
Abstract
Mutation of the Cys1 gene underlies the renal cystic disease in the Cys1cpk/cpk (cpk) mouse that phenocopies human autosomal recessive polycystic kidney disease (ARPKD). Cystin, the protein product of Cys1, is expressed in the primary apical cilia of renal ductal epithelial cells. In previous studies, we showed that cystin regulates Myc expression via interaction with the tumor suppressor, necdin. Here, we demonstrate rescue of the cpk renal phenotype by kidney-specific expression of a cystin-GFP fusion protein encoded by a transgene integrated into the Rosa26 locus. In addition, we show that expression of the cystin-GFP fusion protein in collecting duct cells down-regulates expression of Myc in cpk kidneys. Finally, we report the first human patient with an ARPKD phenotype due to homozygosity for a deleterious splicing variant in CYS1. These findings suggest that mutations in Cys1/CYS1 cause an ARPKD phenotype in mouse and human, respectively, and that the renal cystic phenotype in the mouse is driven by overexpression of the Myc proto-oncogene.
Similar content being viewed by others


Introduction
Autosomal recessive polycystic kidney disease (ARPKD; MIM 263,200) affects 1:26,500 live births1. Cohort studies indicate that 80% or more of patients with typical ARPKD have variants in the Polycystic Kidney and Hepatic Disease 1 (PKHD1) gene2,3,4,5,6. Variants in the DZIP1L gene account for less than 1% of affected patients7, while variants in other hepato-renal fibrocystic disease (HRFD) genes, eg. HNF1B, PKD1, NPHP2, NPHP3, and NPHP13, can phenocopy ARPKD8. For reasons yet to be explained, mice with targeted disruption of Pkhd1 exhibit little or no kidney disease9,10,11,12,13,14,15,16. In the absence of a Pkhd1 mutant mouse model that accurately recapitulates the human disease phenotype, the cpk mouse carrying a spontaneous truncating mutation in Cys1 has been the most widely studied mouse model of ARPKD17,18. Cystin, the Cys1 gene product, is a 145-amino acid cilia-associated protein that is expressed in mouse embryonic kidney and liver ductal epithelium19. Disruption of cystin function results in elevated Myc expression in collecting duct epithelial cells20,21,22,23 and increased cell proliferation20,24. In previous work, we have demonstrated that in renal collecting duct epithelia, cystin physically interacts with necdin in a regulatory complex that modulates Myc expression25.
Cystin deficiency-associated disruption of ciliary signaling and/or overexpression of Myc is associated with aberrant SMAD3 phosphorylation26, overexpression of Fos and Kras proto-oncogenes20,21,22, elevated levels of growth factors27, aberrant localization and abundance of the epidermal growth factor receptor (EGFR) on the apical surface of collecting duct cells28 and altered levels of basement membrane components29,30,31 and epithelial cell adhesion molecules32,33. Until now, the relevance of these effects of cystin deficiency for human disease was unclear in the absence of ARPKD patients with variants in human CYS1. Here we present the first case of human ARPKD due to homozygosity for a CYS1 variant, in this case predicted to cause defective splicing. We also show that complementation of defective Cys1 in cpk mouse kidneys rescues both Myc overexpression and the collecting duct cyst phenotype. These studies suggest that up-regulation of Myc expression in vivo may play a central role in the pathogenesis of mouse recessive polycystic kidney disease (PKD), with important implications for human ARPKD.
Results
Phenotypic rescue of cpk mice by kidney-specific expression of a cystin-GFP fusion protein
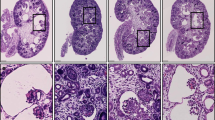
We generated a conditional expression Cys1 transgenic Cys1cpk/cpk (cpk) mouse line carrying a Cys1-GFP transgene knock-in at the Rosa26 locus. In these mice, Cys1-GFP transgene expression is precluded by the presence of a loxP-flanked termination sequence consisting of a PGK-Neo cassette (Fig. 1A, TOFF allele). The Cys1-GFP transgene is expressed by the ROSA26 promoter only after Cre-mediated deletion of the loxP-flanked PGK-Neo cassette (Fig. 1A, TON allele). We crossed Rosa26-Cys1-GFP mice with Cys1cpk/+ mice to generate Cys1cpk/+; Rosa26-Cys1-GFP mice, which were then crossed with Ksp-Cre transgenic mice34 to generate Cys1cpk/+; Rosa26-Cys1-GFP; Ksp-Cre progeny. In these mice, Cre expression, controlled by the Ksp-cadherin regulatory elements, occurs exclusively in the developing distal renal tubular epithelium and the genitourinary tract35, resulting in high level expression of the cystin-GFP fusion protein in the collecting ducts and loops of Henle and low or no expression in the proximal tubules. Finally, the rescue experiments were carried out by crossing Cys1cpk/+; Rosa26-Cys1-GFP; Ksp-Cre mice with Cys1cpk/+ mice. Genotype-confirmed Cys1cpk/cpk; Rosa26-Cys1-GFP; Ksp-Cre experimental “rescue” (R) mice were compared to their Cys1+/+; Rosa26-Cys1-GFP; Ksp-Cre control (C) littermates (Fig. 1B). While cpk mice are characteristically smaller than wild-type littermates and die by 21 days of age36, no differences were observed between R mice and their littermate controls with respect to body size (Fig. 1C, left panel) or viability/lifespan (R mice were routinely euthanized at 12 months of age, as were normal C mice). Kidney sizes at postnatal days 14 and 21 were not significantly different in R and wild-type (WT) mice (Fig. 1C, right panel), while age-matched Cys1cpk/cpk (cpk) mice exhibited the characteristic cystic kidney phenotype. These results indicate that the cpk phenotype was rescued by kidney-specific expression of cystin-GFP.

Rescue of cpk mouse phenotype by kidney-specific expression of Cys1-GFP. (A) Schematic diagram showing the Cys1-GFP transgene knock-in at the Rosa26 locus (Rosa26-Cys1-GFP allele) before (TOFF) and after (TON) deletion of a PGK Neo cassette (yellow rectangle) flanked by LoxP sites (gray triangles). In the TOFF configuration, expression of Cys1-GFP is prevented by the PGK Neo cassette. In cells expressing a Ksp-Cre transgene, Cre-mediated recombination deletes PGK Neo and Cys1-GFP is expressed (TON). SA: splice acceptor, PGK-Neo: Phosphoglycerate kinase promoter driving a neomycin resistance gene followed by 3 polyA signals (3XpA, red rectangle). The purple boxes flanking Cys1-GFP are attB sites. (B) PCR-based genotyping of Rosa26-Cys1-GFP, Ksp-Cre and Cys1 alleles in mice of the indicated genotypes. (C) Cys1-GFP rescue of gross phenotypes in cpk mice. Six-week-old wild-type (WT) and Cys1cpk/cpk; Rosa26-Cys1-GFP; Ksp-Cre/ + rescued (R) mice are of equivalent size. Examination of kidneys from WT, R and cpk mice at 14 and 21 days of age show equivalent sizes of R and WT kidneys, with both markedly smaller than cpk kidneys. (D) Western blot analysis of total kidney protein from 6-week-old mice of the indicated genotypes. Mouse cystin is 145 amino acids long but migrates aberrantly at ~ 25 kDa on SDS-PAGE. Cystin-GFP (arrow, ~ 50 kDa) and endogenous cystin (arrowhead, ~ 25 kDa) were detected using polyclonal rabbit anti-cystin antibody, as previously described19. GAPDH served as an internal protein loading and transfer control. The asterisk indicates non-specific bands.
