
Abstract
Currently, there are increasing concerns about the possibility of a new epidemic due to emerging reports of Mayaro virus (MAYV) fever outbreaks in areas of South and Central America. Haemagogus mosquitoes, the primary sylvan vectors of MAYV are poorly characterized and a better understanding of the mosquito’s viral transmission dynamics and interactions with MAYV and other microorganisms would be important in devising effective control strategies. In this study, a metatranscriptomic based approach was utilized to determine the prevalence of RNA viruses in field-caught mosquitoes morphologically identified as Haemagogus janthinomys from twelve (12) forest locations in Trinidad, West Indies. Known insect specific viruses including the Phasi Charoen-like and Humaiata-Tubiacanga virus dominated the virome of the mosquitoes throughout sampling locations while other viruses such as the avian leukosis virus, MAYV and several unclassified viruses had a narrower distribution. Additionally, assembled contigs from the Ecclesville location suggests the presence of a unique uncharacterized picorna-like virus. Mapping of RNA sequencing reads to reference mitochondrial sequences of potential feeding host animals showed hits against avian and rodent sequences, which putatively adds to the growing body of evidence of a potentially wide feeding host-range for the Haemagogus mosquito vector.
Similar content being viewed by others

Introduction
Mosquitoes serve as important vectors for many infectious agents that contribute to significant morbidity and greater than 1 million human deaths yearly1,2. Viruses transmitted by mosquito vectors include dengue virus, chikungunya virus, yellow fever, and Zika virus, which have all caused major pandemics within recent times. Furthermore, many neglected tropical and other emerging viral diseases transmitted by mosquitoes are still poorly understood. The advent of deep sequencing technologies has now made the detection and quantification of viral agents much easier and affordable. This technological advancement is now contributing to increased discovery rates and a better understanding of uncharacterized viruses, including many insect-associated viruses of human health importance3,4,5,6,7,8,9. Previously uncharacterized viruses with unknown pathogenicity against humans or wildlife have also been detected by RNA techniques10,11. Furthermore, microbiome-based analyses allow for the elucidation of interactions among viruses and other biological systems associated with mosquitoes and their effect on vector competence and transmission of infectious viral agents.
The mosquito Haemagogus janthinomys (Hg. janthinomys) has been reported as the primary vector in the transmission of the Mayaro virus (MAYV), an emerging alphavirus endemic to regions of South and Central America12,13,14,15. The recent detection of MAYV fever cases in previously unreported areas in South America has triggered alarm bells concerning the spread of this virus in the Americas15,16,17,18. Several epidemiological models predict this virus will follow the steps of the Zika and chikungunya viruses in causing a major epidemic in the future19,20. In the event of an epidemic, understanding the virus-host interactions would be critical for developing effective management systems for the MAYV disease. Furthermore, the role of urban mosquitoes such as Aedes and Anopheles species as a potential driving force for spread of MAYV has also been investigated21,22. However, Haemagogus species are the poorly characterized mosquitoes and there is little data available on their viral transmission dynamics.
In this study, we used a transcriptomic-based approach to map the RNA virome of wild caught adult female mosquitoes morphologically identified as Hg. janthinomys from twelve forested locations in Trinidad, West Indies.
Results
RNA Sequencing data from field caught Haemagogus mosquitoes
All mosquitoes from the twelve forested locations included in the RNA sequencing analysis were identified as Hg. janthinomys based on standard taxonomic keys23,24,25 and wing geometric morphology26. However, a subsequent barcoding study conducted on Hg. janthinomys from Trinidad based on sequencing of the cytochrome c oxidase subunit I (COI) gene and the internally transcribed spacer region 2 (ITS2) showed that the population had three distinct genotypes that may represent a species complex, but this needs to be confirmed by additional studies (Ali et al., unpublished data).
Total RNA sequencing of the Haemagagus mosquito pools from the 12 sites yielded a total of 857,590,224 paired end reads after quality control processing, with an overall mean of 71,465,852 and a range of 57,884,090 to 87,768,438 reads as shown in Table 1. Direct Bowtie2 mapping of the total RNA sequences onto representative nucleic acid databases showed average mapping rates of 2415.39 reads per million (RPM) per location against reference viral sequences, 4.60 RPM against potential mosquito feeding host genomes and 1211.60 RPM against the Silva reference 16S bacterial database. The sites with the highest viral species richness were the Ecclesville and Mamon Village locations which included alphaviruses, insect-specific, mosquito-associated, and other viral types, with remaining collection sites having lower viral species richness (Fig. 1).

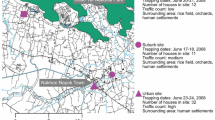
(a) Haemagogus mosquito sampling sites and (b) heatmap of viral reads from twelve forested locations in Trinidad. 1: Chaguaramas (CH), 2: Hollis (H), 3: Cumana (CU), 4: Caroni Swamp (CBS), 5: Mamoral (MO), 6: Mamon village (MA), 7: Ecclesville (E), 8: Claxton Bay (CB), 9: Rousillac (R), 10: Catshill (CA), 11: Morne Diablo (MD), 12: Quinam (Q).
Viral sequences
Mamoral (28,634.83 RPM) overwhelmingly had the highest proportion of mapped viral reads followed by Claxton Bay (203.26 RPM) and Ecclesville (100.06 RPM) (Table 1). The other sites had relatively low proportions of viral reads (0.90–22.83 RMP) except for Catshill, where no viral read was detected. Generally, the majority of the viral RNA reads mapped to known insect specific viruses which included the Phasi Charoen-like and Humaita-Tubiacanga viruses. Additionally, RNA sequencing reads mapped to several viruses associated with a range of insects and feeding hosts and included some of human health importance. Trinity v2.9.1 assembly of mapped viral reads resulted in a total of 171 viral contigs and 4 unassembled reads (Table 2).
