oxidative amidation of benzylic alcohols was catalyzed by a series of transition metals35,36,37,38. Among these metals, copper(II) with low price and high biocompatibility has been considered efficient catalysts for the amidation of alcohols39,40. In this study, CN–Pr–Met–Cu(II) was reported as a recyclable and efficient catalyst for the one-pot conversion of benzylic alcohols to amides. The prepared catalyst contains 7.94% copper(II) and the synthesis of amides was greatly facilitated by this method. Also, this catalyst was used 7 times in the tandem oxidative amidation of benzylic alcohols without significantly reducing the efficiency of the products.
Experimental
Reagents and instruments
All raw materials and solvents used in this study were produced by Merck and Flucka companies and no additional purification process was performed on them. Thin Layer Chromatography (TLC) sheets containing 0.2 mm of F254 silica gel on the aluminum plates were utilized to control the reaction progress. The melting point was investigated using the Electrothermal 9100 apparatus to confirm the accuracy of products formation. FT-IR spectroscopy was used to investigate the catalyst fabrication process and confirm the formation of reaction products. This analysis was accomplished via an AVATAR device manufactured by Thermo in the range 400–4000 cm−1 and by KBr pellets. XRD technique was performed using PANalytical X-PERT-PRO MPD apparatus in the range of 2θ, 5° to 10° and 10° to 90° to study the crystal structures. In this analysis, Cu Kα radiation source with λ = 1.5406 Å and 2θ step size with 0.02° were utilized. FE-SEM and EDS analysis were used to study the catalyst morphology and review the catalyst components respectively, by EM8000 KYKY apparatus. TGA was done by STA504 device to verify the correctness of catalyst synthesis and its thermal stability in the temperature range of 20 °C to 1000 °C at a rate of 5 °C/min under Ar atmosphere. ICP-OES was performed using VISTA-PRO to check the recyclability and reusability of the catalyst. Preparative thin-layer chromatography (PTLC) was used to purify the reaction products, for this purpose, 20 × 20 glass plates with silica gel coating with a thickness of 0.5 mm were used. Carbon and hydrogen nuclear magnetic resonance (NMR) spectroscopy were used to evaluate the structure of some products and for this purpose VARIAN Inova 500 MHz and 125 MHz devices were used.
Preparation of bulk g-C3N4
Bulk g-C3N4 was prepared according to an article published by Zheng et al.41. 10 g of melamine powder was heated in a furnace at 550 °C for 4 h under air atmosphere. Then, the obtained yellow powder was ground for further usage.
Preparation of CN
The procedure utilized for preparation CN was introduced by Tajik et al.9. Initially, 2 g of as-prepared bulk g-C3N4 and 40 mL of sulfuric acid were poured into a 600 mL beaker and the mixture was stirred for 5 h at 90 °C. Then, 400 mL of ethanol was added to the stirring solution and the resulting mixture was stirred at room temperature for 2 h. After the mentioned time, the beaker was remained stationary for 48 h until the precipitate settles well. Subsequently, decantation was done and the solution was sonicated for 6 h. Finally, the precipitate was separated by centrifugation and washed with water and ethanol. The CN sheets were dried in a 60 °C oven for 24 h.
Preparation of CN–Pr–Br
Connection of linker to CN was performed using the method reported by Rashidizadeh et al.15. 40 mL of dry toluene and 1 g of synthesized CN in the previous step were inserted into a 50 mL round bottom flask and placed in an ultrasonic bath for 1 h. Then, 2 mL of 1,3-dibromopropane and 1 mmol NaI were added to the mixture and refluxed for 24 h under an N2 atmosphere. The functionalized precipitate was separated by centrifugation and washed several times with 1 L ethanol and 0.5 L ethyl acetate to completely remove unreacted 1,3-dibromobutane from the CN surface. The final precipitate was dried overnight in an oven at 60 °C to remove residual solvent.
Preparation of CN–Pr–Met
2.5 g of metformin hydrochloride and 3 mL of acetonitrile were poured in a round bottom flask and mixed for 30 min via a magnetic stirrer. Then 0.6 g of NaOH was added to the mixture and stirred for 1 h at room temperature. In the next step, 5 g of KI was added to the reaction mixture under extreme stirring conditions. After 30 min, 3 g of CN–Pr–Br was added to the mixture and the reaction was refluxed for 12 h. The final product was separated by centrifuge, washed with 0.5 L of methanol and 0.5 L of distilled water, and subsequently dried in an oven at 60 °C.