Expression of cystin-GFP fusion protein in the kidneys of rescued cpk mice
We examined the expression of the cystin-GFP fusion protein in the kidneys of R mice. Endogenous cystin was detectable in the kidneys of both WT and C mice and absent from the kidneys of R mice (Fig. 1D). The cystin-GFP fusion protein of ~ 50 kDa was detected in R and C mice (Fig. 1D, lanes 3 and 4). These results demonstrate that cystin-GFP expression was associated with Cre-mediated excision of the PGK-Neo cassette.
Immunofluorescence staining with antibodies against GFP (Fig. 2A–C) and aquaporin-2 (AQP2; Fig. 2D–F) was used to examine cystin-GFP expression in nephron segments of kidneys from R and C mice. AQP2 is expressed primarily on apical cell membranes of collecting duct cells37,38. The cystin-GFP fusion protein was detected in AQP2-positive collecting ducts of R mice (Fig. 2C, I) and C mice (Fig. 2B, H), while cystin-GFP was absent in Rosa26-Cys1-GFP mice, that lack a Ksp-Cre transgene (Fig. 2A, G). Co-localization of AQP2 and cystin-GFP demonstrated cystin-GFP fusion protein expression in the collecting duct cells.

Cystin-GFP fusion protein expression in renal collecting duct. Immunohistochemical detection of cystin-GFP fusion protein (green, A–C), AQP2 (red, D–F) and merged (G–I) in kidney tissues from 6-week-old mice of the indicated genotypes. The Rosa26-Cys1-GFP/+ mice (A, D, G) do not express cystin-GFP fusion protein due to the absence of the Ksp-Cre transgene. The Rosa26-Cys1-GFP/+; Ksp-Cre/+ mice (B, E, H) have a wild-type Cys1 gene and express cystin-GFP fusion protein in Cre-positive cells. The cpk/cpk; Rosa26-Cus1-GFP/+; Ksp-Cre/+ rescue mice (C, F, I) express cystin-GFP fusion protein in Cre-positive cells. Cell nuclei are stained with DAPI (blue). Scale bars equal 50 µm. Images are representative of tissue sections from 3 animals.
Histological evaluation of cystogenesis in rescued cpk mice
The gross evaluation of kidneys from R mice suggested that the renal histology would be normal. Histological evaluation showed that, while the majority of the nephrons in R kidneys appeared to have normal dimensions, occasional cystic structures were present (Fig. 3A–F). Using DBA and LTA lectins, markers of distal and proximal tubules, respectively39,40, we observed (Fig. 3G–I) that the cystic structures stained with LTA (Fig. 3I). These findings suggest that while expression of cystin-GFP in collecting ducts markedly attenuated the cpk renal phenotype, sporadic cyst formation did occur in proximal tubular segments of these kidneys (Fig. 3I). We also showed (Supplementary Fig. S1) that cystin-GFP was expressed in R kidneys throughout the cortical and medullary regions, but was not expressed in glomeruli. These examinations also showed that sporadic cysts formed predominantly along the cortico-medullary junction, corresponding to proximal tubule involvement.

Overall attenuation of renal cystic phenotype in cpk rescue mice, and sporadic proximal tubule cyst formation. Formalin fixed, paraffin embedded 5 µm sections of kidney tissues from 6-week-old mice of the indicated genotypes were stained with H&E and examined by light microscopy (A–F) or stained with lectins LTA (green, proximal tubules) or DBA (red, distal tubules) and examined by immunofluorescence microscopy (G, H, I). Panels (A–C) are ×5 magnification with scale bars equal to 200 µm. Boxed areas are shown at ×40 magnification in corresponding panels (D–F) with scale bars equal to 50 µm. Lectin staining was performed on serial sections corresponding to H&E stained samples. Cell nuclei were stained with DAPI. Scale bars in panels (G–I) are equal to 50 µm. The asterisk (*) identifies a large cyst that was not stained by either LTA or DBA. Images are representative of tissue sections from 3 animals.
Expression of Myc in rescued cpk mice
Myc overexpression in cpk kidneys is well-documented20,21,22,23. In previous work we demonstrated that cystin physically interacts with the DNA-binding protein necdin in a regulatory complex that binds to the Myc P1 promoter25. Necdin enhances Myc promoter activity and cystin antagonizes this effect. In a previous report, we proposed that Myc up-regulation in cpk kidneys results directly from disruption of the cystin-necdin interaction. In the current study, we examined the relative abundance of c-MYC protein in the kidneys of 14-day old R mice as compared to cpk mice. Quantitative immunoblotting revealed comparable levels of c-MYC in WT and R kidneys that were markedly lower than in kidneys of cpk mice (Fig. 4). These results demonstrate that transgene rescue of the cystic kidney phenotype in cpk mice is associated with down-regulation of c-MYC protein expression, suggesting a central role for Myc overexpression in renal cystogenesis in this mouse model.

Decreased c-MYC expression in kidneys of cpk rescue mice. Immunoblot analysis of c-MYC protein expression in kidneys from wild-type (WT), cpk/cpk; Rosa26-Cys1-GFP/+ (cpk) and cpk/cpk; Rosa26-Cys1-GFP/+; Ksp-Cre/+ (Rescue) mice. (A) Total kidney lysates were immunoblotted using anti-c-MYC and anti-GAPDH antibodies. (B) c-MYC band intensity was normalized with GAPDH. Data represents mean ± S.E; Y-axis values indicate c-MYC/GAPDH band intensity ratio; **P < 0.01, versus others, ANOVA, n = 3 each group.
Cystin regulation of Myc gene expression occurs via cystin-necdin mediated repression of Myc transcription25. We therefore used qRT-PCR to assess relative Myc mRNA levels in kidney samples from 14-day-old B6 WT, cpk and R mice (Supplementary Fig. S2). We observed that, consistent with Myc western blotting results, Myc mRNA levels were upregulated in cpk/cpk cystic kidney but normal in kidneys from R mice. These findings further support a central role of Myc gene overexpression in cpk renal cystogenesis.
Recessive CYS1 variant in an individual with childhood cystic kidney disease and liver fibrosis
While the cpk mouse phenotype recapitulates important clinical features of ARPKD, to date no human cases of the disease have been linked to variants in the CYS1 gene. We evaluated a case of a 5-year-old male child, subject B783, born from a consanguineous union, who presented with polyuria, polydipsia and poor growth. Renal ultrasonography showed multiple medullary and cortical cysts concerning for polycystic kidney disease. Renal function was mildly reduced (creatinine 0.6 mg/dL) for age.
To identify a monogenic cause of disease, we performed trio exome sequencing (TRIO-ES) on DNA samples obtained from the proband and parents. Homozygosity mapping from exome variant data for B783 demonstrated 108 Mbp of homozygosity by descent (Fig. 5A), suggesting the parents are approximately fourth degree relatives. Based on this mapping, we hypothesized that a biallelic gene variant residing within a homozygous peak region underlies proband renal disease. To identify the most probable disease-causing variant, we used the following criteria for exome variant filtering41,42,43,44: (1) exclusion of all variants that did not change the amino-acid sequence or affected canonical splice sites (defined as ± 6 nucleotides surrounding the exon–intron boundary), (2) exclusion of variants reported in the homozygous state or with a minor allele frequency greater than 0.1% in a control cohort (ExAC and gnomAD genome databases), (3) inclusion of homozygous bi-allelic variants with appropriate parental segregation consistent with our above hypothesis, and (4) assessment of variants for deleteriousness based on in silico prediction of their impact on protein structure and/or splice site function. This approach failed to identify a strong candidate variant. We, therefore, performed trio genome sequencing (TRIO-GS), which provides increased coverage of non-coding regions.