Known Insect Specific Viruses (ISVs)
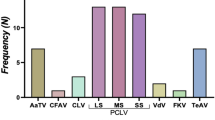
The metaviromes (Fig. S1) were dominated by one ISV, the Phasi Charoen-like virus (PCLV), which has been previously reported in Aedes aegypti (Ae. aegypti) mosquitoes7,27,28. Overall, PCLV was identified in nine locations (1.65–17 064.28 RPM, mean = 1442.02 RPM) while the Humaiata-Tubiacanga virus (HTV), another well-known ISV, was identified in eight (0.09–710.30 RPM, mean = 59.33 RPM) of the twelve locations sampled (Fig. S1). De novo assembly of the viral reads using Trinity v2.9.1 resulted in 3 to 27 contigs (243–6675 bp) of PCLV per site with similarity levels to reference sequences in GenBank ranging from 95.44 to 99.87%. Phylogenetic analysis of segments of the S (551 bp), L (451 bp) and M (439 bp) regions of representative sequences from the different sites showed most of the local sequences clustering into one main branch for all the segments with high bootstrap support (Fig. 2). However, the sequences for all three segments from the Caroni Bird Sanctuary clustered separately from other Trinidad sequences into well-supported clades (81–99% bootstrap support) including reference sequences (Fig. 2a,c), with segments S and M having closest links to sequences from Grenada. Additionally, the local sequences of segment L from Morne Diablo also separated and formed a separate clade (Fig. 2c; 100% bootstrap support).

Maximum-likelihood phylogenetic trees of selected (a) Phasi Charoen-like segment S (551 bp), (b) Phasi Charoen-like segment L (451 bp), (c) Phasi Charoen—like segment M (439 bp) and (d) Humaita-Tubiacanga segment 1 (188 bp) of viral sequences from forest locations in Trinidad. The trees were constructed in MEGA X (Version 10.1), using the Kimura 2-parameter substitution model and 1000 bootstrap pseudoreplicates. Only bootstrap values ≥ 80% are shown. The Trinidad sequences are tagged with the red diamonds.
Assembly of the HTV viral reads showed 1–3 contigs per site (243–1998 bp) with similarities to reference sequences in GenBank ranging from 97.80 to 99.13%. Phylogenetic analysis of a 188 bp region of segment 1 from the different sites showed all local sequences clustering separately from four references from Guadeloupe, with five (5) of the Trinidad sequences in one distinct sub-clade with 91% bootstrap support, the Caroni Bird Sanctuary and Mamoral sequences in a second strongly supported sub-clade (97% bootstrap support), and the Morne Diablo sequence on a weakly-supported branch (Fig. 2d).
Mosquito-borne viruses
A relatively small proportion of viral reads (4.80 RPM) were classified as alphaviruses (Togaviridae family), which accounted for approximately 0.0005% of all viral reads from the twelve locations (Fig. S1). Among these, 10 reads (4 from Mamon Village and 6 from Ecclesville at 0.06 and 0.10 RPM, respectively) mapped onto the genome of the MAYV. Trinity assembly of the mapped MAYV viral reads showed the six reads (three read pairs) from Ecclesville assembled into three contigs 166–277 bp long (Table 3) that aligned to different regions of reference MAYV genomes. However, the four MAYV reads from Mamon Village were unassembled. Blastn analysis revealed, all the contigs and reads had 99.29%-100% similarity to MAYV non-structural (nsp1-3 and nsp4) and structural polyprotein coding regions (Table S1). Most of these sequences had highest similarity to reference South American MAYV sequences, the majority of which were from Venezuela, Bolivia, and Peru (Table 3). Unassembled read M_MAYVF2 from Mamon Village had the highest similarity matches to reference MAYV sequences that originated from Trinidad, and these came from strains isolated in the 1950s, when the first infections by MAYV were reported.
Mosquito-associated viruses
A relatively small number of reads mapped to reference genomes of mosquito-associated viruses (Fig. S1) including the Wuhan mosquito virus 6 (Bunyaviridae) (35.13 RPM), Hubei virga-like virus 2 (Rhabdoviridae) (8.84 RPM) and the Merida virus (Unclassified) (1.86 RPM). These mosquitoes-associated viruses were identified in the Ecclesville, Chaguaramas, Claxton Bay and Mamoral locations. Overall, the Ecclesville metavirome (Fig. S1) was affiliated with a higher number of mosquito-associated viral reads (90.31 RPM) and showed the greatest diversity when compared to other sampling locations including Claxton Bay (1.92 RPM) and Chaguaramas (1.40 RPM).
All mosquito-associated viral reads identified in the Chaguaramas (Fig. 1) and the Claxton Bay metavirome (Fig. S1) belonged to the unclassified viruses. The Guadeloupe mosquito virus sequence (Sobemovirus) was the only mosquito-associated sequence identified in the Mamoral location. Assembly of the mapped viral reads using Trinity v2.9.1 resulted in 1 contig (2162 bp) for the Wuhan mosquito 6 virus, 3 contigs (547–5596 bp) for Hubei virga-like virus, and 10 contigs (234–671 bp) for the Merida virus. Blastn analysis of the contigs of the three viruses showed highest identity of 96.85%, 99.14–99.63% and 97.17–99.15%, respectively, to GenBank reference sequences. Based on phylogenetic analysis for the segment 3 region for the Wuhan mosquito 6 virus (2133 bp), the local Ecclesville sequence diverged from the reference sequences from Australia (Fig. S1a). Furthermore, 1 contig (2978 bp) for the Guadeloupe mosquito virus was identified only in the Mamoral metavirome. Phylogenetic analysis of an 1859 bp region of segment 1 (Fig. S1b) showed the sequence separating from all reference sequences from Guadeloupe. However, it should be noted that bootstrap values were not significant for separation of the Trinidad viral sequences from reference sequences for Wuhan mosquito 6 and the Guadeloupe mosquito virus.