Preparation of CN–Pr–Met–Cu(II)
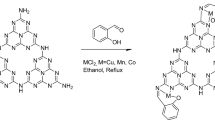
First, 0.5 g of CN–Pr–Met was dispersed in 40 mL of DMF. Then 0.5 g of copper(II) acetate hydrate salt was added to the solution and refluxed for 6 h. The final catalyst was collected by centrifuge and after washing with 200 mL of distilled water and 50 mL ethanol, dried in a 60 °C oven for 12 h. The catalyst synthesis procedure was shown in Fig. 1.

Graphical illustration of CN–Pr–Met–Cu(II) catalyst synthesis.
General procedure for tandem oxidative amidation of benzylic alcohols via amine hydrochloride salts
1 mmol of amine hydrochloride salt, 1.5 mmol of benzyl alcohol derivatives, 20 mg of catalyst, 3 mL of acetonitrile, 1.1 equivalents of calcium carbonate, and 4 equivalents of TBHP are mixed. The reaction mixture was stirred under atmospheric nitrogen for 3 h at 80 °C and the reaction progress was controlled by TLC. After this time, the catalyst was collected by using filter paper, and on the other hand, the filtrate was extracted with water and dichloromethane. The amide precipitates in dichloromethane were formed by using the anti-solvent crystallization method. Finally, the precipitate was purified using PTLC.
Result and discussion
In this research, a novel heterogeneous catalyst was synthesized via functionalization of CN for the tandem oxidative amidation of benzylic alcohols. Figure 1 was shown the preparation of CN–Pr–Met–Cu(II). Initially, bulk g-C3N4 was converted to a CN by chemical exfoliation procedure. In the next step, during a nucleophilic reaction, 1,3-dibromopropane as a linker was attached to the CN. Subsequently, metformin was connected to the linker by a covalent bond, and copper(II) was coordinated to metformin and CN. The as-prepared catalyst was used for the amide synthesis reaction by oxidation of benzylic alcohols in the presence of amine hydrochloride salts. The reaction was investigated in the presence of benzylic alcohol derivatives with electron donor and withdrawing groups as well as different types of amine salts. Furthermore, various analyzes include FT-IR, EDS, XRD, TGA, FE-SEM, and ICP-OES were used to confirm the structure of the catalyst.
Catalyst characterizations
FT-IR analysis
FT-IR spectrum of bulk g-C3N4 was shown in Fig. 2a. a broad peak was observed in the range of 2800 cm−1 to 3640 cm−1, which corresponds to the N–H groups of the molecule, including the –NH2 and =NH groups42. Strong peaks in the region 1240 cm−1 to 1640 cm−1 were identified due to the stretching vibrations of heterocyclic C–N and C=N respectively, and the peak observed in 808 cm−1 was related to triazine units42. Figure 2b was described the CN. Approximately all peaks in the FT-IR spectrum of bulk g-C3N4 are also present in the CN, but the slight difference in their intensity is due to the protonation of bulk g-C3N4 by H2SO443,44. In Fig. 2c, alongside the absorbance bands of the g-C3N4, the peak appears in the range of 2800 cm−1 to 3000 cm−1 was associated with the stretching vibration of the aliphatic C–H in the linker9. Figure 2d illustrates the functionalization of CN–Pr–Br by metformin. The peak in 2940 cm−1 was demonstrated the stretching vibration of the C–H in the CH3 group of the metformin23. Moreover, the peak that observes approximately in 1658 cm−1 and 1580 cm−1 may be related to the stretching vibration of the C=NH and N–H respectively45. According to Fig. 2e which was shown the CN–Pr–Met–Cu(II), the C=NH peak of the metformin shifted to the lower frequencies of the spectrum and proves the coordination of copper(II) to metformin.

FT-IR spectrum of (a) bulk g-C3N4, (b) CN, (c) CN–Pr–Br, (d) CN–Pr–Met and (e) CN–Pr–Met–Cu(II).
EDS analysis
EDS analysis from different stages of catalyst synthesis was shown in Fig. 3. Based on Fig. 3a,b, the presence of carbon and nitrogen in the structure of the CN and its functionalization by 1,3-dibromopropane via the presence of bromine was confirmed. Figure 3c has shown the EDS analysis of CN functionalized by metformin. Due to the similar elements in the structure of metformin and CN, carbon and nitrogen are visible in the EDS spectrum, and the remarkable point is the removal of bromine, which confirms the binding of metformin to the CN–Pr. In Fig. 3d, the presence of C, N and Cu elements was confirmed in the CN-Pr–Met–Cu(II). In addition, EDS mapping of CN–Pr–Met–Cu(II) demonstrated excellent dispersion of C, N, and Cu elements in the structure (Fig. 3e–g).

EDS spectrum of (a) CN, (b) CN–Pr–Br, (c) CN–Pr–Met, (d) CN–Pr–Met–Cu(II) and (e–g) elemental mapping of the CN–Pr–Met–Cu(II).
