
Abstract
Targeted therapy with tyrosine kinase inhibitors (TKI) provides survival benefits to a majority of patients with non-small cell lung cancer (NSCLC). However, resistance to TKI almost always develops after treatment. Although genetic and epigenetic alterations have each been shown to drive resistance to TKI in cell line models, clinical evidence for their contribution in the acquisition of resistance remains limited. Here, we employed liquid biopsy for simultaneous analysis of genetic and epigenetic changes in 122 Vietnamese NSCLC patients undergoing TKI therapy and displaying acquired resistance. We detected multiple profiles of resistance mutations in 51 patients (41.8%). Of those, genetic alterations in EGFR, particularly EGFR amplification (n = 6), showed pronounced genome instability and genome-wide hypomethylation. Interestingly, the level of hypomethylation was associated with the duration of response to TKI treatment. We also detected hypermethylation in regulatory regions of Homeobox genes which are known to be involved in tumor differentiation. In contrast, such changes were not observed in cases with MET (n = 4) and HER2 (n = 4) amplification. Thus, our study showed that liquid biopsy could provide important insights into the heterogeneity of TKI resistance mechanisms in NSCLC patients, providing essential information for prediction of resistance and selection of subsequent treatment.
Similar content being viewed by others


Introduction
Lung cancer is the most common type of cancer and the leading cause of cancer related deaths globally, of which the majority of cases are non-small cell lung cancer (NSCLC)1,2. Clinical studies have shown that patients carrying sensitizing EGFR mutations displayed prolonged progression-free survival upon receiving first generation tyrosine kinase inhibitors (TKI) including erlotinib and gefitinib3,4,5 or the second generation TKI afatinib6,7. Such TKI drugs were shown to either reversibly (first generation TKI, 1G TKI) or irreversibly (second generation TKI, 2G TKI) bind to the ATP binding site of the tyrosine kinase domain of EGFR, thereby inactivate its activity8. Both 1G and 2G TKI drugs have been proposed as the first line treatment for patients with advanced NSCLC who carry TKI targeted mutations9.
Despite the superiority of 1G and 2G TKIs over chemotherapy as first line treatment, almost all patients develop resistance to TKI and show disease progression after a median of 10–14 months of TKI therapy10. Resistance to TKI drugs could either stem from intrinsic or acquired resistance11. Patients with intrinsic resistance fail to respond to initial treatment within less than 3 months12,13. This type of resistance could be due to genetic alterations in driver genes that already exist in tumor cells and confer resistance to therapy14. In contrast, according to the criteria from the World Health Organization (WHO) or Response Evaluation Criteria in Solid Tumours (RECIST), acquired resistance responses are related to secondary genetic alterations that emerge after a complete or partial response to targeted therapies or more than 6 months of stable disease, resulting in tumor survival and continued growth15,16. NSCLC patients could develop a secondary EGFR substitution of threonine to methionine at position 790 (T790M) in exon 20 or EGFR amplification at the time of acquired resistance to erlotinib or gefitinib. In addition to EGFR dependent resistance mechanisms, the activation of bypass pathways through the MET, AXL, IGF1R axis or the phenotypic transformation to small-cell lung cancer (SCLC) has been reported to drive resistant responses to 1G and 2G TKIs17,18,19,20.
Although genetic alterations are known to be associated with TKI resistance, it has become increasingly evident that multiple mechanisms could contribute to the complex process of resistance development21. Epigenetic alterations, particularly DNA methylation, have been reported to affect the sensitivity of cancer cells to TKI drugs through their broad impact on regulation of gene expression22,23,24. Hypermethylation at promoter regions of certain tumor suppressor genes (TSG) was associated with increased resistance to gefitinib in lung cancer cell lines through silencing their expression25. Moreover, global hypomethylation has recently been proposed as a hallmark of cancer, which tends to occur in promoter regions of oncogenes, leading to activation of these genes26,27. It is thought that the abnormal hypomethylation at genome-wide level is linked with copy number aberration (CNA), resulting in genome instability, an important tumorigenic event28,29. How methylation changes are linked with genetic mutations during the acquisition of TKI resistance remains unknown. Most current knowledge of TKI resistance mechanisms is based on in vitro cell line models that focus on elucidating the contribution of either mutation or methylation changes of specific genes. As such, findings from such models may under-represent the complexity of resistance mechanisms in NSCLC patients.
In this study, we examined the application of plasma cell free DNA (cfDNA) to simultaneously profile genetic and methylation landscapes in 122 Vietnamese NSCLC patients who developed acquired resistance to TKI drugs. Our study showed that genetic mutations, genome-wide and hotspot methylation states, and genome instability are important signals associated with acquired resistance to TKI drugs. The combination of these signals provides essential information for prediction of resistance and selection of subsequent treatment.