Other viruses
RNA sequencing reads from Eccleville also mapped to the picorna-like virus (44.40 RPM) and reads from the Chaguaramas site mapped to unclassified viruses including Kaiowa virus (1.16 RPM), Cumabru-like virus (0.08 RPM) and Guato-like virus (0.16 RPM). The Kaiowa virus (1.30 RPM) and Guato-like virus (0.62 RPM) were also detected in mosquitoes from Claxton Bay. Additionally, reads mapped to other viruses including the avian leukosis virus in Ecclesville (1.05 RPM), Mamon Village (2.57 RPM), Claxton Bay (0.58 RPM), Cumana (1.40 RP), Hollis (1.43 RPM), Morne Diablo (1.57 RPM) and Rousillac (0.69 RPM); the black queen cell virus in Quinam (0.90 RPM); and the pepper mild mottle virus in Claxton Bay (0.06 RPM).
Trinity v2.9.1 assembly of the reads resulted in 2 contigs (826–1172 bp) of the unclassified Kaiowa virus (99 to 100% similarity levels) in the Claxton Bay and Chaguaramas sites, as well as 1 contig (203 bp) of the Cumbaru-like virus (similarity level = 93%) and 4 contigs (256–300 bp) of the Guato-like viruses (similarity level = 98–99%) from the Chaguaramas location (Table S1). Kaiowa and Guato viruses were previously found in mosquitoes from Brazil29.
Furthermore, 5 contigs (320–4519 nt) from Ecclesville were unique, with highest similarity levels (94.94–96.94%) to the Atrato picorna-like virus. Phylogenetic analysis of a 350 bp region of the polyprotein segment (Fig. S1c) resulted in all Ecclesville contigs forming a single clade that diverged from the GenBank reference Atrato picorna-like virus sequence. A relatively large number (33) of contigs (217–1020 nt) from 7 locations (Hollis, Cumana, Mamon Village, Ecclesville, Claxton Bay, Rousillac and Morne Diablo) aligned with high nucleotide identity (99–100%) to the avian leukosis virus strains from China. Maximum likelihood phylogeny of the avian leukosis viral contigs resulted in local sequences separating into two main clades with sequences from Cumana and Hollis clustering together (Fig. S1d) and sequences from Rousillac and Mamon village forming a strongly supported clade. This suggests the local avian leukosis viruses are highly diverse and may include unique lineages as compared to the reference sequences available in the GenBank database. Four contigs (289–442 bp) from the Ecclesville location aligned with 94–96% nucleotide identity to unclassified bat sobemoviruses and luteoviruses (Table S1).
Potential feeding host sequences
Mapping of RNA reads to potential mosquito blood-meal host mitochondrial sequences showed that, although levels were relatively low, there were hits to the avian species, Gallus gallus, at four locations (Claxton Bay- 0.61 RPM, Mamon Village- 1.85 RPM, Rousillac-0.49 RPM, and Hollis-0.75 RPM) and to the rodent species, Mus musculus, at two locations (Chaguaramas-17.73 RPM and Catshill-3.72 RPM). Assembly of the avian reads resulted in 17 contigs (203–982 nt) with > 99% nucleotide identity to Gallus gallus and assembly of the rodent reads resulted in 25 contigs (240–966 nt) with > 99% identity to Mus musculus (Table S1). The contig sequences are available at: https://zenodo.org/record/4932469#.YMQbQKhKi01.
Bacterial sequences
In all twelve locations, a relatively moderate level of reads (717.28 RPM) mapped to the SILVA bacterial 16S rRNA gene sequence database (Version 132). As seen in Fig. 3, the majority of mapped reads from all locations were classified as Proteobacteria (relative abundance = 96.5–57.3%), and within this phylum, the Gammaproteobacteria class dominated at all locations (relative abundance = 61.6–97.0%) except Chaguaramas (7.4%), Claxton Bay (37.9%) and Rousillac (36.6%). Mapping of the RNA sequences against 301 representative 16S rRNA gene sequences of Wolbachia spp. showed a mapping rate ranging from 0.92 Log10 RMP to 3.6 Log10 RPM. Assembly of reads mapped to the Wolbachia 16S rRNA genes resulted in 7 contigs (234–1569 nt) from four sites (Claxton Bay, Chaguaramas, Ecclesville, Rousillac) that showed 96.04–100% similarity to Wolbachia species based on Blastn analysis. The Wolbachia contig sequences are available at https://zenodo.org/record/4932469#.YMQbQKhKi01.

Plots of relative abundance of Haemagogus RNA sequence reads mapped to the Silva bacterial database showing phyla diversity and domination of Proteobacteria throughout all locations.
Discussion
This is the first report on the virome of any Haemagogus mosquito species and provides important baseline data on a range of viruses associated with these mosquitoes. The data shows the presence of nucleic acid sequences from arboviruses of human and animal health significance in Haemagogus mosquitoes found in Trinidad, which may have epidemiological implications for this region. Viral sequences identified include those that belong to the families Bunyaviridae, Togaviridae and Rhabdoviridae, which have been previously reported to be a part of the viromes of other mosquito species3,4,5,6,11. The insect specific viruses (ISVs) characterized were generally similar for most of the sampling locations on the island apart from Catshill and Chaguaramas, where no ISV’s were identified. PCLV and HTV, the two most common ISVs, have also been identified in similar studies associated with field-caught Ae. aegypti mosquitoes from the Caribbean region as well as in Asia and Australia7,28,30,31.