A recessive variant in CYS1 associated with polycystic kidney disease and liver fibrosis. (A) Genome-wide homozygosity mapping of B783 identifies homozygous peak regions. CYS1 is located within a peak region of chromosome 2 (arrowhead). (B) Exon structure of CYS1 cDNA. The locations of start codon, stop codons, and affected splice site (boundary exon 1–2) are indicated. The region of exon 1 deleted in the cpk mouse is also indicated. (C) Protein domain structure of cystin-1. A myristoylation site (M), polybasic region (P) and AxEGG motif required for targeting cystin-1 to the cilium (C) are indicated. Arrow indicates the boundary between protein regions encoded by exons 1 and 2. Abbreviations: AA, amino acid; bp, base pairs; C, cilium trafficking domain; M, myristoylation site; P, polybasic region.
Using the same filtering approach as described above for TRIO-ES, we identified a homozygous splice-site variant (c.318 + 5G > A) in the exon 1 donor site in the CYS1 gene (Fig. 5B, C). Conceivably, this variant was not detected by TRIO-ES because CYS1 exon 1 was under-represented due to its high GC-content, which can result in low capture efficiency during the generation of sequence libraries for WES (and see Discussion)45. Consistent with our hypothesis, this variant resides within a region of homozygosity of descent (Fig. 5A). It is deleterious based on the following criteria: (i) the splice variant is extremely rare, as it is absent in the gnomAD database (gnomAD, version 2.1.1), and (ii) the variant was predicted to have deleterious effect on splicing using four independent in silico prediction tools (Table 1). Importantly, in both TRIO-ES and TRIO-GS, we did not identify causative variants in 100 cystic kidney disease genes including PKHD1 or DZIP1L. Allele-specific PCR confirmed the variant was homozygous in the affected child and heterozygous in the parents (Supplementary Fig. S3).
Based upon these findings, the clinical data of subject B783 was further queried. Prenatal ultrasonography showed polyhydramnios and echogenic, but normal sized, kidneys. B783 did not develop respiratory distress after birth and had no subsequent pulmonary problems. Similarly to the established phenotype observed in cpk mice46,47,48 (on some genetic backgrounds), he exhibited mild congenital liver fibrosis (Fig. 5B) but did not have abnormalities in liver function tests. Moreover, his renal function worsened. By age 11 years, he had progressed to end-stage kidney disease and was on dialysis. Overall, the renal-hepatic syndromic features of subject B783 further support that the homozygous CYS1 variant c.318 + 5G > A is likely pathogenic.
Based on this unique case, we analyzed ES data from 521 individuals with pediatric onset cystic kidney disease to identify additional families with deleterious variants in the CYS1 locus but did not identify any additional cases. Our findings strongly suggest the first identification of a causative variant in CYS1 in a human patient with an ARPKD phenotype.
Minigene assay confirms an effect of CYS1 variant c.318 + 5G > A on splicing
We were unable to obtain samples of patient-derived mRNA needed to directly ascertain effects of the variant on CYS1 gene splicing. As an alternative strategy, we performed minigene-based assays49 that allowed comparison of RNA splicing products derived from CYS1 WT to those from the c.318 + 5G > A allele minigene constructs transfected into human renal collecting duct cells. As illustrated in Fig. 6A, CYS1 DNA sequences were cloned between vector pSpliceExpress exons A and B such that: 20 bp of CYS1 exon 1 3’ sequence, including either the WT (construct CYS1-ex1DNR-ex2, in which “DNR” denotes “WT donor”) or the c.318 + 5G > A patient-derived variant splice donor site (CYS1-ex1DNRMUT-ex2), replaced 3’ exon A vector sequences, followed by 200 bp of adjacent CYS1 intron 1 sequence, followed by CYS1 sequence consisting of the entire CYS1 exon 2 (53 bp) flanked on either side by 200 bp of adjacent CYS1 intronic sequences. A positive control construct was also generated (CYS1-ex2), consisting of CYS1 exon 2, flanked on either side by 200 bp of native CYS1 intron sequences, ligated between vector exons A and B. The constructs were transfected into human renal collecting duct cells, RT-PCR (using the Fwd & Rev primer pair) was performed on extracted mRNA, amplicons were resolved by electrophoresis through agarose gels and analyzed by restriction digestion and DNA sequencing to resolve the composition of relevant mRNA splicing products derived from the transfected plasmids (Fig. 6B). RT-PCR product sizes from parent vector, CYS1-ex2 positive control, and CYS1-ex1DNR-ex2 containing WT donor sequence yielded amplicons equivalent in size to the expected splice products, verified by subcloning and DNA digestion and sequencing (Fig. 6C and Supplementary Data). Higher molecular weight amplicons correspond in size to anticipated unspliced or intermediate splice forms of construct-derived mRNAs. The CYS1-ex1DNRMUT-ex2 construct, by contrast, generated a collection of amplicons lacking the PCR fragment corresponding to the normal, WT splice product (Fig. 6B, C and Supplementary Data). Instead, this construct generated a novel lower MW fragment and a spectrum of abnormal higher MW amplicons. The variant caused a shift to the use of an alternative upstream splice donor site as well as an alternative downstream splice acceptor site (Fig. 6 and Supplementary Data). Higher MW bands corresponding to incorrectly spliced transcripts from the mutation-bearing construct are experimental results consistent with in silico based predictions, and independently support the c.318 + 5G > A variant as deleterious. Furthermore, it is known that mutations of the type identified in the patient can lead to diverse, abnormal splicing products50.

CYS1 variant c.318 + 5G > A caused abnormal splicing of RNA expressed from a transfected minigene in human renal cells. (A) Experimental minigene splicing constructs were generated by cloning the following CYS1 sequences between vector exons A and B: 20 bp of CYS1 exon 1 3’ sequence including either the WT or the c.318 + 5G > A patient-derived variant splice donor site; then 200 bp of adjacent intron1 sequence; then CYS1 sequence consisting of the entire CYS1 exon 2 (53 bp) flanked by 200 bp of adjacent CYS1 intronic sequences. Minigene constructs were transfected into human renal collecting duct (CD-hTERT) cells85. Normal splice donor (Do) and acceptor (Ac) sites are indicated by arrows. Forward (Fwd) and reverse (Rev) primers corresponding to sequences in vector exons A and B, respectively, were used in RT-PCR. The position of the c.318 + 5G > A patient-derived variant is indicated with a red X. (B) RT-PCR reaction products (using Fwd & Rev primer pair) from CD-hTERT RNA following transfection with the indicated plasmids. Amplicons were resolved by agarose gel electrophoresis. RT+, addition of reverse transcriptase; RT-, negative control reactions lacking reverse transcriptase. CD-hTERT indicates template RNA from untransfected cells used as a negative control. NTC, no template control. The vector positive control generated an amplicon of approximately 200 bp, as expected (purple asterisk). CYS1-ex2 and CYS1-ex1DNR-ex2 generated amplicons of the expected 250 bp size (red asterisks). In contrast, the c.318 + 5G > A variant showed loss of the 250 bp amplicon and the appearance of a novel, shorter amplicon (blue asterisk). The identities of the bands marked with asterisks were confirmed by cloning of excised DNA and subsequent restriction and sequence analysis (Supplementary Data). (C) RT-PCR amplicon composition and size (bp) corresponding to splicing products generated from transfected minigene constructs amplified using the Fwd and Rev primer pair (shown in A). Outcomes for the variant construct, assuming an effect of the CYS1 variant on splicing, could not be predicted a priori. The composition of the splicing product shown here is from the actual sequence of the cloned amplicon (see Supplementary Data). Sequences of cloned splice product amplicons from all other constructs corresponded precisely to anticipated outcomes.