Results
Clinical features of patient cohort
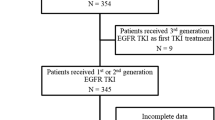
In this study, a total of 122 patients with advanced NSCLC were recruited from three hospitals in Vietnam (Vietnam National Cancer Hospital, Hanoi Oncology Hospital and Thu Duc District Hospital). The majority were treated with first generation TKI erlotinib (82 cases, 67.2%) and gefitinib (14 cases, 11.5%) while 3 patients (2.5%) received second generation TKI afatinib as first-line treatment (Table 1). Five patients were given erlotinib as an initial treatment, followed by a second-line treatment with either gefitinib (2 cases, 1.6%) or afatinib (3 cases, 2.5%). The remaining patients (18 cases, 14.8%) were confirmed to be administered with either first or second generation TKI but the specific drug name was not recorded (Table 1). Liquid biopsy from all 122 patients were sampled at the time of disease progression after achieving partial or stable disease (> 3 months) or at least 6 months post TKI treatment, satisfying the criteria of acquired resistance proposed by RECIST (Response Evaluation Criteria in Solid Tumours) or WHO15,16. The majority of patients were diagnosed at advanced stages (III-IV) (82%) with metastatic adenocarcinoma cancer (69.7%) (Table 1). Additional clinical characteristics and complete mutation results of all 122 patients are summarized in Table S1.
Heterogeneous mutational profiles of NSCLC patients undergoing acquired resistance to 1G or 2G TKI drugs
To profile the genetic alterations associated with TKI resistance, we used a liquid biopsy based assay to identify mutations, either single base substitutions or amplifications, in 9 major driver genes including EGFR, KRAS, BRAF, NRAS, ALK, ROS1, PIK3CA, MET and HER2. We detected multiple mutations that could potentially drive acquired resistance responses to TKI drugs in 51 patients (41.8%) (Fig. 1A).

The heterogeneity of mutation profiles of 122 Vietnamese NSCLC patients with acquired resistance to first and second generation EGFR TKI. (A) Distribution of TKI resistance associated mutations of 122 Vietnamese NSCLC. (B) Frequencies of EGFR sensitizing mutations (del19, L858R, or rare EGFR mutations) in different resistance mutation profiles. (C) Variant allele frequency (VAF, %) of EGFR sensitizing mutations in different resistance mutation profiles. Data are presented as median VAF, each data point represents one patient. **p < 0.01; ***p < 0.001 (one-way ANOVA, Kruskal–Wallis test).
Of the known resistance mutations, EGFR T790M was detected in 39 cases (32%), representing the most common resistance mutation among the Vietnamese NSCLC patients (Fig. 1A). Of those cases, the majority (23 cases, 18.8%) carried only the T790M mutation while the remaining 16 cases (13.2%) harbored extra co-occurring mutations. Seven of them (5.7%) carried a variety of rare EGFR mutations that were defined as non-classical mutations in the tyrosine kinase-domain (exons 18–21) including S768I (1 case), co-occurring S768I and G719C (2 cases), A750P (2 cases), V834L (1 case), and E709K (1 case). In addition, all 4 cases (2.5%) with PIK3CA mutations including 3 with mutation in codon 545 (E545K) and 1 with mutation in codon 1047 (H1047R) co-occurred with T790M (Figs. 1A and S1). The co-occurrence of T790M and EGFR amplification was detected in 5 (4.1%) out of 8 cases with EGFR amplification (Figs. 1A and S1).
In addition to mutations in EGFR, we detected resistance variants in off target genes including MET and HER2 amplification in 4 (3.3%) and 5 cases (4.1%), respectively (Fig. 1A). MET and HER2 amplification mutations appeared to be mutually exclusive with EGFR mutations (Figs. 1A and S1). No mutation was detected in KRAS, NRAS, BRAF, ALK or ROS1.
At the time of acquired resistance, the primary sensitizing mutations, either del19 or L858R were retained in almost all cases (46/51, 90.2%) carrying known resistance mutations, except for those with HER2 and MET applications showing either complete loss of sensitizing mutations or markedly lower VAFs (ranging from 1 to 2%) than cases with mutations in EGFR (Fig. 1B,C). By contrast, cases with EGFR amplification or those with co-occurrence of T790M and EGFR amplification harbored significantly higher sensitizing mutation VAFs than other cases without amplification (p < 0.01, Fig. 1C). We further found a significant positive correlation between levels of EGFR amplification and sensitizing VAFs (r = 0.94, 95% CI 0.68–0.99, p = 0.0006, Fig. S2A). These results suggested that EGFR amplification might occur in the primary EGFR sensitizing clones while HER2 and MET amplification could arise from de novo resistance tumor clones. For the majority of T790M + cases (33/39 cases, 84.6%), T790M had VAF lower than their corresponding sensitizing mutations (Fig. S2B), indicating that T790M might be acquired by resistance sub-clones.
Taken together, the analysis of genetic alterations in plasma samples revealed both inter- and intra-tumor heterogeneity of mutational profiles in Vietnamese NSCLC patients with acquired resistance to TKI drugs.