FE-SEM imaging
FE-SEM imaging was evaluated in Fig. 4 to study the morphology of synthesized CN and CN–Pr–Met–Cu(II). According to Fig. 4a,b, the sheets in the structure of CN and change during the functionalization process, suggesting that surface of the CN sheets were covered by synthesized ligand.

FE-SEM image of (a) CN and (b) CN–Pr–Met–Cu(II).
XRD analysis
The XRD pattern of the synthesized CN–Pr–Met–Cu(II) was shown in Fig. 5a. Peaks related to CN, metformin, and Cu(II) acetate hydrate were observed in the XRD pattern of the prepared catalyst. XRD pattern of bulk g-C3N4 was shown in Fig. 5b. The peak observed in 2θ about 26.8° is related to the interplanar stacking of the conjugated aromatic systems for (002) reflection. Also, the observed peak in 2θ about 13.1° corresponds to the in-plane structural packing motif of repeated tri-s-triazine units for (100) reflection15,43. Comparing the XRD pattern in Fig. 5b,c shows that bulk g-C3N4 has been converted to CN. After exfoliation, the (002) peak intensity was decreased, and on the other hand, this peak appeared at a slightly higher diffraction angle15. Moreover, the peak at 2θ about 13.1° has almost disappeared. These observations are due to the shortening of the interplanar stacking space in CN15. Observation of all metformin peaks at 2θ around 12.2°, 17.7°, 22.4°, 24.5°, 31.1°, and 39.4° can confirm the maintenance of the crystalline structure of the metformin in the final catalyst (Fig. 5d)46. Cu(II) acetate hydrate peaks are also visible in the final CN–Pr–Met–Cu(II) pattern in 2θ around 12.75°, 14.2°, 15.37°, 15.53°, 16.37°, 25.9, 37.81°, 38.82°, 39.4°, 40.79°,46.35°, 47.37°, and 48.16° (Fig. 5e).

XRD pattern of (a) prepared CN–Pr–Met–Cu(II), (b) bulk g-C3N4, (c) CN, (d) Metformin, and (e) Cu(II) acetate hydrate. (f) TG analysis of the CN–Pr–Met–Cu(II) and CN.
TG analysis
The thermal stability of the catalyst and CN were investigated by TG analysis in Fig. 5f. The synthesized catalyst was stable up to 400 °C, although its partial mass loss in the range of 50 °C to 100 °C was due to the evaporation of adsorbed water or air contaminants collected by the solids. The observed mass reduction in the range of 100 °C to about 500 °C can be related to the destruction of organic parts and thermal deformation/collapse of the CN23,47. The CN–Pr–Met–Cu(II) main mass loss occurred at 500 °C to 700 °C, which was related to the thermal decomposition or deformation of heptazine units in the structure of the CN47. The CN–Pr–Met–Cu(II) has higher thermal stability than CN, and this may be due to the synergistic effect and supramolecular interactions between carbon nitride and other components of the catalyst48.
Catalytic properties
The catalytic performance of CN–Pr–Met–Cu(II) was explored for the tandem oxidative amidation of benzylic alcohols. In this regard, optimization was performed to evaluate the best conditions for the reaction (Table 1). Initially, the reaction was performed in the presence of 20 mg of CN–Pr–Met–Cu(II) catalyst, with 1.1 equivalent CaCO3 as a base and 4 equivalent TBHP as oxidant at ambient temperature under inert N2 atmosphere. In this case, 35% yield of the product was observed (Table 1, entry 1). Then, the effect of temperature, solvent, oxidant, base, the amount of catalyst, and time were examined. Temperatures from 25 to 120 °C were tested (Table 1, entries 1–6). With the increasing temperature up to 80 °C, the reaction efficiency increases, but in the next step with increasing temperature up to 120 °C, a decrease in reaction efficiency was observed. This observation is due to the oxidation of benzyl alcohols to benzoic acid49. In addition, the reaction was checked in different amounts of CN–Pr–Met–Cu(II) and it was observed that the optimized amount of catalyst was 20 mg (Table 1, entries 4, 8–9). Subsequently, the reaction was checked in the presence of CaCO3, K2CO3, and Na2CO3 as a base and the CaCO3 has the best performance (Table 1, entries 4, 10–11). Low solubility and weak basicity of CaCO3 caused slow deprotonation of the amine hydrochloride salt. Bases such as K2CO3 and Na2CO3, by rapid deprotonation of the amine hydrochloride salt and the immediate production of free amines, cause the formation of undesirable intermediates (imine) instead of the intermediate (IV) (Fig. 6). Therefore, the use of K2CO3 and Na2CO3 reduced the reaction