Discussion
In the current study, we demonstrate that expression of the Cys1 transgene in renal collecting ducts of Cys1cpk/cpk mice rescues the cystic phenotype and down-regulates expression of Myc in vivo at the protein and mRNA levels. We also report the first ARPKD patient with a homozygous CYS1 variant, a c.318 + 5G > A variant predicted to disrupt splicing, which we validated in an in vitro minigene splicing assay. This variant affects the +5 position of the canonical donor splice site, AG/GURAGU. Pathogenic variants affecting +5 position of the donor splice site have been reported in other genes51 including the OXCT1 gene in succinyl-CoA:3-ketoacid CoA transferase (SCOT) deficiency52 and the gene encoding fumarylacetoacetate hydrolase in hereditary tyrosinaemia53. Detailed analysis of G-to-A sequence changes at the +5 position have revealed disrupted base pairing between donor splice site of a pre-mRNA and the U1snRNP of the spliceosome leading to decreased efficiency of the splice site recognition and exon skipping that can result in frameshifting and premature protein termination54. However, this ex vivo system does not allow us to predict whether the variant leads to structural protein alterations (e.g. truncation) and/or decreased abundance of the CYS1 gene product in vivo.
The identification of a pathogenic CYS1 variant in a HRFD patient confirms the importance of the CYS1 gene product for normal function of human collecting duct cells. While it is surprising that CYS1 deficiency causing ARPKD has been observed in only one family, it is important to note that this gene is GC-rich, particularly its first exon. Such GC-rich regions can be difficult to amplify and sequence using Sanger methodology55 and can be missed in next-generation sequencing because low sequence complexity prevents efficient capture prior to library construction45. As described in this report, the CYS1 exon 1 variant was not detected in our initial TRIO-ES analysis but was revealed by WGS. Our recruitment approach for cystic kidney disease patients (pediatric onset of echogenic kidneys and/or 2 + cysts) yielded a cohort, in which 50/79 (63.3%) subjects had a detectable monogenic cause in a cystic kidney disease gene or phenocopy. Among these 50 patients, 32 (64%) had causative variants detected in a NPHP-renal ciliopathy disease gene, while 2/50 (4%) had a causative variant in PKHD1. As WGS comes to be more routinely applied in sequencing of patients’ DNA, we speculate that more ARPKD-associated CYS1 variants will be identified.
Development of collecting duct cysts in humans with presumed PKHD1 variants and mice with cystin deficiency suggests a shared pathobiology and possibly similar molecular mechanisms underlying cyst formation. In the cpk mouse model, renal cysts initially develop at embryonic day 15.5 (E15.5) and are restricted to proximal tubules56,57. As cpk mice develop and disease progresses, cysts predominantly affect the distal collecting duct region56. Specific expression of cystin-GFP in developing ureteric bud-derived collecting ducts rescued the renal cystic phenotype. Transgenic cpk R mice expressing the fusion protein only in AQP2-positive collecting duct cells exhibited survival rates and kidney sizes similar to WT mice. Interestingly, while collecting duct cysts were absent in R mice, these animals did develop proximal tubular cysts suggesting that the initial phase of proximal tubular cystogenesis was not rescued. Proximal tubule cysts have also been observed in human ARPKD fetal specimens between 14 and 26 weeks of gestation, but not in the kidneys of fetuses older than 34 weeks of gestation40. These observations suggest a gradual shift of cyst formation from proximal tubules to collecting ducts during early fetal development in both human and mouse ARPKD. Consistent with previously published observations58 that the cpk mutation does not lead to liver pathology on a B6 strain genetic background, examination of liver sections from cpk and R mice did not reveal evidence of hepatic lesions.
Cystin is a cilium-associated protein that localizes to the basal bodies and the ciliary axoneme18,19,59. Treatment of cpk mice with paclitaxel, which promotes microtubule assembly, prevents renal cyst formation, suggesting that cystin may stabilize microtubule assembly within the ciliary axoneme60. The primary cilia of the collecting duct epithelium function as transmitters of mechano- and chemosensory stimuli to signaling pathways that regulate multiple key cellular processes including differentiation, proliferation, apoptosis, tissue homeostasis and cell polarity61. We have previously demonstrated that cystin, with two functional nuclear localization signals, can be released from the ciliary membrane through a myristoyl-electrostatic switch and translocate to the nucleus where it forms a regulatory complex with necdin to modulate Myc expression25.
The Myc proto-oncogene plays a critical role in normal kidney development62 and several lines of evidence suggest a central role for dysregulated Myc expression in the pathophysiology of polycystic kidney disease. First, overexpression of Myc in the kidneys of SBM transgenic mice causes polycystic kidney disease63 and renal cystic disease remitted in a subset of SBM mice that underwent spontaneous reversion to normal kidney Myc expression64. Second, treatment of cpk mice with antisense Myc oligonucleotides mitigated the cystic phenotype23. Third, Myc is overexpressed in mouse models of autosomal dominant polycystic kidney disease (ADPKD)65,66 and Myc expression appears to be tightly regulated by PC1, the product of the Pkd1 gene67. Fourth, pharmacological inhibition of glucogen synthase kinase 3 beta (GSK3beta), which accelerates cyst formation in cpk mice, leads to decreased Myc expression and amelioration of the cystic phenotype68. Similarly, Myc is down-regulated in Cys1cpk/cpk; Smad3+/− mice and these double mutants have a milder phenotype than cpk mice26. Our findings that complementation of mutant cpk with cystin-GFP rescues the cystic phenotype and restores normal Myc expression provides further evidence that cystin acts in vivo as a negative regulator of Myc.
In summary, we demonstrate that cystin deficiency causes ARPKD in humans and mice, and that targeted renal expression of a cystin-GFP fusion protein prevents cyst formation in the cpk mouse, most probably by downregulating Myc expression in collecting duct cells. Our identification of the first case of human ARPKD caused by a CYS1 variant confirms the relevance of the cpk mouse as an ARPKD model yielding important insights into molecular mechanisms underlying disease pathobiology.
Materials and methods
Animal study approvals
All mouse experiments were approved by the Institutional Animal Care and Use Committees of Children’s National Research Institute and the University of Alabama at Birmingham (UAB), and experiments were carried out in accordance with relevant guidelines and regulations. The study was carried out in compliance with ARRIVE guidelines. Knock-in transgenic mice were generated at the University of Alabama at Birmingham (UAB) Transgenic & Genetically Engineered Models Core facility. Ksp-Cre mice were obtained from Jackson Laboratory (Bar Harbor, ME). Mouse colonies were maintained in the animal facility at Children’s National Research Institute.
Antibodies and lectins
Anti-GAPDH antibody was purchased from Cell Signaling Technologies (# 2118). Anti-AQP2 antibody was purchased from Santa Cruz Biotechnologies (# SC9882). Alexa Fluor 488 conjugated anti-GFP antibody was obtained from Life Technologies (# A21311). Polyclonal rabbit anti-cystin antibody (70,053) was generated in our lab and described previously19. Rabbit monoclonal anti-c-Myc antibody was purchased from Abcam (# ab32072). Goat anti-rabbit HRP conjugated secondary antibody was purchased from American Qualex Solution Products (# A102PS). Donkey anti-Goat IgG Alexa Fluor 555 was obtained from Life Technologies (# A21432). Lectins LTA-FITC (# W0909) and DBA-Rhodamine (# Y0828) were obtained from Vector Laboratories.
Vector cloning
Cys1-GFP cDNA was amplified from previously described pEGFP-N119. Gateway PCR primers were used to add flanking attB sites. A one-tube Gateway reaction was performed using pDonr221 and pRosa26 Dest69. The reaction product was used to transform competent STBL3 cells (Life Technologies # C7373-03) that were plated on Ampicillin and Kanamycin to select for destination and entry clones, respectively. Destination clones were screened by restriction enzyme digest prior to sequencing. The pRosa26 Dest Cys1-GFP targeting vector was linearized with Kpn1 and electroporated into ES cells. G418 resistant colonies were screened by long-range PCR as described70 and positive clones were used to produce chimeric founder mice.