Distinct genome-wide methylation and copy number alterations in patients with EGFR dependent and independent resistance mechanisms
Genome-wide epigenetics, particularly DNA methylation changes, have been implicated in the acquisition of drug resistance by cancer cells30. However, the relationship between DNA methylation and mutation alterations in mediating resistance to TKI drugs remains largely unknown. To profile the methylation changes at the genome-wide level of different resistance mutation groups, we employed the workflow previously reported by Jiang et al.31. The assay was based on the combination of bisulfite conversion and massively parallel sequencing to analyze methylation density (MD) in bins of 1 Mb across 22 chromosomes. The analysis was performed for all cases with EGFR amplification (3 cases) and MET amplification (4 cases). For the remaining cases, only 3 out of 5 cases with co-occurring T790M and EGFR amplification and 4 out of 5 cases with HER2 amplification had sufficient cell free DNA for the analysis. Among 23 patients carrying T790M mutation, we randomly selected 5 cases with sufficient plasma. The total 19 chosen cases were grouped into either EGFR dependent resistance mutations (on-target) or EGFR independent mutations (off-target) which have comparable median age (69 versus 64.5 years, p > 0.05) (Fig. S3A). Strikingly, cases with resistance mutations in EGFR including T790M only, EGFR amplification and co-occurrence of EGFR amplification and T790M displayed extensive reduction in methylation density at genome-wide level compared to that of healthy subjects. By contrast, cases with off-target resistance mutations including HER2 and MET amplification showed comparable methylation density to healthy control subjects (Fig. 2A). We further quantified the proportion of hypo-methylated bins that have the MD lower than 3 SDs below the mean of the corresponding bins of the healthy subjects. We found a significantly higher proportion of hypo-methylated bins in cases with on target resistance mutations compared to those with off-target mutations (38.9% versus 8.3%, p < 0.01, Fig. 2B). There was no statistically significant difference in the proportion of hypo-methylated bins among the on-target mutation groups (Fig. 2B).

EGFR dependent (on-target) and independent (off-target) resistance mechanisms showed distinct landscapes of genome-wide methylation and copy number changes. (A, C) Methylation density (MD) (A) and DNA copy number (C) alterations for each 1 Mb bin along the chromosomes (chr1-22). Cases with T790M (n = 5), EGFR amplification (n = 3) and T790M and EGFR amplification co-occurrence (n = 3) were grouped into the on-target group while HER2 amplification (n = 4) and MET amplification (n = 4) cases were grouped into the off-target group. Healthy individuals (n = 20) were used as a reference group to determine the baseline levels of MD and CNA. (B, D) Percent of hypo-methylated bins (B) and CNA bins (D) in cases with different resistance mutation profiles grouped into the on-target group or the off-target group. The component bar graphs showed the comparison between the two groups. Data are shown as mean ± SEM. **p < 0.01, ****p < 0.0001 (Mann–Whitney test). (E) Linear regression and Pearson’s correlation between proportion of hypo-methylated bins and CNA bins in 19 patients from both on-target and off-target groups.
Genome-wide hypo-methylation has been associated with genome instability in cancer cells as reflected in DNA copy number aberrations (CNA)28. Hence, we further explored CNA in different resistance mutation groups. Consistent with the trend of hypo-methylation, we detected pronounced alterations in DNA copy number at multiple regions in cases with on-target resistance mutations, which were not observed in those carrying off-target mutations (Fig. 2C,D). As expected, we observed a significant correlation between numbers of hypo-methylated bins and CNA bins in all tested cases (R = 0.82, 95% CI 0.60–0.93%, p < 0.0001, Fig. 2E) and such alterations tended to occur across 22 chromosomes instead of localizing to any particular chromosomes (Fig. S3B,C).
These results showed that cases with on-target resistance mutations carried remarkable genome-wide hypo-methylation and CNA while not much changes were detected in off-target resistance mutation groups.
Genome-wide hypo-methylation and CNA correlated with the duration of drug response in EGFR amplification cases
The extensive changes in global hypo-methylation and CNA detected in cases with on-target mutations could be accumulated during the response to TKI drug treatment and may play important roles in the acquisition of resistance. Thus, we examined the correlation between these changes and the time to treatment resistance (TTTR). Since the levels of hypo-methylation and CNA were similar between EGFR amplification cases and those carrying co-occurring T790M and EGFR amplification, we put these samples into the same EGFR amplification group for the correlation analysis. Although both EGFR amplification and T790M cases displayed extensive changes in hypo-methylation and CNA, only EGFR amplification cases showed significantly positive correlations between CNA or hypo-methylation changes with TTTR (r = 0.94, 95% CI 0.6–0.99, p = 0.004 for CNA, Fig. 3A; r = 0.9 95% CI 0.3–0.98, p = 0.02 for hypo-methylation, Fig. 3B; insignificant correlations, p > 0.05 in T790M cases, Fig. S4A,B). Moreover, we found that the abundance of EGFR amplification mutations but not T790M in plasma significantly correlated with TTTR (r = 0.92, 95% CI: 0.5–0.99, p = 0.008, Fig. 3C for EGFR amplification cases and r = -0.18, p > 0.05; insignificant correlation, p > 0.05 for T790M cases, Fig. S4C). These results suggested that changes in DNA hypo-methylation and CNA concurrently occurred with EGFR amplification and were gradually accumulated during the treatment with TKI drugs. This process may be required to ensure the survival of tumor cells before acquiring the resistance.

Accumulation of genetic and epigenetic changes in cases with EGFR amplification correlated with the duration of response to TKI treatment. (A–C): Linear regression and Pearson’s correlation between proportion of CNA bins (A), hypo-methylated bins (B), EGFR amplification scores (C) and time to treatment resistance (TTTR) in 6 cases with EGFR amplification including 3 cases with co-occurring T790M.