yield50. The effect of solvents (Table 1, entries 4, 12–13) and oxidants (Table 1, entries 4, 14–15) was evaluated and the best result was provided in acetonitrile solvent by TBHP. Then for optimization reaction time, the reaction was carried out in 2 to 6 h, and the best efficiency was provided in about 3 h (Table 1, entries 4, 16–18). The reaction did not carry out by removing each of the catalyst, oxidant, and base (Table 1, entries 19–21). In addition in Table 2, to confirm the performance synthesized catalyst and the effect of its components in the reaction, CN (Table 2, entry 1), copper(II) acetate hydrate (Table 2, entry 2), metformin (Table 2, entry 3), a mixture of metformin and copper(II) acetate hydrate (1:1) (Table 2, entry 4), a mixture of CN and copper(II) acetate hydrate (1:1) (Table 2, entry 5), CN–Pr–Br (Table 2, entry 6), CN–Pr–Met (Table 2, entry 7), and CN–Pr–Met–Cu(II) (Table 2, entry 8) were added separately to the reaction as a catalyst and the efficiency of the obtained product was evaluated. As predicted, CN–Pr–Met–Cu(II) catalyst had a higher ability to perform the reaction. Based on the results, it was determined that the main component to catalyze the reaction is copper ions. Table 3 summarizes the products synthesized by CN–Pr–Met–Cu(II) catalyst. The formation of products was confirmed by melting point. Moreover, FT-IR, 1H-NMR and 13C-NMR of selected amides are shown in the supplementary information file (Figs. S1–S6).

Proposed mechanism for tandem oxidative amidation reaction performed using the novel CN–Pr–Met–Cu(II) catalyst.
Various benzylic alcohols and amines were used to synthesize amides via a tandem oxidative amidation reaction. In this regard, benzyl alcohols with donor groups (CH3, and OCH3) and withdrawing groups (Cl, Br, and NO2), respectively increase and decrease the product’s efficiency. Benzylic alcohols with electron-withdrawing groups; caused the stability of the radical intermediate during the conversion of alcohol to aldehydes and reduce aldehyde production. On the other hand, electron donor groups on the benzylic alcohols made the intermediate unstable and increases its conversion to aldehydes (Fig. 7)51. In addition, the efficiency was not significantly different in the presence of aromatic and aliphatic amines.

The effect of electron-withdrawing and electron-donating groups on aldehyde formation.
According to the previous studies and observed results, the radical mechanism has been proposed for this reaction (Fig. 6)65. In step 1, intermediate (II) was formed during the separation of a hydrogen from benzyl alcohol (I). In step 2, intermediate (II) was converted to an aldehyde (III) by losing hydrogen and aldehyde formation was confirmed using TLC66. In step 3, the aldehyde reacts with the amine in the presence of CaCO3 and forms the hemiaminal intermediate (IV)67. Subsequently in step 4, tert-butylperoxyl radical was produced by the reaction of the catalyst with TBHP oxidant. The active tert-butylperoxyl radical separates the hydrogen from the hemiaminal (IV) and formed the intermediate (V). Finally, in step 5, the desired amide (VI) was formed via oxidation of intermediate (V). A radical scavenger was used to confirm the radical mechanism. In this regard, 1 equivalent of 2,6-di-t-butyl-4-methylphenol (BHT) was added to the model reaction and no product was formed49.
In addition, the reusability of the CN–Pr–Met–Cu(II) was evaluated under the optimized reaction. The efficiency of the catalytic reaction did not decrease significantly after seven reaction cycles (Fig. 8a). Also, FT-IR, FE-SEM, and XRD results indicated that the structure of the catalyst has not changed (Fig. 8b–d). Catalyst leaching was also evaluated and according to the obtained results from ICP-OES analysis, Cu% decreased from 7.94 to 7.88% after seven cycles.

(a) Recyclability of the CN–Pr–Met–Cu(II) in tandem oxidative amidation of benzyl alcohol with benzylamine hydrochloride salt. (b) FT-IR, (c) XRD analysis of fresh and used CN–Pr–Met–Cu(II). (d) FE-SEM image of used CN–Pr–Met–Cu(II).
Following the investigation of the catalyst properties from different points of view, a comparison was made between the performance of the synthesized catalyst and the catalysts reported in the literature. According to Table 4, CN–Pr–Met–Cu(II) was acceptable in terms of efficiency, recyclability, and reaction time.