Although, the average read depth for the two ISV’s were relatively low in comparison to Ae. aegypti from other studies, this is the first report of ISV’s identified in Haemagogus mosquito species. Furthermore, the broad distribution of both viruses in Haemagogus mosquitoes collected from the majority of sites was similar to observations of other researchers working with in field caught Ae aegypti mosquitoes7,30. This suggests that these ISVs may be naturally abundant and have a broad mosquito host range in the wild. However, there is need for further characterization of ISV’s from other mosquito species in the wild. The phylogenetic analyses showed relatively distinct sequences for all PCLV segments from Trinidad, except for the sequences from the Caroni Bird Sanctuary which clustered with the reference sequence from Grenada, an island close to Trinidad. This suggests a broad regional distribution for at least some of the PCLV viral lineages. Maximum likelihood phylogeny also showed sequences of Trinidad HTV as a distinct lineage that was separate from reference sequences from Guadeloupe. Due to the limited sample size and the fact that the study was only limited to the island of Trinidad, it is not possible to conclude whether the distinct lineages of viral sequences found were due to evolutionary processes in an isolated ecosystem. Additionally, several reports have shown that viral heterogeneity can be influenced by vector and host interactions19,32 but the potential role of these types of interactions on the occurrence of distinct clades of the PCLV and HTV sequences in Trinidad needs to be established.
The RNA sequence data from this study shows that the ISVs found in Haemagogus were similar to those reported in other Culicidae vectors and hence they may be broadly associated with mosquitoes. Furthermore, although this report has revealed ISVs known to be associated with other competent disease vectors, there is need for further characterization of ISV’s from other mosquito species in the wild.
MAYV was only identified at very low levels in the Ecclesville and Mamon Village locations. This finding was similar to previous studies which also reported isolation of the virus at low infection rates in pools of field caught Hg. janthinomys mosquitoes from South America33,34,35. Ecclesville and Mamon Village sites are in areas known to have primate troops (the Red Howler Monkey, Alouatta macconnelli), which are the main reservoir and host feeding preference reported for Haemagogus mosquitoes in the sylvatic cycle of this emerging alphavirus13,16,19,35. Hence, the non-human primates could be the possible source of the MAYV in Haemagogus from these sites. Most of the other sampling locations are not associated with non-human primates, which could have accounted for the absence of MAYV. There is limited information on the prevalence of MAYV in mosquitoes or potential hosts present in the island, but it must be noted that the first report of MAYV infections included four out of five patients who were forest workers in Trinidad, and three of these individuals were stationed at Catshill and Moruga sites that are relatively close to Ecclesville and Mamon Village36. However, the possibility of other feeding hosts at Ecclesville and Mamon Village sites being the source of MAYV also cannot be discounted since avian and small mammalian species have also been reported to be associated with MAYV isolation in previous studies37,38. Haemagogus mosquitoes have been shown to be very attracted to humans as a blood feeding source33,35,39 and have become well adapted to breeding in artificial containers40,41,42. The forest peripherals in Mamon Village and Ecclesville are used for housing and agricultural activities42 where humans may also serve as potential intermediary host when MAYV is in circulation.
RNA sequencing of the Haemagogus mosquito pool from Chaguaramas resulted in detection of unclassified Kaiowa, Guato and Cumbaru viruses (Fig. 1a) that were recently isolated from the salivary glands in Culex and other medically important mosquito species29,43. Additionally, other mosquito-associated viral sequences were identified in the Ecclesville location including a picorna-like virus, Merida virus and Hubei-virga like 2 virus. The Merida virus was previously initially isolated from the Yucatan’s capital state44 and Hubei virga like 2 from Hubei region in China45 which confirms a wide distribution and multiple Culicidae hosts for these viruses.
A unique unclassified picorna-like virus (Ecclesville picorna like virus) was also identified from Ecclesville, with the Atrato picorna-like virus 1 (Table S1) isolated from field Psorophora species along the Columbian river bank46 as its closest relative. Potential new lineages of the Wuhan mosquito virus 6 were also identified in Ecclesville whose location is ecologically disparate to the Hubei, China location where the virus was initially isolated from Culex mosquito species47. The Guadeloupe mosquito virus lineage (Sobemovirus) was also found to be associated with Haemagogus mosquitoes from Mamoral only, which suggests this virus may be regionally distributed and may have multiple mosquito hosts occupying different ecosystems since it was previously identified from urban Aedes aegypti collected from households in Guadeloupe30.
The findings of this study also showed the association of other viruses with Haemagogus that have not been reported before in any other mosquito species. The Pepper mild mottle virus was the only plant virus identified in this study and was found in the Claxton Bay Haemagogus mosquito pool. Culicidae mosquitoes are known to feed on nectar, plant fluids and fruit sap48,49, hence the uptake of plant associated viruses is not surprising and has also been reported in previous metagenomic mosquito studies5,6,7,50. The Pepper mild mottle virus is a capsicum virus51 known to be affiliated with insects such as aphids49,52, but this is the first record of the virus being associated with a mosquito species. Similarly, small numbers of viral reads were identified as Black queen cell virus (BQCV) from the Quinam location which may have been due to acquisition from environmental sources. Mosquitoes require carbohydrates as an energy source53 and nectar and plant secretions contains glucose and fructose sugars which are attractive to mosquitoes. Some Haemagogus mosquitoes may have fed on nectar or sugar exudates from plant flowers that infected bees may have previously fed on, resulting in the mosquito ingesting the virus. This insect associated virus has only been reported to be infective to honeybees around the world, including Australia and parts of South Africa54,55. This is the first study reporting the association of the BQCV with Haemagogus or any mosquito species. Further, this virus has not been previously reported in Trinidad and Tobago. The apiary industry has been in decline in the country due to the poor health status of hives56 and the role of the BQCV in this decline needs to be investigated.
The avian leukosis virus57 was also found throughout several sampling sites. This retrovirus is known to be associated with birds and is vertically transmitted (adult to baby chicks)58. Although some strains (Type E) are known to be endogenous in the genomes of birds, the sequences obtained in this study were highly similar to exogenous strains (Type J and RSA), suggesting that the mosquitoes may be acquiring the virus from feeding on host animals. The detection of this virus in Haemagogus from several locations strongly suggests birds as potential feeding hosts for these mosquitoes. Previous studies have shown that a mosquito’s blood-meal can provide evidence of potential feeding hosts using RNA-Seq data analysis7,59. Birds as potential feeding hosts for Haemagogus is further supported by the fact that bird sequences were the most common hits when RNA reads were mapped against common animals from Trinidad. Birds have also been previously suggested to be feeding hosts for Haemagogus in the MAYV sylvatic cycle33,60.