Distinct hyper-methylation of transcriptional regulatory regions of cancer related genes in patients with EGFR dependent and independent resistance mutations
The acquisition of resistance to TKI might be associated with changes in expression of many genes, which could be affected by the methylation states of their transcriptional regulatory regions23,24. To identify such genes, we designed a panel of capture probes targeting 450 regions (Table S2) which were selected based on their significant contributions to cancer carcinogenesis reported by previous publications32,33,34,35,36,37. The majority of regions contain CpG islands overlapping or located in close proximity to promoter regions of cancer related genes including oncogenes and tumor suppressor genes. Distinct methylation patterns were observed in cases with on-target resistance mutations compared to those carrying off-target mutations, which showed methylation profiles similar to healthy control subjects (Fig. 4A). A total of 260 differentially methylated regions (DMR) out of 450 regions (57.8%) were identified when comparing on-target and off-target mutation groups. Of those, the majority (202 DMR) were significantly hyper-methylated (log2FC > 1, FDR < 0.05, Table S3) while the remaining 58 DMR were found hypo-methylated in cases with on-target resistance mutations compared to those with off-target mutations. Pathway enrichment analysis by gProfiler was performed on the set of genes mapped by those DMR. We found that 202 hyper-methylated regions were enriched in 5 pathways from the REACTOME and KEGG database including pathways regulating activation of HOX genes (2 pathways), the transcriptional regulation by TFAP2, the pluripotency of stem cells and the maturity of onset diabetes of the young (Fig. 4B). The sets of genes associated with those pathways were listed in Table S4. The activation of HOX genes and the regulation of transcription by TFAP2 pathways consisted of several component genes that were tumor suppressor genes involved in the regulation of tumor differentiation. The hyper-methylation at regulatory regions of such genes in cases with on-target resistance mutations could lead to the suppression of their expression, resulting in tumor progression. The functional contribution of the remaining two pathways to cancer tumorigenesis is not well understood. There was no significant pathway enriched by the 58 hypo-methylated DMR. The majority of those DMR were found to be fully methylated in cases with MET and HER2 amplification as well as healthy individuals, suggesting the re-activation of such genes in the on-target groups via reduced methylation at their promoter regions. These results showed that cases carrying resistance mutations in EGFR accumulated abundant hyper-methylation changes in regulatory regions of tumor suppressor genes such as HOX genes to inhibit the differentiation of cancer cells during the acquisition of TKI resistance. By contrast, cases with HER2 and MET amplification developed resistance to TKI via mechanisms independent of the acquisition of hyper-methylation of such genes.

EGFR dependent resistance mechanisms displayed markedly high levels of hyper-methylation in the transcriptional regions of genes involved in regulation of differentiation. (A) Heat-map showed differentially methylated regions (DMRs) among 450 target regions mapped to regulatory regions of cancer related genes across samples within the on-target group (T790M, T790M-EGFR amplification and EGFR amplification) and off-target group (HER2 and MET amplification). Healthy subjects were used as a reference group to determine the basal levels of methylation at target regions. Heatmap visualization was analyzed with ComplexHeatmap package version 2.8.0 (https://www.bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html). Hierarchical clustering was done using Euclidean distance. Red in the scale bar represents a high level of methylation. (B) KEGG and Reactome pathway enrichment analysis using g: Profiler (https://biit.cs.ut.ee/gprofiler) for genes associated with 202 DMRs showing hyper-methylation in the on-target group compared to the off-target group.
Discussion
Acquiring resistance to TKI drugs is a complex and dynamic process possibly governed by both genetic and epigenetic changes38. Profiling mutation and methylation landscapes simultaneously could lead to comprehensive understanding of the mechanisms for acquiring resistance and providing essential information for designing secondary treatment to overcome resistance. In patients with advanced NSCLC, liquid biopsy has become the only ethically feasible method for sampling and profiling these changes, highlighting its role in both academic research and clinical application. Here we took advantage of liquid biopsy to sample a large number of patients, and used ctDNA extracted from their blood to detect both genetic and epigenetic changes by massively parallel sequencing.
We uncovered the heterogeneity of both mutation and methylation alterations in 122 Vietnamese NSCLC patients who developed acquired resistance to 1G and 2G TKI drugs (Fig. 1). Multiple resistance mutations were detected in 51/122 patients (41.8%). Of those, T790M was identified as being the most frequent resistance mutation, detected in 32% of all cases, which is consistent with previous studies reporting a range of 30% to 50% from ctDNA39,40. In addition to mutations in the on-target gene EGFR, we detected substitution mutations in PIK3CA (E545K and H1047R) and amplification of MET and HER2, which have been reported to activate the bypass pathways of EGFR signaling41,42. These findings suggested that multiple resistance mutations could be associated with acquired resistance to TKI drugs, highlighting the inter-individual heterogeneity of resistance mechanisms. Our analysis also revealed the intra-tumor heterogeneity of resistance mutation profiles. Indeed, we detected multiple co-occurring resistance mutations present in the background of tumors carrying EGFR mutations in our cohort. It is thought that a single substitution in T790 codon, a key position in the ATP binding pocket, is sufficient to drive acquired resistance to first generation TKIs43. However, the co-occurrence of multiple resistance mutations in individual patients has been reported to be responsible for the failure of treatment targeting a single driver mutation in heterogeneous tumors41. This point was clearly demonstrated in our data where 33.3% (13/39) of T790M positive cases co-existed with other resistance-associated mutations including EGFR amplification and PIK3CA mutations. In addition, we detected a variety of rare EGFR mutations in T790M positive cases including mutations in codon E709, G719, A750, S768, V834L and L861 in 10 cases, with 8/10 cases also carrying T790M. Although these rare EGFR mutations are known to drive conformational changes in the drug binding site of EGFR, their contributions to TKI drugs responses are still under debate and warrant future clinical validation44.