Conclusion
In this study, we synthesized a promising heterogeneous catalyst made of CN, metformin, 1,3-dibromopropane, and copper(II) acetate hydrate. The CN–Pr–Met–Cu(II) catalyst exhibited distinguished catalytic activity for tandem oxidative amidation of benzylic alcohols in the presence of amine hydrochloride salt. Amides with good to excellent yields were synthesized in 3 h with 0.02 g of CN–Pr–Met–Cu(II) catalyst. Moreover, the fabricated catalyst showed high thermal resistance, and about 40% of the catalyst weight was maintained up to 1000 °C. The recyclability of CN–Pr–Met–Cu(II) catalyst is a remarkable feature that could be reused 7 times without a significant reduction in its efficiency.
References
Wen, J., Xie, J., Chen, X. & Li, X. A review on g-C3N4-based photocatalysts. Appl. Surf. Sci. 391, 72–123 (2017).
Nagal, V. et al. Emerging Applications of g-C3N4 films in Perovskite-based solar cells. ECS J. Solid State Sci. Technol. 10, 065001 (2021).
Chang, X. et al. MnO2/g-C3N4 nanocomposite with highly enhanced supercapacitor performance. Nanotechnology 28, 135705 (2017).
Zou, J. et al. An ultra-sensitive electrochemical sensor based on 2D g-C3N4/CuO nanocomposites for dopamine detection. Carbon 130, 652–663 (2018).
Patnaik, S., Sahoo, D. P. & Parida, K. An overview on Ag modified g-C3N4 based nanostructured materials for energy and environmental applications. Renew. Sustain. Energy Rev. 82, 1297–1312 (2018).
Ren, Y., Zeng, D. & Ong, W. J. Interfacial engineering of graphitic carbon nitride (g-C3N4)-based metal sulfide heterojunction photocatalysts for energy conversion: A review. Chin. J. Catal. 40, 289–319 (2019).
Bahuguna, A., Kumar, A., Kumar, S., Chhabra, T. & Krishnan, V. 2D–2D nanocomposite of MoS2-graphitic carbon nitride as multifunctional catalyst for sustainable synthesis of C3-functionalized indoles. ChemCatChem 10, 3121–3132 (2018).
Xiao, J. et al. Super synergy between photocatalysis and ozonation using bulk g-C3N4 as catalyst: A potential sunlight/O3/g-C3N4 method for efficient water decontamination. Appl. Catal. B Environ. 181, 420–428 (2016).
Tajik, Z., Ghafuri, H., Ghanbari, N. & Hanifehnejad, P. Preparation and characterization Of g-C3N4@ L-arginine as a highly-efficient and recyclable catalyst for the synthesis of 1, 4-dihydropyridine, 4H-chromene, and 2, 3-dihydro quinazoline derivatives. Sci. Rep. 11, 1–14 (2021).
Yang, H. et al. Effect of mesoporous g-C3N4 substrate on catalytic oxidation of CO over Co3O4. Appl. Surf. Sci. 401, 333–340 (2017).
Zarei, M., Mohammadzadeh, I., Saidi, K. & Sheibani, H. g-C3N4 quantum dot decorated MoS2/Fe3O4 as a novel recoverable catalyst for photodegradation of organic pollutant under visible light. J. Mater. Sci. Mater. Electron. 32, 26213–26231 (2021).
Jiang, Z., Zhang, X., Chen, H. S., Hu, X. & Yang, P. Formation of g-C3N4 nanotubes towards superior photocatalysis performance. ChemCatChem 11, 4558–4567 (2019).
Majdoub, M., Anfar, Z. & Amedlous, A. Emerging chemical functionalization of g-C3N4: Covalent/noncovalent modifications and applications. ACS Nano 14, 12390–12469 (2020).
Ye, X., Zheng, Y. & Wang, X. synthesis of ferrocene-modified carbon nitride photocatalysts by surface amidation reaction for phenol synthesis. Chin. J. Chem. 32, 498–506 (2014).
Rashidizadeh, A., Ghafuri, H., Esmaili Zand, H. R. & Goodarzi, N. Graphitic carbon nitride nanosheets covalently functionalized with biocompatible vitamin B1: Synthesis, characterization, and its superior performance for synthesis of quinoxalines. ACS Omega 4, 12544–12554 (2019).
Rahmati, M., Ghafuri, H., Ghanbari, N. & Tajik, Z. 1,4-butanesultone functionalized graphitic carbon nitride: efficient catalysts for the one-pot synthesis of 1,4-dihydropyridine and polyhydroquinoline derivative through hantzsch reaction. Polycycl. Aromat. Compd. https://doi.org/10.1080/10406638.2020.1852583 (2020).
Chen, X. et al. A bio-inspired strategy to enhance the photocatalytic performance of g-C3N4 under solar irradiation by axial coordination with hemin. Appl. Catal. B Environ. 201, 518–526 (2017).