Mus musculus RNA was also identified in Haemagogus samples collected from Chaguaramas and Catshill locations suggesting rodents as an alternative feeding host for these mosquitoes. A previous entomological survey42 found Haemagogus mosquitoes present in high densities at locations in Trinidad with no known non-human primates, the reported major host of these mosquitoes13,14,15. Hence, the data from the current study supports previous suggestion that other animals may be serving as feeding hosts61,62. Some of the alternative hosts may also be potentially involved in the MAYV sylvatic cycle or may, in time, adapt to serve as reservoirs, which has major epidemiological implications for MAYV fever. Although MAYV has been detected in non-primate animals including birds, rodents and reptiles33,37,60,63, the ability of these animals to serve as strong reservoirs of the virus is not known and currently primates are still considered the primary reservoir15,16,37. Future outbreaks can be more widespread if the virus evolves to utilize animals like birds or rodents as efficient reservoirs since these animals are present within or are close to major human population centers. Epidemics can be further exacerbated by the widespread occurrence of Haemagogus mosquitoes, including in areas close to human communities in some countries like Trinidad and Tobago, as was noted by Ali et al. (2019).
This study is also the first report on the microbiome of the Haemagogus mosquito species inferred from the RNA-seq data. Although the Ribo-ZeroTM Magnetic kit (Illumina Inc.) was used to deplete ribosomal RNA, the depletion was evidently incomplete since a significant number of reads mapped to the Silva bacterial 16S rRNA database that showed dominance of the Gammaproteobacteria class within the Proteobacteria phylum. The high prevalence of this bacterial class has similarly been reported in other mosquito species including Culex and Anopheles64,65,66,67,68. More importantly, mapping against representative Wolbachia 16S rRNA gene sequences provided strong evidence for the association of this bacterial genus with Haemagogus. The presence of Wolbachia species is known to negatively affect insect vector competence, as was demonstrated for Aedes mosquito species transmission of mosquito-borne pathogens such as the Zika virus and dengue virus67. Additional studies are needed to determine if members of this bacterial genus may have similar effects in reducing vector competence of Haemagogus and transmissibility of MAYV and other Haemagogus transmitted arboviruses.
The lack of availability of whole genome sequences for any Haemagogus mosquito species made data analysis challenging since it was not possible to filter out the host genome sequences before analysis of sequences of taxonomic importance. Despite the challenges, this study has added valuable baseline data on Haemagogus spp. and highlights the need for further work on characterizing the virome and microbiome of these medically important mosquitoes.
Methods
Mosquito collection
Adult female mosquitoes were collected in twelve forested areas in Trinidad previously described by Ali et al. 2019 (Fig. 1) during the rainy season over the period June to December 2018 using the human bait adult catching method at ground level69. Haemagogus mosquitoes (n = 205) were collected and transported to the lab on dry ice. The specimens were morphologically identified using taxonomic keys23,24,25, pooled (3–5 mosquitoes) and stored at − 80 °C until further use. Mosquitoes with distinctive Hg. janthinomys morphological features were pooled and used for RNA sequencing. Subsequent to RNA sequencing, DNA bar-coding analysis of mosquitoes from Trinidad based on sequencing of the mitochondrial cytochrome oxidase I (COI) gene and the ITS2 region showed major sequence variations in these genes that suggest the possibility of the occurrence of a species complex (Ali et al., unpublished data). The mosquitoes analyzed in this study were subsequently referred to as Haemagogus spp.
Nucleic acid preparation and sequencing
Mosquito pools were homogenized in 1 ml TRIzol LS reagent (Invitrogen) using a handheld cordless tissue homogenizer (VWR). Total RNA was extracted using the TRIzol LS / mosquito homogenate according to the manufacturers’ instructions. The RNA samples from the twelve locations were purified using the RNeasy MinElute Cleanup Kit (Qiagen) and shipped to Novogene Corporation Inc (Sacramento, California, U.S.A) for library construction and sequencing. The Ribo-ZeroTM Magnetic kit (Illumina Inc.) was used to deplete the ribosomal RNA. Libraries were sequenced from both ends (150 bp) on an Illumina platform (Illumina Inc, San Diego, U.S.A.) and the lncRNA pipeline was used to extract 150 bp paired end reads into FASTQ files.
Viral read and contig analysis
See Fig. 4 for workflow of bioinformatic analysis used to analyze the Illumina sequencing data. Host filtering was not conducted for contig generation since there is no reference genome available for Haemagogus. Raw paired-end reads were quality filtered (Q < 20) and trimmed for adaptor sequences in the Galaxy platform70 (www.usegalaxy.org) using Trimmomatic v0.36.671. FastQC reports were generated for both paired files using FastQC v0.7272; the paired end reads were mapped against the RVDB: Reference Viral Database73 for a broader exploratory search and then against single reference genomes using Bowtie 2 v2.3.4.274. Using BAM files generated from Bowtie 2 mapping from single reference genomes, Qualimap BamQC reports were generated using Qualimap BamQC v2.2.2d75. The BAM files generated from the Bowtie2 step were further filtered to a minimum MAPQ quality score of 20 using Filter SAM or BAM tool v1.1.176 and the filtered reads were then extracted as FASTQ files using SAMtools fastx files v1.976 for contig assembly. Trinity v2.9.177 was used for de novo assembly of the generated FASTQ reads. Nucleotide similarity of viral contigs to published sequences (nucleotide collection nt/nr) was determined using the blastn suite v2.10.078 from NCBI (www.ncbi.nlm.nih.gov). The distribution of viral identified reads was visualized using the Krona tool79.