PCR genotyping
PCR conditions are described in Supplementary Table S1.
Immunoblotting
Kidney tissue was collected, homogenized, and processed for immunoblotting as previously described25. For cystin and control western blots, immuno-reactive protein bands were visualized using SuperSignal West Dura chemiluminescent substrate (Thermo Fisher Scientific # 34076) and exposed to film. For c-MYC and control western blots, images were obtained with a ChemiDoc Imaging System (Bio-Rad laboratory, Inc.) Densitometry was analyzed using Image Lab (Bio-Rad laboratory, Inc., Version 6.0).
Kidney histology
Tissue samples were collected and fixed in 10% formalin (Fisher Scientific # 23-245-684) for 2 days, then stored in 70% ethanol. The samples were dehydrated, paraffin embedded, cut into 5 µm sections and stained with hematoxylin and eosin (H&E) and slide-mounted for examination by the UAB Comparative Pathology Laboratory.
Immunofluorescence analysis
Tissue samples were collected from 6 week-old mice and processed, using published methods34. Immunofluorescence detection and image acquisition were performed using an Olympus FLUOVIEW FV1000 confocal laser scanning microscope configured with both an Argon Laser (488 nm) and a Laser diode (405 nm, 440 nm, and 559 nm). Images were analyzed using Olympus FV10-ASW 3.0 Viewer software.
Lectin staining
Five µm sections of fixed paraffin embedded kidney tissues (prepared as described for histopathology) were stained with Rhodamine labeled DBA (Vector Laboratory # RL-1032) and Fluorescein labeled LTA (Vector Laboratory # FL-1321)71. Immunofluorescence detection, image acquisition, and analysis were performed as described above.
Research subjects and study approval
Cystic kidney disease cases with pediatric onset of echogenic kidneys and/or 2 + renal cysts were recruited as previously described72. We obtained blood samples and pedigrees following informed consent from individuals with cystic kidney disease or their legal guardians. Clinical data were obtained using a standardized questionnaire (http://www.renalgenes.org). Approval for human subjects research was obtained from Institutional Review Boards of the University of Michigan, Boston Children’s Hospital, and local IRB equivalents, and all procedures were carried out in accordance with relevant guidelines and regulations.
Exome/genome sequencing and variant calling
For subject B783, TRIO-ES and data processing were performed by the Genomics Platform at the Broad Institute of Harvard and MIT (Broad Institute, Cambridge, MA). Exome sequencing (> 250 ng of DNA, at > 2 ng/μl) was performed using Illumina exome capture (38 Mb target). Single nucleotide polymorphisms (SNPs) and insertions/deletions (indels) were jointly called across all samples using the Genome Analysis Toolkit (GATK) HaplotypeCaller. Default filters were applied to SNP and indel calls using the GATK Variant Quality Score Recalibration approach. Lastly, variants were annotated using the Variant Effect Predictor. For additional information, please refer to the Supporting Information Section S1 in the exome aggregation consortium (ExAC) study73. The variant call set was uploaded on to Seqr (https://seqr.broadinstitute.org) and analysis of the entire ES output was performed. TRIO-GS and data processing were performed by the Genomics Platform at the Broad Institute of MIT and Harvard. PCR-free preparation of sample DNA (350 ng input at > 2 ng/µl) is accomplished using Illumina HiSeq X Ten v2 chemistry. Libraries are sequenced to a mean target coverage of > 30×. Genome sequencing data was processed through a pipeline based on Picard, using base quality score recalibration and local realignment at known indels. The BWA aligner was used for mapping reads to the human genome build 38. Single Nucleotide Variants (SNVs) and insertions/deletions (indels) are jointly called across all samples using Genome Analysis Toolkit (GATK) HaplotypeCaller package version 3.4. Default filters were applied to SNV and indel calls using the GATK Variant Quality Score Recalibration (VQSR) approach. Annotation was performed using Variant Effect Predictor (VEP). Lastly, the variant call set was uploaded to seqr for collaborative analysis between the CMG and investigator.
For the additional 521 individuals with cystic kidney disease, ES was performed by either the Broad Institute as above or alternatively the Yale Center for Genomics as follows. Sequence reads were mapped against the human reference genome (NCBI build 37/hg19) using CLC Genomics Workbench (version 6.5.1) (CLC bio). Genetic location information is according to the February 2009 Human Genome Browser data, hg19 assembly (http://www.genome.ucsc.edu). Downstream processing of aligned BAM files were done using Picard and Samtools74, and SNV calling was done using GATK5.
From both platforms, variant calling was performed in line with proposed guidelines42, and the following criteria were employed as previously described43,44. The variants included were rare in the population with mean allele frequency < 0.1% and with 0 homozygotes in the adult reference genome databases ExAC and gnomAD. Additionally, variants were non-synonymous and/or located within splice-sites. Based on an autosomal homozygous recessive hypothesis, homozygous variants were evaluated. Subsequently, variant severity was classified based on prediction of protein impact (truncating frameshift or nonsense variants, essential or extended splice-site variants, and missense variants). Splice-site variants were assessed by in silico tools MaxEnt, NNSPLICE, HSF, and CADD splice-site variant prediction scores75,76,77,78. Missense variants were assessed based on SIFT, MutationTaster and PolyPhen 2.0 conservation prediction scores79,80,81 and evolutionary conservation based on manually derived multiple sequence alignments.
Homozygosity mapping (HM)
Homozygosity mapping was calculated based on exome sequencing data. In brief, aligned BAM files were processed using Picard and SAMtools4 as described74. Single nucleotide variant calling was performed using Genome Analysis Tool Kit (GATK)82. The resulting VCF files were used to generate homozygosity mapping data and visual outputs using the program Homozygosity Mapper83.
Web resources used
UCSC Genome Browser, http://genome.ucsc.edu.
Ensembl Genome Browser, www.ensembl.org.
gnomAD browser 2.0.3., https://gnomad.broadinstitute.org.
Polyphen2, https://genetics.bwh.harvard.edu/pph2.
Sorting Intolerant From Tolerant (SIFT), http://sift.jcvi.org.
MutationTaster, www.mutationtaster.org.
Combined Annotation Dependent Depletion, https://cadd.gs.washington.edu.
NNSPLICE splice-site variant prediction, www.fruitfly.org/seq_tools/splice.html.
MaxEnt splice prediction, http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html.
Human Splice Finder, www.umd.be/HSF/.