Chen, H., Lei, M. & Hu, L. Synthesis of 1-aryl indoles via coupling reaction of indoles and aryl halides catalyzed by CuI/metformin. Tetrahedron 70, 5626–5631 (2014).
Morales, D. R. & Morris, A. D. Metformin in cancer treatment and prevention. Annu. Rev. Med. 66, 17–29 (2015).
Soukas, A. A., Hao, H. & Wu, L. Metformin as anti-aging therapy: Is it for everyone? Trends Endocrinol. Metab. 30, 745–755 (2019).
Sharma, S., Ray, A. & Sadasivam, B. Metformin in COVID-19: A possible role beyond diabetes. Diabetes Res. Clin. Pract. 164, 108183 (2020).
Fortun, S., Beauclair, P. & Schmitzer, A. R. Metformin as a versatile ligand for recyclable palladium-catalyzed cross-coupling reactions in neat water. RSC Adv. 7, 21036–21044 (2017).
Hamed, A. S. & Ali, E. M. Cu(II)–metformin immobilized on graphene oxide: An efficient and recyclable catalyst for the Beckmann rearrangement. Res. Chem. Intermed. 46, 701–714 (2020).
Yang, P., Ma, R. & Bian, F. Palladium supported on metformin-functionalized magnetic polymer nanocomposites: A highly efficient and reusable catalyst for the suzuki–miyaura coupling reaction. ChemCatChem 8, 3746–3754 (2016).
Veisi, H., Mirshokraie, S. A. & Ahmadian, H. Synthesis of biaryls using palladium nanoparticles immobilized on metformine-functionalized polystyrene resin as a reusable and efficient nanocatalyst. Int. J. Biol. Macromol. 108, 419–425 (2018).
Kojoori, R. K. Synthesis of highly selective nano-structured functionalized SBA-15 metformin palladium composite catalyst in partialal hydrogenation of alkynes. J. Chil. Chem. Soc. 61, 3144–3149 (2016).
Raoufi, F., Monajjemi, M., Aghaei, H., Zare, K. & Ghaedi, M. Preparation, characterization and first application of graphene oxide-metformin-nickel for the suzuki cross-coupling reaction. ChemistrySelect 5, 211–217 (2020).
Köhn, M. & Breinbauer, R. Die Staudinger-Ligation–ein Geschenk für die chemische biologie. Angew. Chem. 116, 3168–3178 (2004).
Owston, N. A., Parker, A. J. & Williams, J. M. Highly efficient ruthenium-catalyzed oxime to amide rearrangement. Org. Lett. 9, 3599–3601 (2007).
Gololobov, Y. G. & Kasukhin, L. F. Recent advances in the Staudinger reaction. Tetrahedron 48, 1353–1406 (1992).
Choudary, B. M., Bhaskar, V., Kantam, M. L., Rao, K. K. & Raghavan, K. V. Acylation of amines with carboxylic acids: The atom economic protocol catalysed by Fe (III)-montmorillonite. Catal. Lett. 74, 207–211 (2001).
Severin, R. & Doye, S. The catalytic hydroamination of alkynes. Chem. Soc. Rev. 36, 1407–1420 (2007).
Allen, C. L., Atkinson, B. N. & Williams, J. M. Transamidation of primary amides with amines using hydroxylamine hydrochloride as an inorganic catalyst. Angew. Chem. Int. Ed. 51, 1383–1386 (2012).
Kamble, R. B. et al. Ti-superoxide catalyzed oxidative amidation of aldehydes with saccharin as nitrogen source: Synthesis of primary amides. RSC Adv. 10, 724–728 (2020).
Oldenhuis, N. J., Dong, V. M. & Guan, Z. Catalytic acceptorless dehydrogenations: Ru-Macho catalyzed construction of amides and imines. Tetrahedron 70, 4213–4218 (2014).
Watson, A. J., Maxwell, A. C. & Williams, J. M. Ruthenium-catalyzed oxidation of alcohols into amides. Org. Lett. 11, 2667–2670 (2009).
Soulé, J. F., Miyamura, H. & Kobayashi, S. Powerful amide synthesis from alcohols and amines under aerobic conditions catalyzed by gold or gold/iron,-nickel or-cobalt nanoparticles. J. Am. Chem. Soc. 133, 18550–18553 (2011).
Owston, N. A., Parker, A. J. & Williams, J. M. Iridium-catalyzed conversion of alcohols into amides via oximes. Org. Lett. 9, 73–75 (2007).
Azizi, K., Karimi, M., Nikbakht, F. & Heydari, A. Direct oxidative amidation of benzyl alcohols using EDTA@ Cu(II) functionalized superparamagnetic nanoparticles. Appl. Catal. A Gen. 482, 336–343 (2014).