Workflow for bioinformatic analysis for viral, bacteria and feeding host read and contig analysis. All analyses were conducted using the Galaxy server (https://usegalaxy.eu/) except NCBI BLAST and generation of phylogenetic trees.
Bacterial read and contig analysis
Using the reference ARB/SILVA Version v138 SSU database80,81,82, ribosomal RNA sequences were mapped and extracted using the Bowtie 2 software v2.3.4.274. The identity of reads was determined using mothur v.1.42.083 after aligning to the mothur formatted RDP database (https://rdp.cme.msu.edu/) trainset16. Data visualization was done using the phyloseq package (v1.34.0) in R. The RNA sequences were further mapped directly onto 301 reference Wolbachia 16S rRNA gene sequences downloaded from the ARB/Silva ribosomal database (https://www.arb-silva.de/). The mapped reads were then assembled using Trinity (Galaxy version v2.9.1) and contigs generated were identified by Blastn.
Feeding host read and contig analysis
Trimmed and filtered reads were mapped against 142 sequences of mitochondrial bar-coding genes and three full mitochondrial genomes of 116 animals using the Bowtie 2 software v2.3.4.274 (Table S1). The animals included were 27 mammals, 10 reptiles and 79 bird species which are commonly found in Trinidad84. The sequences used for mapping are available at https://zenodo.org/record/4932469#.YMQbQKhKi01.
Relative abundance of taxonomic groups
The relative abundance of the different taxonomic groups was expressed in reads per million (RPM) of total number of filtered reads per sample. The RPM was calculated using the formula:
Phylogenetic analysis
The phylogenetic relationships of selected viral assembled viral contigs and representative reference sequences from GenBank were determined using the MEGA X 10.0.5 software85. The contigs of all coding regions were aligned with the closest reference sequence from GenBank and amino acid sequences generated to confirm accuracy of the nucleotide sequences. Sequences were aligned using Clustal W and trimmed before phylogenetic trees were generated using the Maximum Likelihood method (Kimura 2-parameter substitution model) with 1000 bootstrap replications.
Data availability
The datasets analyzed during this study are available on the NCBI GenBank SRA database accession PRJNA689580 (https://www.ncbi.nlm.nih.gov/sra/PRJNA689580). The sequences are available in the NCBI GenBank database with accession numbers MW650653-MW650790, MW727408-MW727425, MW773204-MW773213 and MW779452-MW779457 (Table S1). All the sequence data generated during this study are also available at: https://zenodo.org/record/4932469#.YMQbQKhKi01.
References
National Academies of Sciences, E. & Medicine. Global Health Impacts of Vector-Borne Diseases: Workshop Summary. (The National Academies Press, 2016).
WHO. Key Facts, Vector-borne diseases, http://www.who.int/news-room/fact-sheets/detail/vector-borne-diseases (2017).
Auguste, A. J. et al. Isolation and characterization of sylvatic mosquito-borne viruses in Trinidad: Enzootic transmission and a new potential vector of Mucambo virus. Am. J. Trop. Med. Hyg. 83, 1262–1265 (2010).
Frey, K. G. et al. Bioinformatic characterization of mosquito viromes within the Eastern United States and Puerto Rico: discovery of novel viruses. Evolut. Bioinform. 12, EBO. S38518 (2016).
Atoni, E. et al. Metagenomic virome analysis of Culex mosquitoes from Kenya and China. Viruses 10, 30 (2018).
Xia, H. et al. Comparative metagenomic profiling of viromes associated with four common mosquito species in China. Virol. Sin. 33, 59–66 (2018).
Zakrzewski, M. et al. Mapping the virome in wild-caught Aedes aegypti from Cairns and Bangkok. Sci. Rep. 8, 4690 (2018).
Belda, E. et al. De novo profiling of RNA viruses in Anopheles malaria vector mosquitoes from forest ecological zones in Senegal and Cambodia. BMC Genom. 20, 1–27 (2019).
Nanfack-Minkeu, F. et al. Interaction of RNA viruses of the natural virome with the African malaria vector, Anopheles coluzzii. Sci. Rep. 9, 6319 (2019).
Delwart, E. L. Viral metagenomics. Rev. Med. Virol. 17, 115–131 (2007).
Cholleti, H. et al. Discovery of novel viruses in mosquitoes from the Zambezi Valley of Mozambique. PLoS ONE 11, e0162751 (2016).
Izurieta, R. O. et al. Hunting in the rainforest and Mayaro virus infection: An emerging alphavirus in Ecuador. J. Glob. Infect. Dis. 3, 317 (2011).
Mota, M. T. d. O., Ribeiro, M. R., Vedovello, D. & Nogueira, M. L. Mayaro virus: A neglected arbovirus of the Americas. Future Virol. 10, 1109–1122 (2015).
Acosta-Ampudia, Y. et al. Mayaro: An emerging viral threat?. Emerg. Microbes Infect. 7, 163 (2018).
PAHO/WHO. Epidemiological Alert: Mayaro Fever (Pan American Health Organization/World Health Organization, 2019).
Pego, P. N., Gomes, L. P., Provance Jr, D. W. & De Simone, S. G. Mayaro virus disease. (2014).
Hozé, N. et al. Reconstructing Mayaro virus circulation in French Guiana shows frequent spillovers. Nat. Commun. 11, 1–9 (2020).
Santos, F. M., Dias, R. S., de Souza Fernandes, L., da Silva, C. C. & de Paula, S. O. Mayaro virus infection: Clinical features and global threat. Curr. Treat. Opt. Infect. Dis. 1–11 (2020).
Hotez, P. J. & Murray, K. O. Dengue, West Nile virus, chikungunya, Zika—And now Mayaro? PLoS Negl. Trop. Dis. (2017).
Cella, E. et al. Mayaro virus infection, the next epidemic wave after Zika? Evolutionary and structural analysis. Asian Pac J Trop Med 11, 194 (2018).