Mini-gene splicing assay
The minigene assay was used as previously described49 to confirm the predicted effect of the patient-derived CYS1 c.318 + 5G > A variant on splicing. Minigene constructs were engineered in the vector pSpliceExpress84 (a kind gift of Dr. Stefan Stamm). CYS1 DNA insert sequences were synthesized and ligated between pSpliceExpress exons A and B as follows (see Fig. 6): 20 bp of CYS1 exon 1 3’ sequence, including either the WT or the c.318 + 5G > A patient-derived variant splice donor site, replaced 3’ exon A vector sequences, followed by 200 bp of adjacent CYS1 intron1 sequence, followed by CYS1 sequence consisting of the entire CYS1 exon 2 (53 bp) flanked on either side by 200 bp of adjacent CYS1 intronic sequences. A positive control construct was also generated, consisting of CYS1 exon 2, flanked on either side by 200 bp of native CYS1 intron sequences, ligated between vector exons A and B. Plasmid constructs, including the parent pSpliceExpress vector, were prepared using QIAGEN Plasmid Plus Midi Kit (QIAGEN # 12943) and were eluted in water. Plasmids were transfected into immortalized normal human kidney collecting duct (CD-hTERT) cells (a kind gift of Dr. D. Bell)85 using Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific # 11668019) according to the manufacturer’s instructions. CD-hTERT cells were cultured in DMEM/F-12 medium (Thermo Fisher Scientific # 11330-057) supplemented with 5% heat-inactivated fetal bovine serum (Thermo Fisher Scientific # 10437-028), 1% penicillin/streptomycin (Thermo Fisher Scientific # 15140-122), 1% L-Glutamine 200 mM (100×, Thermo Fisher Scientific # 25030-081), 200 ng/ml dexamethasone (Sigma-Aldrich # D2915), 1% Insulin-Trasferrin-Selenium solution (Thermo Fisher Scientific # 41400-045), and 1.3 ng/ml 3,3′,5-Triiodo-L-thyronine sodium salt (Sigma-Aldrich # T6397). Following plasmid transfection, cells were maintained in culture for 48 h. Total RNA was then extracted from the cells using RNeasy Mini Kit (QIAGEN # 74104) according to the manufacturer’s instructions. Following treatment with RQ1 RNase-Free DNase (Promega # M6101) for 30 min., the total RNA was re-purified using the RNeasy Mini Kit. cDNA was generated using SuperScript III First-Strand Synthesis SuperMix (Thermo Fisher Scientific # 18080-400) according to manufacturer’s instructions. cDNAs generated from transfected plasmid constructs were amplified by PCR using GoTaq Master Mixes (Promega # M7123) and the primers “Fwd” (5′-CCTGCTCATCCTCTGGGAGC-3′) and “Rev” (5′-AGGTCTGAAGGTCACGGGCC-3′) (see Fig. 6). PCR cycling used an initial denaturation at 95 °C for 2 min, followed by 33 cycles of 95 °C for 30 s, 56 °C for 30 s, 72 °C for 1 min, and final extension at 72 °C for 5 min. The PCR products were resolved by agarose gel electrophoresis. Amplicons of interest (marked by asterisks in Fig. 6) were extracted from agarose gels using QIAquick Gel Extraction Kit (QIAGEN # 28704), TA-cloned with TOPO TA Cloning Kit for Subcloning, and used to transform One Shot TOP10 Chemically Competent E. coli (Thermo Fisher Scientific # K450001) according to manufacturer’s instructions. Individual clones were analyzed by restriction digestion and DNA sequencing.
References
Alzarka, B., Morizono, H., Bollman, J. W., Kim, D. & Guay-Woodford, L. M. Design and implementation of the hepatorenal fibrocystic disease core center clinical database: a centralized resource for characterizing autosomal recessive polycystic kidney disease and other hepatorenal fibrocystic diseases. Front. Pediatr. 5, 80. https://doi.org/10.3389/fped.2017.00080 (2017).
Szabo, T. et al. Comprehensive genetic testing in children with a clinical diagnosis of ARPKD identifies phenocopies. Pediatric. Nephrol. (Berl., Ger.) 33, 1713–1721. https://doi.org/10.1007/s00467-018-3992-5 (2018).
Melchionda, S. et al. Expanding the mutation spectrum in 130 probands with ARPKD: identification of 62 novel PKHD1 mutations by sanger sequencing and MLPA analysis. J. Hum. Genet. 61, 811–821. https://doi.org/10.1038/jhg.2016.58 (2016).
Adeva, M. et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore) 85, 1–21. https://doi.org/10.1097/01.md.0000200165.90373.9a (2006).
Bergmann, C. et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 67, 829–848. https://doi.org/10.1111/j.1523-1755.2005.00148.x (2005).
Sharp, A. M. et al. Comprehensive genomic analysis of PKHD1 mutations in ARPKD cohorts. J. Med. Genet. 42, 336–349. https://doi.org/10.1136/jmg.2004.024489 (2005).
Lu, H. et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 49, 1025–1034. https://doi.org/10.1038/ng.3871 (2017).
Bergmann, C. Early and severe polycystic kidney disease and related ciliopathies: an emerging field of interest. Nephron 141, 50–60. https://doi.org/10.1159/000493532 (2019).
Moser, M. et al. A mouse model for cystic biliary dysgenesis in autosomal recessive polycystic kidney disease (ARPKD). Hepatology 41, 1113–1121. https://doi.org/10.1002/hep.20655 (2005).
Garcia-Gonzalez, M. A. et al. Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum. Mol. Genet. 16, 1940–1950. https://doi.org/10.1093/hmg/ddm141 (2007).
Woollard, J. R. et al. A mouse model of autosomal recessive polycystic kidney disease with biliary duct and proximal tubule dilatation. Kidney Int. 72, 328–336. https://doi.org/10.1038/sj.ki.5002294 (2007).
Gallagher, A. R. et al. Biliary and pancreatic dysgenesis in mice harboring a mutation in Pkhd1. Am. J. Pathol. 172, 417–429. https://doi.org/10.2353/ajpath.2008.070381 (2008).
Kim, I. et al. Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J. Am. Soc. Nephrol. 19, 455–468. https://doi.org/10.1681/ASN.2007070770 (2008).
Williams, S. S., Cobo-Stark, P., James, L. R., Somlo, S. & Igarashi, P. Kidney cysts, pancreatic cysts, and biliary disease in a mouse model of autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 23, 733–741. https://doi.org/10.1007/s00467-007-0735-4 (2008).
Bakeberg, J. L. et al. Epitope-tagged Pkhd1 tracks the processing, secretion, and localization of fibrocystin. J. Am. Soc. Nephrol. 22, 2266–2277. https://doi.org/10.1681/ASN.2010111173 (2011).
Outeda, P. et al. A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C-terminus in disease mechanism. Kidney Int. 92, 1130–1144. https://doi.org/10.1016/j.kint.2017.04.027 (2017).
Nagao, S., Kugita, M., Yoshihara, D. & Yamaguchi, T. Animal models for human polycystic kidney disease. Exp. Anim. 61, 477–488. https://doi.org/10.1538/expanim.61.477 (2012).
Hou, X. et al. Cystin, a novel cilia-associated protein, is disrupted in the cpk mouse model of polycystic kidney disease. J. Clin. Invest. 109, 533–540. https://doi.org/10.1172/JCI14099 (2002).
Tao, B. et al. Cystin localizes to primary cilia via membrane microdomains and a targeting motif. J. Am. Soc. Nephrol. 20, 2570–2580. https://doi.org/10.1681/ASN.2009020188 (2009).
Cowley, B. D. Jr., Smardo, F. L. Jr., Grantham, J. J. & Calvet, J. P. Elevated c-myc protooncogene expression in autosomal recessive polycystic kidney disease. Proc. Natl. Acad. Sci. U. S. A. 84, 8394–8398. https://doi.org/10.1073/pnas.84.23.8394 (1987).
Cowley, B. D. Jr., Chadwick, L. J., Grantham, J. J. & Calvet, J. P. Elevated proto-oncogene expression in polycystic kidneys of the C57BL/6J (cpk) mouse. J. Am. Soc. Nephrol. 1, 1048–1053 (1991).
Harding, M. A., Gattone, V. H. 2nd., Grantham, J. J. & Calvet, J. P. Localization of overexpressed c-myc mRNA in polycystic kidneys of the cpk mouse. Kidney Int. 41, 317–325 (1992).
Ricker, J. L., Mata, J. E., Iversen, P. L. & Gattone, V. H. c-myc antisense oligonucleotide treatment ameliorates murine ARPKD. Kidney Int. 61, S125–S131. https://doi.org/10.1046/j.1523-1755.2002.0610s1125.x (2002).
Gattone, V. H. et al. Autosomal recessive polycystic kidney disease in a murine model. A gross and microscopic description. Lab. Invest. 59, 231–238 (1988).