Bantreil, X., Fleith, C., Martinez, J. & Lamaty, F. Copper-catalyzed direct synthesis of benzamides from alcohols and amines. ChemCatChem 4, 1922–1925 (2012).
Zheng, Y., Lin, L., Ye, X., Guo, F. & Wang, X. Helical graphitic carbon nitrides with photocatalytic and optical activities. Angew. Chem. 126, 12120–12124 (2014).
Wang, Q., Guan, S. & Li, B. 2D graphitic-C3N4 hybridized with 1D flux-grown Na-modified K2Ti6O13 nanobelts for enhanced simulated sunlight and visible-light photocatalytic performance. Catal. Sci. Technol. 7, 4064–4078 (2017).
Zou, L. R. et al. A facile and rapid route for synthesis of gC3N4 nanosheets with high adsorption capacity and photocatalytic activity. RSC Adv. 6, 86688–86694 (2016).
Tong, J. et al. Rapid and high-yield production of gC3N4 nanosheets via chemical exfoliation for photocatalytic H2 evolution. RSC Adv. 5, 88149–88153 (2015).
Hemmati, S., Mehrazin, L., Pirhayati, M. & Veisi, H. Immobilization of palladium nanoparticles on metformin-functionalized graphene oxide as a heterogeneous and recyclable nanocatalyst for Suzuki coupling reactions and reduction of 4-nitrophenol. Polyhedron 158, 414–422 (2019).
An, K., Guan, L., Kang, H. & Tian, D. Zipper-like thermosensitive molecularly imprinted polymers based on konjac glucomannan for metformin hydrochloride. Iran. Polym. J. 30, 331–342 (2021).
Elshafie, M., Younis, S. A., Serp, P. & Gad, E. A. Preparation characterization and non-isothermal decomposition kinetics of different carbon nitride sheets. Egypt. J. Pet. 29, 21–29 (2020).
Shi, Y. et al. Influence of g-C3N4 nanosheets on thermal stability and mechanical properties of biopolymer electrolyte nanocomposite films: A novel investigation. ACS Appl. Mater. Interfaces 6, 429–437 (2014).
Rashidizadeh, A., Ghafuri, H., Goodarzi, N. & Azizi, N. Tandem oxidative amidation of alcohols catalyzed by copper modified well-ordered mesoporous graphitic carbon nitride. Solid State Sci. 109, 106427 (2020).
Ghosh, S. C. et al. Iron-catalyzed efficient synthesis of amides from aldehydes and amine hydrochloride salts. Adv. Synth. Catal. 354, 1407–1412 (2012).
da Silva, G. & Bozzelli, J. W. Benzoxyl radical decomposition kinetics: Formation of benzaldehyde+ H, phenyl+ CH2O, and benzene+ HCO. J. Phys. Chem. A 113, 6979–6986 (2009).
Yu, P., Wang, Y., Zeng, Z. & Chen, Y. Metal-free C-N or C–C bond cleavages of α-azido ketones: An oxidative-amidation strategy for the synthesis of α-ketothioamides and amides. J. Org. Chem. 84, 14883–14891 (2019).
Feng, C., Yin, G., Yan, B., Chen, J. & Ji, M. Convenient synthesis of amides by Zn(ClO4)2·6H2O catalysed Ritter reaction with nitriles and halohydrocarbons. J. Chem. Res. 42, 383–386 (2018).
Zhu, L. et al. Carbon–carbon bond formation of trifluoroacetyl amides with grignard reagents via C (O)–CF3 bond cleavage. J. Org. Chem. 84, 5635–5644 (2019).
Manasa, K. L., Tangella, Y., Krishna, N. H. & Alvala, M. A metal-free approach for the synthesis of amides/esters with pyridinium salts of phenacyl bromides via oxidative C-C bond cleavage. Beilstein J. Org. Chem. 15, 1864–1871 (2019).
Sharley, D. D. & Williams, J. M. Acetic acid as a catalyst for the N-acylation of amines using esters as the acyl source. Chem. Commun. 53, 2020–2023 (2017).
Koroleva, E. V., Bondar, N. F., Katok, Y. M., Chekanov, N. A. & Chernikhova, T. V. 2-Isoxazolines with an electron-acceptor substituent at C (5) in reactions with nucleophilic reagents. Chem. Heterocycl. Compd. 43, 362–369 (2007).
Behrouz, S., Rad, M. N. & Forouhari, E. Highly efficient preparation of amides from aminium carboxylates using N-(p-toluenesulfonyl) imidazole. J. Chem. Res. 40, 101–106 (2016).
Sathe, P. A. et al. Tandem synthesis of aromatic amides from styrenes in water. Tetrahedron Lett. 59, 2820–2823 (2018).