We observed that the majority of cases carrying EGFR on-target mutations retained the primary sensitizing mutations, either del19 or L858R at the time of resistance. This suggested that EGFR on-target resistance mutations might arise from the same primary tumor clones. Among those cases, EGFR amplification cases displayed remarkably high levels of sensitizing VAFs which were significantly correlated with amplification scores (Fig. 3C), suggesting that the amplification occurred in the primary sensitizing clones and affected the copy number of the allele containing sensitizing EGFR mutations. In contrast to the on-target mutation cases, those with MET or HER2 amplification showed loss of primary sensitizing mutations and did not carry any other known resistance mutations. This suggested that MET and HER2 amplification could be de novo mutations and present as the sole drivers of resistance to TKI drugs.
Interestingly, our global methylation investigation also revealed the heterogeneity of methylation profiles in cases harboring different resistance mutation profiles. Cases with on-target mutations including T790M, EGFR amplification and their co-occurrence showed extensive hypo-methylation and CNA at genome-wide level which were not detected in off-target mutations including MET and HER2. This highlighted that different resistance mutation profiles were associated with different methylation changes. The interrelationship between DNA methylation and genetic changes is complex and possibly bidirectional21,45. It remains unknown whether mutations and methylation changes have causative roles or bidirectional relationship45.
We further detected the correlations between the levels of hypo-methylation or CNA and the duration of drug response for cases with EGFR amplification. Such correlations suggested that the amplification of EGFR and genome instability might have occurred concurrently and that they were accumulated gradually and possibly required for tumor cells to sustain their growth under the pressure of drug selection. In support to this notion, a previous study using both in vitro and in vivo models showed that amplification of the wild-type EGFR allele conferred resistance to TKI drugs through alleviating the inhibitor pressure of TKI drugs46. In contrast, T790M cases did not show correlation with the duration of drug response, suggesting that the changes in hypo-methylation and CNA in those cases could be passenger events that might not have a functional role during the response to 1G or 2G TKIs. Our finding was consistent with a recent study showing that genome-wide CNA was detected in patients carrying T790M at the time of resistance to gefitinib and that the presence of CNA was associated with poor response to subsequent treatment with 3G TKI, osimertinib47. MET and HER2 amplification did not show noticeable changes in both global methylation and CNA, suggesting that the acquisition of genome instability during the response to TKI drugs might be unique to the EGFR dependent resistance mechanisms.
In addition to changes at genome-wide levels, we also detected hyper-methylation of regulatory regions of multiple genes that might be involved in TKI resistance. Of those, a group of Homeobox (HOX) genes involved in regulating growth and differentiation of cancer cells were found to be hyper-methylated. The hyper-methylation status at transcriptional regulatory regions of those genes indicated their expression was suppressed in cases with EGFR dependent resistance mutations. Our finding was consistent with a recent study by Shu et al.48 showing that increased methylation of HOXB9 correlated with higher rate of resistance to EGFR-TKI. Thus, such consistent findings indicated that the hyper-methylation status of HOX genes could be exploited to predict and treat EGFR amplification mediated resistance.
There were several limitations in our study. First, this was a retrospective collection of cases for which the cumulative changes of genetic and methylation information were analysed at the time of TKI resistance. Therefore, our analysis was unable to elucidate how mutation or methylation changes evolved during the treatment process. A longitudinal analysis is warranted to understand the relationship of such changes. Second, a main limitation of our study is the small sample size. Due to low frequencies of certain mutation subtypes and insufficient plasma samples, only a small number of samples in each group were subjected to methylation and CNA analysis. Thus, our current study might be considered as exploratory analyses and future studies with a larger cohort are required for robust analysis of resistance mechanisms among different mutation profiles. Third, due to the lack of paired tumor biopsies, the amplifications detected by our liquid biopsy method could not be validated by a gold standard technique such as FISH. However, the higher VAF of sensitizing mutation or T790M in cases with EGFR amplification compared to those without EGFR amplification suggested that the amplification events detected were true positives. Lastly, we could not detect any known resistance mechanisms for 71 patients (58.2%) in our cohort, suggesting that there were additional mechanisms driving acquired resistance and not captured by our assay. The additional mechanisms could include changes in tumor histology with tumor cells displaying features of small-cell lung cancer (SCLC) or epithelial mesenchymal transition (EMT), or alterations in other novel genes involved in the MAP kinase and mTOR pathway41. Therefore, an extended panel covering additional driver genes or higher resolution of methylation and CNA analysis will be required to identify new resistance mechanisms.
In conclusion, we have shown a clinical application of liquid biopsy in profiling both mutation and methylation alterations in patients with advanced NSCLC who developed resistance to TKI drugs. Our findings uncovered distinct patterns of DNA methylation and copy number changes between patients harboring either EGFR dependent and independent resistance mutations (Fig. S5), highlighting the heterogeneity of acquired resistance to TKI drugs for which several pathways may be involved.
Methods
Patient recruitment
A total of 122 patients from Vietnam National Cancer hospital, Ha Noi Oncology hospital and Thu Duc district hospital were recruited to this study between 2018 and 2020. 54 out of 122 patients were from the research funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED). The recruitment criteria are patients with advanced NSCLC (stage III or IV) who had received either first (gefitinib or erlotinib) or second generation (afatinib) EGFR-TKI therapy and showed acquired resistance responses (according to WHO or RECIST criteria)16,49. We also recruited 20 healthy individuals to obtain cfDNA for validation of our MET amplification detection pipeline. Plasma samples were collected at the time of disease progression and after at least 6 months following initial TKI drug treatment. This study was approved by the Ethic Committee of University of Medicine and Pharmacy at Ho Chi Minh City, Vietnam. All patients and healthy individuals were given written informed consent prior to participation in the study. All methods were performed in accordance with the relevant guidelines and regulations.