Brustolin, M., Pujhari, S., Henderson, C. A. & Rasgon, J. L. Anopheles mosquitoes may drive invasion and transmission of Mayaro virus across geographically diverse regions. PLoS Negl. Trop. Dis. 12, e0006895 (2018).
Dieme, C., Ciota, A. T. & Kramer, L. D. Transmission potential of Mayaro virus by Aedes albopictus, and Anopheles quadrimaculatus from the USA. Parasit. Vectors 13, 1–6 (2020).
Arnell, J. H. Mosquito Studies (Diptera, Culicidae). XXXII. A revision of the genus Haemagogus. Estudios sobre zancudos (Diptera, Culicidae). XXXII. Una revisión del género Haemagogus. Contrib. Am. Entomol. Inst. 10, 1–174 (1973).
Darsie , R. Mosquitoes of Argentina. I. Keys for identification of adult females and fourth stage larvae in English and Spanish (Diptera, Culicidae). Mosquito systematics (USA) (1985).
WRBU. Medically Important Arthropod Species—Southcom-Haemagogus., http://www.wrbu.org/mqID/mq_medspc/AD/HGjan_hab.html (2012).
Silva, S. O. F., Mello, C. F. d. & Alencar, J. Morphological differentiation between seven Brazilian populations of Haemagogus capricornii and Hg. janthinomys (Diptera: Culicidae) using geometric morphometry of the wings. Revista da Sociedade Brasileira de Medicina Tropical 52 (2019).
Bolling, B. G., Weaver, S. C., Tesh, R. B. & Vasilakis, N. Insect-specific virus discovery: significance for the arbovirus community. Viruses 7, 4911–4928 (2015).
Zhang, X. et al. Discovery and high prevalence of Phasi Charoen-like virus in field-captured Aedes aegypti in South China. Virology 523, 35–40 (2018).
Pauvolid-Corrêa, A. et al. Novel viruses isolated from mosquitoes in Pantanal, Brazil. Genome Announc. 4 (2016).
Shi, C. et al. Stable distinct core eukaryotic viromes in different mosquito species from Guadeloupe, using single mosquito viral metagenomics. Microbiome 7, 121 (2019).
Ramos-Nino, M. E. et al. High prevalence of Phasi Charoen-like virus from wild-caught Aedes aegypti in Grenada, WI as revealed by metagenomic analysis. PLoS ONE 15, e0227998 (2020).
Simpson, J. E. et al. Vector host-feeding preferences drive transmission of multi-host pathogens: West Nile virus as a model system. Proc. R. Soc. B Biol. Sci. 279, 925–933 (2012).
Hoch, A. L., Peterson, N. E., LeDuc, J. W. & Pinheiro, F. P. An outbreak of Mayaro virus disease in Belterra, Brazil. Am. J. Trop. Med. Hyg. 30, 689–698 (1981).
Dégallier, N. et al. Estimation of the survival rate, the relative density and the infection rate of a population of Haemagogus janthinomys Dyar (Diptera, Culicidae) from which strains of yellow fever were isolated in Brazilian Amazon. Bull. Soc. Pathol. Exot. 84, 386–397 (1991).
Azevedo, R. S. et al. Mayaro fever virus, Brazilian amazon. Emerg. Infect. Dis. 15, 1830 (2009).
Anderson, C. R., Downs, W. G., Wattley, G. H., Ahin, N. W. & Reese, A. A. Mayaro virus: A new human disease agent. Am. J. Trop. Med. Hyg. 6, 1012–1016 (1957).
de Thoisy, B., Gardon, J., Salas, R. A., Morvan, J. & Kazanji, M. Mayaro virus in wild mammals, French Guiana. Emerg. Infect. Dis. 9, 1326 (2003).
Figueiredo, L. T. M. Emergent arboviruses in Brazil. Rev. Soc. Bras. Med. Trop. 40, 224–229 (2007).
De Abreu, F. V. S. et al. Haemagogus leucocelaenus and Haemagogus janthinomys are the primary vectors in the major yellow fever outbreak in Brazil, 2016–2018. Emerg. Microbes Infect. 8, 218 (2019).
Zavortink, T. J. Mosquito studies (Diptera, Culicidae). XXVIII. The new world species formerly placed in Aedes (Finlaya). (1972).
Chadee, D. D. & Tikasingh, E. S. Observations on the seasonal incidence and diel oviposition periodicity of Haemagogus mosquitoes (Diptera: Culicidae) in Trinidad, WI: Part I. Haemagogus janthinomys Dyar. Ann. Trop. Med. Parasitol. 83, 507–516 (1989).
Ali, R. et al. Changing patterns in the distribution of the Mayaro virus vector Haemagogus species in Trinidad, West Indies. Acta Tropica 199, 105108 (2019).
de Lara Pinto, A. Z. et al. Novel viruses in salivary glands of mosquitoes from sylvatic Cerrado, Midwestern Brazil. PLoS ONE 12, e0187429 (2017).
Charles, J. et al. Merida virus, a putative novel rhabdovirus discovered in Culex and Ochlerotatus spp. mosquitoes in the Yucatan Peninsula of Mexico. J. Gener. Virol. 97, 977 (2016).
Shi, M. et al. Redefining the invertebrate RNA virosphere. Nature 540, 539–543 (2016).
Nitsche, A., Hankeln,T., Acosta,O., Velez,I.D. and Schiemann,D.J. (Division of Highly Pathogenic Viruses, Robert Koch Institute, Seestrasse 10, Berlin 13353, Germany, 2016).
Li, C.-X. et al. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. elife 4, e05378 (2015).
Rozendaal, J. A. Vector Control: Methods for Use by Individuals and Communities. (World Health Organization, 1997).
Whitfield, A. E., Falk, B. W. & Rotenberg, D. Insect vector-mediated transmission of plant viruses. Virology 479, 278–289 (2015).
Ng, T. F. F. et al. Broad surveys of DNA viral diversity obtained through viral metagenomics of mosquitoes. PLoS ONE 6, e20579 (2011).