Wu, M., Yang, C., Tao, B., Bu, S. & Guay-Woodford, L. M. The ciliary protein cystin forms a regulatory complex with necdin to modulate Myc expression. PLoS ONE 8, e83062. https://doi.org/10.1371/journal.pone.0083062 (2013).
Hama, T. et al. Aberrant Smad3 phosphoisoforms in cyst-lining epithelial cells in the cpk mouse, a model of autosomal recessive polycystic kidney disease. Am. J. Physiol. Renal. Physiol. 313, F1223–F1231. https://doi.org/10.1152/ajprenal.00697.2016 (2017).
Nakamura, T. et al. Growth factor gene expression in kidney of murine polycystic kidney disease. J. Am. Soc. Nephrol. 3, 1378–1386 (1993).
Orellana, S. A., Sweeney, W. E., Neff, C. D. & Avner, E. D. Epidermal growth factor receptor expression is abnormal in murine polycystic kidney. Kidney Int. 47, 490–499 (1995).
Ebihara, I. et al. Altered mRNA expression of basement membrane components in a murine model of polycystic kidney disease. Lab. Invest. 58, 262–269 (1988).
Taub, M., Laurie, G. W., Martin, G. R. & Kleinman, H. K. Altered basement membrane protein biosynthesis by primary cultures of cpk/cpk mouse kidney. Kidney Int. 37, 1090–1097 (1990).
Rankin, C. A. et al. Matrix metalloproteinases and TIMPS in cultured C57BL/6J-cpk kidney tubules. Kidney Int. 50, 835–844 (1996).
Harding, M. A., Chadwick, L. J., Gattone, V. H. 2nd. & Calvet, J. P. The SGP-2 gene is developmentally regulated in the mouse kidney and abnormally expressed in collecting duct cysts in polycystic kidney disease. Dev. Biol. 146, 483–490 (1991).
Rocco, M. V., Neilson, E. G., Hoyer, J. R. & Ziyadeh, F. N. Attenuated expression of epithelial cell adhesion molecules in murine polycystic kidney disease. Am. J. Physiol. 262, F679–F686. https://doi.org/10.1152/ajprenal.1992.262.4.F679 (1992).
Shao, X., Somlo, S. & Igarashi, P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 13, 1837–1846. https://doi.org/10.1097/01.asn.0000016444.90348.50 (2002).
Shao, X., Johnson, J. E., Richardson, J. A., Hiesberger, T. & Igarashi, P. A minimal Ksp-cadherin promoter linked to a green fluorescent protein reporter gene exhibits tissue-specific expression in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 13, 1824–1836 (2002).
Mandell, J., Koch, W. K., Nidess, R., Preminger, G. M. & McFarland, E. Congenital polycystic kidney disease. Genetically transmitted infantile polycystic kidney disease in C57BL/6J mice. Am. J. Pathol. 113, 112–114 (1983).
Chou, C. L. et al. Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct. Roles of ryanodine-sensitive Ca2+ stores and calmodulin. J. Biol. Chem. 275, 36839–36846. https://doi.org/10.1074/jbc.M005552200 (2000).
Elliot, S., Goldsmith, P., Knepper, M., Haughey, M. & Olson, B. Urinary excretion of aquaporin-2 in humans: a potential marker of collecting duct responsiveness to vasopressin. J. Am. Soc. Nephrol. 7, 403–409 (1996).
Holthofer, H. Lectin binding sites in kidney: a comparative study of 14 animal species. J. Histochem. Cytochem. 31, 531–537. https://doi.org/10.1177/31.4.6827083 (1983).
Nakanishi, K., Sweeney, W. E. Jr., Zerres, K., Guay-Woodford, L. M. & Avner, E. D. Proximal tubular cysts in fetal human autosomal recessive polycystic kidney disease. J. Am. Soc. Nephrol. JASN 11, 760–763 (2000).
Lovric, S., Ashraf, S., Tan, W. & Hildebrandt, F. Genetic testing in steroid-resistant nephrotic syndrome: when and how?. Nephrol. Dial. Transplant. 31, 1802–1813. https://doi.org/10.1093/ndt/gfv355 (2016).
MacArthur, D. G. et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476. https://doi.org/10.1038/nature13127 (2014).
van der Ven, A. T. et al. Whole-exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. J. Am. Soc. Nephrol. 29, 2348–2361. https://doi.org/10.1681/ASN.2017121265 (2018).
Vivante, A. & Hildebrandt, F. Exploring the genetic basis of early-onset chronic kidney disease. Nat. Rev. Nephrol. 12, 133–146. https://doi.org/10.1038/nrneph.2015.205 (2016).
Meienberg, J. et al. New insights into the performance of human whole-exome capture platforms. Nucleic Acids Res. 43, e76. https://doi.org/10.1093/nar/gkv216 (2015).
Gattone, V. H. 2nd., MacNaughton, K. A. & Kraybill, A. L. Murine autosomal recessive polycystic kidney disease with multiorgan involvement induced by the cpk gene. Anat. Rec. 245, 488–499. https://doi.org/10.1002/(SICI)1097-0185(199607)245:3%3c488::AID-AR5%3e3.0.CO;2-O (1996).
Ricker, J. L., Gattone, V. H. 2nd., Calvet, J. P. & Rankin, C. A. Development of autosomal recessive polycystic kidney disease in BALB/c-cpk/cpk mice. J. Am. Soc. Nephrol. 11, 1837–1847 (2000).
Fry, J. L. Jr. et al. A genetically determined murine model of infantile polycystic kidney disease. J. Urol. 134, 828–833. https://doi.org/10.1016/s0022-5347(17)47448-9 (1985).
Boddu, R. et al. Intragenic motifs regulate the transcriptional complexity of Pkhd1/PKHD1. J. Mol. Med. (Berl.) 92, 1045–1056. https://doi.org/10.1007/s00109-014-1185-7 (2014).
De Crescenzo, A. et al. A splicing mutation of the HMGA2 gene is associated with Silver-Russell syndrome phenotype. J. Hum. Genet. 60, 287–293. https://doi.org/10.1038/jhg.2015.29 (2015).
De Conti, L., Skoko, N., Buratti, E. & Baralle, M. Complexities of 5’splice site definition: implications in clinical analyses. RNA Biol. 9, 911–923. https://doi.org/10.4161/rna.20386 (2012).
Hori, T. et al. Molecular basis of two-exon skipping (exons 12 and 13) by c.1248+5g>a in OXCT1 gene: study on intermediates of OXCT1 transcripts in fibroblasts. Hum. Mutat. 34, 473–480. https://doi.org/10.1002/humu.22258 (2013).
Poudrier, J. et al. Frequency of the IVS12 + 5G–>A splice mutation of the fumarylacetoacetate hydrolase gene in carriers of hereditary tyrosinaemia in the French Canadian population of Saguenay-Lac-St-Jean. Prenat. Diagn. 16, 59–64. https://doi.org/10.1002/(SICI)1097-0223(199601)16:1%3c59::AID-PD810%3e3.0.CO;2-D (1996).
BinEssa, H. A. et al. Functional analysis of 22 splice-site mutations in the PHEX, the causative gene in X-linked dominant hypophosphatemic rickets. Bone 125, 186–193. https://doi.org/10.1016/j.bone.2019.05.017 (2019).
Benita, Y., Oosting, R. S., Lok, M. C., Wise, M. J. & Humphery-Smith, I. Regionalized GC content of template DNA as a predictor of PCR success. Nucleic Acids Res. 31, e99. https://doi.org/10.1093/nar/gng101 (2003).
Avner, E. D. et al. Congenital murine polycystic kidney disease. I. The ontogeny of tubular cyst formation. Pediatr. Nephrol. 1, 587–596 (1987).