Plasma cell free DNA isolation
Peripheral blood (10 mL) was drawn in Streck tubes (Cell-free DNA BCT, Streck) and subject to two rounds of centrifugation (2000×g for 10 min then 16,000×g for 10 min) to separate plasma from blood cells. The plasma fraction (4–6 mL) was collected, aliquoted (2 mL per aliquot) and stored at − 80 °C until cell free DNA extraction.
Cell free DNA was extracted from an aliquot of 2 mL of plasma using the MagMAX Cell-Free DNA Isolation kit (Thermo Fisher, USA) following the manufacturer’s instructions.
Ultra-deep massively parallel sequencing (MPS) with unique molecular identifier tagging
Cell free DNA was prepared and sequenced as previously described50. Briefly, a library with unique molecular identifier tagging was prepared from 2 ng of cfDNA using the Accel-NGS 2S Plus DNA library kit (Swift Biosciences, USA) following the manufacturer's instructions. Library concentrations were quantified using QuantiFluor dsDNA system (Promega, USA). Equal amounts of libraries (150 ng per sample) were pooled together and hybridized with xGen Lockdown probes for 9 targeted genes EGFR, KRAS, NRAS, BRAF, ALK, ROS1, MET, HER2 and PIK3CA (IDT DNA, USA). For ALK and ROS1, customized probes for intron regions were designed and mixed with probes for exon regions at equal concentrations. For detection of plasma EGFR, HER2 and MET amplification, circulating tumor DNA copy number variation pipeline was used as previously described51. The pipeline was evaluated using cfDNA sequencing data of 20 healthy people and positive MET amplification control DNA (Horizon, USA).
Sequencing was run using NextSeq 500/550 High output kits v2 (150 cycles) on Illumina NextSeq 550 system (Illumina, USA) with minimum target coverage of 10,000 ×. A total on-target fraction of 20–40% was detected.
Variant calling using Mutect2
Each sample was barcoded with a single 8-bp index in the P7 primer and each DNA fragment was tagged with a unique identifier consisting of a random 9-bp sequence within the P5 primer. Pair-end (PE) reads and the corresponding unique identifier sequences were generated using the bcl2fastq package (Illumina). The reads were aligned to the human genome (hg38) using BWA and then grouped by the unique identifier in order to determine a consensus sequence for each fragment, eliminating sequencing and PCR errors that account for less than 50% of reads per fragment. The consensus reads were used for final variant calling using Mutect252. A custom pipeline with call to BWA 0.7.153, Picard 2.18.23 (Broad Institute, GitHub Repository), Samtools 1.954 and Fulcrum genomic analysis packages51 were built to perform the above-mentioned analysis steps. Unique molecular identifier (UMI) grouping was performed by using the fgbio 0.8.0 package (Fulcrum Genomics)51. For detection of ALK and ROS1 rearrangement, fusion variant calling was analyzed using Factera v1.4.4 with default parameters54. For detection of EGFR, MET and HER2 amplification, FACETS V0.5.6 was used to analyse copy number variation55.
Bisulfite treatment and sequencing library preparation
Among cases with known mutational profiles, 19 cases had sufficient extracted cell free DNA were analysed for methylation changes at both genome-wide and targeted regional levels. A minimum of 2 ng isolated cfDNA of those samples was subjected to bisulfite conversion using EZ DNA Methylation-Gold™ Kit (ZYMO RESEARCH, D5006, USA). Bisulfite-converted DNA was subject to dual indexed library preparation using Accel-NGS® Methyl-Seq DNA Library Kit (Swift Biosciences, 30024, USA) and the library products were subsequently divided into 2 portions for target and genome wide sequencing.
Genome wide methylation and CNA analysis
One half of the DNA library was directly sequenced on MGI DNBSEQ-G400 system in a 100 bp paired end format and at a sequencing depth of 20 million reads per case (equivalent to 0.6 ×). The analysis of methylation and CNA was performed using the Methy-Pipe31. Briefly, the genome was divided into multiple 1 Mb bins. The methylation density (MD) for each bin was calculated as the number of methylated cytosines in the context of CpG dinucleotides divided by the total number of cytosines at CpG positions. Hypomethylated bins are defined as those with MD lower than 3 SDs (standard deviation) below the mean of the corresponding bin of the healthy subjects. For CNA analysis, sequenced reads aligned to each bin were counted. In order to prevent GC bias, sequence counts were corrected by GC-content and normalized with the median read counts of all bins (QDNAseq)56. The z-score of each bin was then calculated based on the mean and SD of the corresponding bin in the healthy subjects. The bin with aberration in copy number was defined as having the z-score > 3 or < − 3. Subsequently, the proportion of CNA bins for each sample was calculated.