Antoniak, D., Sakowicz, A., Loska, R. & Mąkosza, M. Direct conversion of aromatic aldehydes into benzamides via oxidation with potassium permanganate in liquid ammonia. Synlett 26, 84–86 (2015).
Firouzabadi, H. & Adibi, M. Methyltriphenylphosphonium tetrahydroborate (MePh3PBH4). A stable, selective and versatile reducing agent. Phosphorus Sulfur Silicon Relat. Elem. 142, 125–147 (1998).
Gockel, S. N. & Hull, K. L. Chloroform as a carbon monoxide precursor: In or ex situ generation of CO for Pd-catalyzed aminocarbonylations. Org. Lett. 17, 3236–3239 (2015).
Patel, O. P., Anand, D., Maurya, R. K. & Yadav, P. P. Copper-catalyzed highly efficient oxidative amidation of aldehydes with 2-aminopyridines in an aqueous micellar system. Green Chem. 17, 3728–3732 (2015).
Yang, S. et al. Copper-catalyzed dehydrogenative reaction: Synthesis of amide from aldehydes and aminopyridine. Tetrahedron 69, 6431–6435 (2013).
Arefi, M., Saberi, D., Karimi, M. & Heydari, A. Superparamagnetic Fe(OH)3@Fe3O4 nanoparticles: an efficient and recoverable catalyst for tandem oxidative amidation of alcohols with amine hydrochloride salts. ACS Comb. Sci. 17, 341–347 (2015).
Sepahvand, H., Bazgir, A. & Shaabani, A. Cu-catalyzed oxidative-reaction of tosylmethylisocyanide and benzyl alcohols: Efficient synthesis of 4-(tert-butylperoxy)-5-aryloxazol-2 (3 H)-ones and 5-aryloxazol-2 (5 H)-ones. Catal. Lett. 150, 2068–2075 (2020).
Yoo, W. J. & Li, C. J. Highly efficient oxidative amidation of aldehydes with amine hydrochloride salts. J. Am. Chem. Soc. 128, 13064–13065 (2006).
Reddy, K. R., Maheswari, C. U., Venkateshwar, M. & Kantam, M. L. Oxidative amidation of aldehydes and alcohols with primary amines catalyzed by KI-TBHP. Eur. J. Org. Chem. 2008, 3619–3622 (2008).
Wang, Y., Zhu, D., Tang, L., Wang, S. & Wang, Z. Highly efficient amide synthesis from alcohols and amines by virtue of a water-soluble gold/DNA catalyst. Angew. Chem. Int. Ed. 50, 8917–8921 (2011).
Zhang, L. et al. Aerobic oxidative coupling of alcohols and amines over Au–Pd/resin in water: Au/Pd molar ratios switch the reaction pathways to amides or imines. Green Chem. 15, 2680–2684 (2013).
Sutar, Y. B., Bhagat, S. B. & Telvekar, V. N. General and efficient oxidative amidation of benzyl alcohols with amines using diacetoxyiodobenzene and TBHP. Tetrahedron Lett. 56, 6768–6771 (2015).
Ghosh, S. C. et al. Tandem oxidative amidation of benzyl alcohols with amine hydrochloride salts catalysed by iron nitrate. Tetrahedron Lett. 54, 4922–4925 (2013).
Fu, R. et al. An efficient, eco-friendly and sustainable tandem oxidative amidation of alcohols with amines catalyzed by heteropolyanion-based ionic liquids via a bifunctional catalysis process. Tetrahedron 72, 8319–8326 (2016).
Acknowledgements
The authors gratefully acknowledge the partial support from the Research Council of the Iran University of Science and Technology.
Author information
Authors and Affiliations
Contributions
H.G. Supervision, Conceptualization, Writing—Review & Editing. M.G.G. Investigation, Methodology, Writing—original draft, Formal analysis. H.D. Methodology, Project administration.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ghafuri, H., Ghafori Gorab, M. & Dogari, H. Tandem oxidative amidation of benzylic alcohols by copper(II) supported on metformin-graphitic carbon nitride nanosheets as an efficient catalyst. Sci Rep 12, 4221 (2022). https://doi.org/10.1038/s41598-022-07543-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-07543-3
This article is cited by
-
Heteroployacid on the composite of boehmite and polyionic liquid as a catalyst for alcohol oxidation and tandem alcohol oxidation Knoevenagel condensation reactions
Scientific Reports (2022)
-
Coupling photocatalytic water oxidation with reductive transformations of organic molecules
Nature Communications (2022)
-
In situ biosynthesis of palladium nanoparticles on banana leaves extract-coated graphitic carbon nitride: An efficient and reusable heterogeneous catalyst for organic transformations and antimicrobial agent
Biomass Conversion and Biorefinery (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