Nauta, J., Ozawa, Y., Sweeney, W. E. Jr., Rutledge, J. C. & Avner, E. D. Renal and biliary abnormalities in a new murine model of autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 7, 163–172 (1993).
Lee, C. H. et al. Magnetic resonance microscopy of renal and biliary abnormalities in excised tissues from a mouse model of autosomal recessive polycystic kidney disease. Physiol. Rep. https://doi.org/10.14814/phy2.12517 (2015).
Yoder, B. K., Hou, X. & Guay-Woodford, L. M. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 13, 2508–2516. https://doi.org/10.1097/01.asn.0000029587.47950.25 (2002).
Woo, D. D., Tabancay, A. P. Jr. & Wang, C. J. Microtubule active taxanes inhibit polycystic kidney disease progression in cpk mice. Kidney Int. 51, 1613–1618 (1997).
Kathem, S. H., Mohieldin, A. M. & Nauli, S. M. The roles of primary cilia in polycystic kidney disease. AIMS Mol. Sci. 1, 27–46. https://doi.org/10.3934/molsci.2013.1.27 (2014).
Trudel, M. in Polycystic Kidney Disease (ed Li, X.) (2015).
Trudel, M., Barisoni, L., Lanoix, J. & D’Agati, V. Polycystic kidney disease in SBM transgenic mice: role of c-myc in disease induction and progression. Am. J. Pathol. 152, 219–229 (1998).
Trudel, M., Chretien, N. & D’Agati, V. Disappearance of polycystic kidney disease in revertant c-myc transgenic mice. Mamm. Genome 5, 149–152 (1994).
Burtey, S. et al. Overexpression of PKD2 in the mouse is associated with renal tubulopathy. Nephrol. Dial. Transplant. 23, 1157–1165. https://doi.org/10.1093/ndt/gfm763 (2008).
Kurbegovic, A. & Trudel, M. Progressive development of polycystic kidney disease in the mouse model expressing Pkd1 extracellular domain. Hum. Mol. Genet. 22, 2361–2375. https://doi.org/10.1093/hmg/ddt081 (2013).
Parrot, C. et al. c-Myc is a regulator of the PKD1 gene and PC1-induced pathogenesis. Hum. Mol. Genet. 28, 751–763. https://doi.org/10.1093/hmg/ddy379 (2019).
Tao, S. et al. Glycogen synthase kinase-3beta promotes cyst expansion in polycystic kidney disease. Kidney Int. 87, 1164–1175. https://doi.org/10.1038/ki.2014.427 (2015).
Hohenstein, P. et al. High-efficiency Rosa26 knock-in vector construction for Cre-regulated overexpression and RNAi. PathoGenetics 1, 3. https://doi.org/10.1186/1755-8417-1-3 (2008).
O’Connor, A. K. et al. An inducible CiliaGFP mouse model for in vivo visualization and analysis of cilia in live tissue. Cilia 2, 8. https://doi.org/10.1186/2046-2530-2-8 (2013).
Patel, V. et al. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum. Mol. Genet. 17, 1578–1590. https://doi.org/10.1093/hmg/ddn045 (2008).
Braun, D. A. et al. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int. 89, 468–475. https://doi.org/10.1038/ki.2015.317 (2016).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. https://doi.org/10.1038/nature19057 (2016).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. https://doi.org/10.1093/bioinformatics/btp352 (2009).
Reese, M. G., Eeckman, F. H., Kulp, D. & Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 4, 311–323. https://doi.org/10.1089/cmb.1997.4.311 (1997).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394. https://doi.org/10.1089/1066527041410418 (2004).
Desmet, F. O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67. https://doi.org/10.1093/nar/gkp215 (2009).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. https://doi.org/10.1093/nar/gky1016 (2019).
Sim, N. L. et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452-457. https://doi.org/10.1093/nar/gks539 (2012).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. https://doi.org/10.1038/nmeth.2890 (2014).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. https://doi.org/10.1038/nmeth0410-248 (2010).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 111011–111033. https://doi.org/10.1002/0471250953.bi1110s43 (2013).
Seelow, D., Schuelke, M., Hildebrandt, F. & Nurnberg, P. HomozygosityMapper: an interactive approach to homozygosity mapping. Nucleic Acids Res. 37, W593-599. https://doi.org/10.1093/nar/gkp369 (2009).
Kishore, S., Khanna, A. & Stamm, S. Rapid generation of splicing reporters with pSpliceExpress. Gene 427, 104–110 (2008).
Steele, S. L. et al. Telomerase immortalization of principal cells from mouse collecting duct. Am. J. Physiol. Renal. Physiol. 299, F1507-1514. https://doi.org/10.1152/ajprenal.00183.2010 (2010).
Acknowledgements
The authors thank Gene Siegel, MD, PhD (UAB) for histological analysis; the Cellular Imaging and Analysis Core at Children’s National Research Institute for assistance with microscopy; Ljubica Caldovic, PhD and Adam Richman for contributions to writing, editing and revising the manuscript; and Zsuzsanna Bebok (UAB) for reviewing and constructively commenting on the work. This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant P30 DK074038. The authors would like to thank members of the University of Alabama at Birmingham Transgenic & Genetically Engineered Models (TGEMs) facility for creating the Cys1-GFP transgene knock-in animals. TGEMs is supported by NIH National Cancer Institute Grant P30CA13148, NIH NIAMS Grant P30AR048311, and NIH NIDDK Grants P30 DK074038, P30 DK05336, and P60 DK079626 (to RAK). F.H. is the William E. Harmon Professor of Pediatrics. This research is supported by a Grant from the National Institutes of Health to F.H. (DK-076683-13). A.J.M. was supported by an NIH Training Grant (T32DK-007726), by the 2017 Post-doctoral Fellowship Grant from the Harvard Stem Cell Institute, and by the American Society of Nephrology Lipps Research Program 2018 Polycystic Kidney Disease Foundation Jared J. Grantham Research Fellowship. D.B. was supported by a fellowship Grant (404527522) from the German Research Foundation (DFG). Sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute Grant UM1 HG008900 and in part by National Human Genome Research Institute Grant R01 HG009141. The Yale Center for Mendelian Genomics (UM1HG006504) is funded by the National Human Genome Research Institute. The GSP Coordinating Center (U24 HG008956) contributed to cross-program scientific initiatives and provided logistical and general study coordination. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
C.Y., N.H., A.K.O., and J.A.W. generated DNA constructs, performed laboratory experiments and mouse breeding, analyzed data, prepared figures and tables, and contributed to the writing of relevant sections of the manuscript. R.A.K. supervised the production of transgenic mice. D.A.B. and S.S. prepared subject clinical samples, and performed pedigree and genetic analysis. H.M.F. performed subject clinical assessments, study recruitment, sample collection and distribution. M.L. and K.M.L. contributed quality control of exome sequencing results, initial analysis and annotation, and function-based interpretation of variant calling. S.M. performed exome sequencing, exome quality control assessment, and variant calling. A.J.M. collected and prepared subject clinical samples, performed pedigree and genetic analysis, designed figures and tables, and wrote relevant sections of the manuscript. F.H. and L.M.G.-W. were responsible for study conception and overall organization of the work. F.H., and L.M.G.-W. affirmed the accuracy of the reported findings, oversaw manuscript preparation and reviewed and approved the final version.
Corresponding author
Ethics declarations
Competing interests
F.H. is a co-founder of Goldfinch Biopharma Inc. All other authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, C., Harafuji, N., O’Connor, A.K. et al. Cystin genetic variants cause autosomal recessive polycystic kidney disease associated with altered Myc expression. Sci Rep 11, 18274 (2021). https://doi.org/10.1038/s41598-021-97046-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-97046-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