Target methylation analysis
Half of the DNA library was enriched using a customized panel of xGen Lockdown Probes targeting 450 regions and covering 9593 CpG sites (IDT, USA). The panel was constructed based on previous publications and the 450 regions were mapped to regulatory regions including promoters or enhancers of tumor suppressor genes involving cancer progression (Table S2)32,33,34,35,36,37. The hybridization was performed using XGen hybridization and wash kit (IDT, 1072281, USA). Each sample was sequenced at 20 million reads on the MGI DNBSEQ-G400 system. After trimming sequencing adapters, sequenced reads were aligned to reference genome and methylation calling was performed using Bismark aligner57. The methylation patterns were analyzed to find the differences among sample groups based on the methylation percentage of each region examined. The methylation percentage of each region is calculated by the formula: %Methylation = methylated cytosine (C)/(methylated C + un-methylated C). The methylated cytosines were those cytosines recovered among the CpG dinucleotides on the sequenced reads mapped to the target regions whereas un-methylated C were recovered thymine among the CpG dinucleotides on the sequenced reads mapped to the target regions.
Statistical analysis
Wilcoxon Rank Sum test and was performed for identifying the regions that were differentially methylated between EGFR dependent and independent groups. The p values were corrected by the Benjamini Hochberg method for multiple comparisons. Pearson’s correlation analysis was performed to analyze the correlations between levels of hypo-methylation or CNA and time to treatment resistance or the correlation of mutation abundance between the resistance and sensitizing mutations. Pathway analysis was carried out using gProfiler to identify biological pathways significantly enriched based on the set of genes associated with DMR58.
Data availability
All data generated or analyzed during this study are included in this published article. Sequencing data will be deposited in a public portal database (NCBI SRA) upon acceptance.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Sher, T., Dy, G. K. & Adjei, A. A. Small cell lung cancer. Mayo Clin. Proc. 83, 355–367 (2008).
Mok, T. S. et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957 (2009).
Shepherd, F. A. et al. Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 353, 123–132 (2005).
Maemondo, M. et al. First-line gefitinib in patients aged 75 or older with advanced non-small cell lung cancer harboring epidermal growth factor receptor mutations: NEJ 003 study. J. Thorac. Oncol. 7, 1417–1422 (2012).
Yang, J.C.-H. et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 16, 141–151 (2015).
Sequist, L. V. et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 31, 3327–3334 (2013).
Passaro, A., Jänne, P. A., Mok, T. & Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nature Cancer 2, 377–391 (2021).
Solassol, I., Pinguet, F. & Quantin, X. FDA- and EMA-approved tyrosine kinase inhibitors in advanced EGFR-mutated non-small cell lung cancer: safety, tolerability, plasma concentration monitoring, and management. Biomolecules 9, 1–10 (2019).
Rosell, R. et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 361, 958–967 (2009).
Stewart, E. L., Tan, S. Z., Liu, G. & Tsao, M. S. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations: A review. Transl Lung Cancer Res 4, 67–81 (2015).
Wang, J., Wang, B., Chu, H. & Yao, Y. Intrinsic resistance to EGFR tyrosine kinase inhibitors in advanced non-small-cell lung cancer with activating EGFR mutations. Onco. Targets. Ther. 9, 3711–3726 (2016).
Santoni-Rugiu, E. et al. Intrinsic resistance to EGFR-tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: Differences and similarities with acquired resistance. Cancers 11, 923 (2019).
Kubo, T. et al. MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int. J. Cancer 124, 1778–1784 (2009).
Camidge, D. R., Pao, W. & Sequist, L. V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 11, 473–481 (2014).
Jackman, D. et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 28, 357–360 (2010).
Engelman, J. A. et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043 (2007).
Zhang, Z. et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 44, 852–860 (2012).
Terai, H. et al. Activation of the FGF2-FGFR1 autocrine pathway: A novel mechanism of acquired resistance to gefitinib in NSCLC. Mol. Cancer Res. 11, 759–767 (2013).
Oser, M. G., Niederst, M. J., Sequist, L. V. & Engelman, J. A. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 16, e165–e172 (2015).
Glasspool, R. M., Teodoridis, J. M. & Brown, R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br. J. Cancer 94, 1087–1092 (2006).
Hou, T. et al. Decitabine reverses gefitinib resistance in PC9 lung adenocarcinoma cells by demethylation of RASSF1A and GADD45β promoter. Int. J. Clin. Exp. Pathol. 12, 4002–4010 (2019).
Maeda, M. et al. CpG hypermethylation contributes to decreased expression of PTEN during acquired resistance to gefitinib in human lung cancer cell lines. Lung Cancer 87, 265–271 (2015).
Yang, B., Yang, Z.-G., Gao, B., Shao, G.-G. & Li, G.-H. 5-Aza-CdR can reverse gefitinib resistance caused by DAPK gene promoter methylation in lung adenocarcinoma cells. Int. J. Clin. Exp. Pathol. 8, 12961–12966 (2015).
Blair, L. P. & Yan, Q. Epigenetic mechanisms in commonly occurring cancers. DNA Cell Biol. 31(Suppl 1), S49-61 (2012).
Flavahan, W. A., Gaskell, E. & Bernstein, B. E. Epigenetic plasticity and the hallmarks of cancer. Science 357, 6348 (2017).
Darwiche, N. Epigenetic mechanisms and the hallmarks of cancer: an intimate affair. Am. J. Cancer Res. 10, 1954–1978 (2020).
Brennan, K. & Flanagan, J. M. Is there a link between genome-wide hypomethylation in blood and cancer risk?. Cancer Prev. Res. 5, 1345–1357 (2012).