Colson, P. et al. Pepper mild mottle virus, a plant virus associated with specific immune responses, fever, abdominal pains, and pruritus in humans. PLoS ONE 5 (2010).
Dietzgen, R. G., Mann, K. S. & Johnson, K. N. Plant virus–insect vector interactions: Current and potential future research directions. Viruses 8, 303 (2016).
Barredo, E. & DeGennaro, M. Not just from blood: Mosquito nutrient acquisition from nectar sources. Trends Parasitol. (2020).
Leat, N., Ball, B., Govan, V. & Davison, S. Analysis of the complete genome sequence of black queen-cell virus, a picorna-like virus of honey bees. J. Gen. Virol. 81, 2111–2119 (2000).
Spurny, R. et al. Virion structure of black queen cell virus, a common honeybee pathogen. J. Virol. 91, e02100-02116 (2017).
Inalsingh, S. The Business of Honey: Rebuilding the Apiculture Industry of Trinidad & Tobago. (American College of Healthcare Science, 2014).
Payne, L. et al. A novel subgroup of exogenous avian leukosis virus in chickens. J. Gen. Virol. 72, 801–807 (1991).
Cui, Z., Sun, S., Zhang, Z. & Meng, S. Simultaneous endemic infections with subgroup J avian leukosis virus and reticuloendotheliosis virus in commercial and local breeds of chickens. Avian Pathol. 38, 443–448 (2009).
Logue, K. et al. Unbiased characterization of Anopheles mosquito blood meals by targeted high-throughput sequencing. PLoS Negl. Trop. Dis. 10, e0004512 (2016).
Calisher, C. H., Gutierrez, E., Maness, K. & Lord, R. D. Isolation of Mayaro virus from a migrating bird captured in Louisiana in 1967. Bull. Pan Am. Health Org. (PAHO); 8 (3), 1974 (1974).
Alencar, J. et al. Feeding patterns of Haemagogus janthinomys (Diptera: Culicidae) in different regions of Brazil. J. Med. Entomol. 42, 981–985 (2005).
Mucci, L. F. et al. Feeding habits of mosquitoes (Diptera: Culicidae) in an area of sylvatic transmission of yellow fever in the state of São Paulo, Brazil. J. Venom. Anim. Toxins Incl. Trop. Dis. 21, 6 (2015).
Talarmin, A. et al. Mayaro virus fever in French Guiana: Isolation, identification, and seroprevalence. Am. J. Trop. Med. Hyg. 59, 452–456 (1998).
Zouache, K. et al. Bacterial diversity of field-caught mosquitoes, Aedes albopictus and Aedes aegypti, from different geographic regions of Madagascar. FEMS Microbiol. Ecol. 75, 377–389 (2011).
Minard, G., Mavingui, P. & Moro, C. V. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit. Vectors 6, 146 (2013).
Baldini, F. et al. Evidence of natural Wolbachia infections in field populations of Anopheles gambiae. Nat. Commun. 5, 3985 (2014).
Hegde, S., Rasgon, J. L. & Hughes, G. L. The microbiome modulates arbovirus transmission in mosquitoes. Curr. Opin. Virol. 15, 97–102 (2015).
Baldini, F. et al. First report of natural Wolbachia infection in the malaria mosquito Anopheles arabiensis in Tanzania. Parasit. Vectors 11, 635. https://doi.org/10.1186/s13071-018-3249-y (2018).
Chadee, D. D., Tikasingh, E. S. & Ganesh, R. Seasonality, biting cycle and parity of the yellow fever vector mosquito Haemagogus janthinomys in Trinidad. Med. Vet. Entomol. 6, 143–148 (1992).
Afgan, E. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544 (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Andrews, S. (Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom, 2010).
Goodacre, N., Aljanahi, A., Nandakumar, S., Mikailov, M. & Khan, A. S. A reference viral database (RVDB) to enhance bioinformatics analysis of high-throughput sequencing for novel virus detection. mSphere 3, e00069–00018 (2018).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294. https://doi.org/10.1093/bioinformatics/btv566 (2015).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. https://doi.org/10.1093/bioinformatics/btp352 (2009).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644 (2011).
Zhang, Z., Schwartz, S., Wagner, L. & Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7, 203–214 (2000).
Ondov, B. D., Bergman, N. H. & Phillippy, A. M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 12, 385 (2011).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Yilmaz, P. et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. https://doi.org/10.1093/nar/gkt1209 (2013).
Glöckner, F. O. et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 261, 169–176 (2017).
Schloss, P. D. et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Rutherford, M. G., Landstrom, L., M. Wainwright, Leal, E. & Dean, R. Trinidad and Tobago Wildlife Guide, Rainforest Publications, 2012.
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Acknowledgements
We would like to thank the Insect Vector Control Division (IVCD), Ministry of Health, Trinidad and Tobago for field assistance. The work was supported by The Campus Research & Publication Fund, School of Graduate Studies, University of the West Indies, St. Augustine Campus. CRP.5. DEC18.58.
Author information
Authors and Affiliations
Contributions
R.A. collected the mosquitoes and performed nucleic acid extraction; R.A. and A.R. undertook bioinformatic analysis, attained funding and wrote manuscript with input from all authors. A.M., J.J., C.C., C.V.F.C. and D.W.S. provided advise, logistic support and wrote manuscript with all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ali, R., Jayaraj, J., Mohammed, A. et al. Characterization of the virome associated with Haemagogus mosquitoes in Trinidad, West Indies. Sci Rep 11, 16584 (2021). https://doi.org/10.1038/s41598-021-95842-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-95842-6
This article is cited by
-
Tips and tools to obtain and assess mosquito viromes
Archives of Microbiology (2024)
-
Bacterial diversity in Haemagogus leucocelaenus (Diptera: Culicidae) from Vale do Ribeira, São Paulo, Brazil
BMC Microbiology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