Zhang, W. et al. Global DNA hypomethylation in epithelial ovarian cancer: Passive demethylation and association with genomic instability. Cancers 12, 764 (2020).
Niu, X. et al. Genome-wide DNA methylation analysis reveals GABBR2 as a novel epigenetic target for EGFR 19 deletion lung adenocarcinoma with induction erlotinib treatment. Clin. Cancer Res. 23, 5003–5014 (2017).
Jiang, P. et al. Methy-Pipe: an integrated bioinformatics pipeline for whole genome bisulfite sequencing data analysis. PLoS ONE 9, e100360 (2014).
Chen, X. et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 11, 1–10 (2020).
Zhang, C. et al. DNA methylation analysis of the SHOX2 and RASSF1A panel in bronchoalveolar lavage fluid for lung cancer diagnosis. J. Cancer 8, 3585–3591 (2017).
Tsou, J. A. et al. DNA methylation profile of 28 potential marker loci in malignant mesothelioma. Lung Cancer 58, 220–230 (2007).
Hulbert, A. et al. Early detection of lung cancer using DNA promoter hypermethylation in plasma and sputum. Clin. Cancer Res. 23, 1998–2005 (2017).
Selamat, S. A. et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 22, 1197–1211 (2012).
Nelson, H. H. et al. Key epigenetic changes associated with lung cancer development: results from dense methylation array profiling. Epigenetics 7, 559–566 (2012).
Wen, J., Fu, J., Zhang, W. & Guo, M. Genetic and epigenetic changes in lung carcinoma and their clinical implications. Mod. Pathol. 24, 932–943 (2011).
Jin, Y. et al. Mutational profiling of non-small-cell lung cancer patients resistant to first-generation EGFR tyrosine kinase inhibitors using next generation sequencing. Oncotarget 7, 61755–61763 (2016).
Liam, C. K., Wahid, M. I., Rajadurai, P., Cheah, Y. K. & Ng, T. S. Epidermal growth factor receptor mutations in lung adenocarcinoma in Malaysian patients. J. Thorac. Oncol. 8, 766–772 (2013).
Rotow, J. & Bivona, T. G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 17, 637–658 (2017).
Sequist, L. V. et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 3, 7526 (2011).
Kobayashi, S. et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792 (2005).
Harrison, P. T., Vyse, S. & Huang, P. H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 61, 167–179 (2020).
You, J. S. & Jones, P. A. Cancer genetics and epigenetics: Two sides of the same coin?. Cancer Cell 22, 9–20 (2012).
Nukaga, S. et al. Amplification of EGFR wild-type alleles in non-small cell lung cancer cells confers acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors. Cancer Res. 77, 2078–2089 (2017).
Buder, A. et al. Somatic copy-number alterations in plasma circulating tumor DNA from advanced EGFR-mutated lung adenocarcinoma patients. Biomolecules 11, 618 (2021).
Su, S.-F. et al. Genome-wide epigenetic landscape of lung adenocarcinoma links HOXB9 DNA methylation to intrinsic EGFR-TKI resistance and heterogeneous responses. JCO Precision Oncol. 5, 418–431 (2021).
Wu, S. G. & Shih, J. Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 17, 38 (2018).
Tran, L. S. et al. Ultra-deep massively parallel sequencing with unique molecular identifier tagging achieves comparable performance to droplet digital PCR for detection and quantification of circulating tumor DNA from lung cancer patients. PLoS ONE 14, e0226193 (2019).
Burriesci, M. S., Lehnert, E. M. & Pringle, J. R. Fulcrum: Condensing redundant reads from high-throughput sequencing studies. Bioinformatics 28, 1324–1327 (2012).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Newman, A. M. et al. FACTERA: A practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics 30, 3390–3393 (2014).
Shen, R. & Seshan, V. E. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016).
Scheinin, I. et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 24, 2022–2032 (2014).
Krueger, F. & Andrews, S. R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011).
Raudvere, U. et al. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Acknowledgements
This research is funded by The Vietnam National Foundation for Science and Technology Development (NAFOSTED, https://nafosted.gov.vn/) under Grant Number 108.01-2017.306 (to HNN).
Author information
Authors and Affiliations
Contributions
Conceptualization: H.N.N., D.M.D., S.D.N., A.V.H., K.T.D., M.D.P., H.G., L.S.T. Data curation: H.N.N., Q.T.T.N., T.H.T.N., C.V.N., K.N.D.L., H.T.L., M.L.T.N., T.V.N., V.U.T., B.A.L., L.G.H.L., Q.C.H., H.A.T.P., B.T.V., L.T.N., T.C.V.N., A.T.H.D., D.T.N., L.S.T. Formal analysis: H.N.N., N.P.T.C., M.D.P., H.G., L.S.T. Funding acquisition: H.N.N., D.M.D., A.V.H. Methodology: H.G., H.N.N. Project administration: T.T.T.D., K.T.D. Software: N.P.T.C., H.G. Supervision: H.N.N. Writing-original draft: L.S.T. Writing-review & editing: M.D.P., H.G., H.N.N.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nguyen, HN., Cao, NP.T., Van Nguyen, TC. et al. Liquid biopsy uncovers distinct patterns of DNA methylation and copy number changes in NSCLC patients with different EGFR-TKI resistant mutations. Sci Rep 11, 16436 (2021). https://doi.org/10.1038/s41598-021-95985-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-95985-6
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
