
Abstract
Kyasanur forest disease virus (KFDV) causing tick-borne hemorrhagic fever which was earlier endemic to western Ghats, southern India, it is now encroaching into new geographic regions, but there is no approved medicine or effective vaccine against this deadly disease. In this study, we did in-silico design of multi-epitope subunit vaccine for KFDV. B-cell and T-cell epitopes were predicted from conserved regions of KFDV envelope protein and two vaccine candidates (VC1 and VC2) were constructed, those were found to be non-allergic and possess good antigenic properties, also gives cross-protection against Alkhurma hemorrhagic fever virus. The 3D structures of vaccine candidates were built and validated. Docking analysis of vaccine candidates with toll-like receptor-2 (TLR-2) by Cluspro and PatchDock revealed strong affinity between VC1 and TLR2. Ligplot tool was identified the intermolecular hydrogen bonds between vaccine candidates and TLR-2, iMOD server confirmed the stability of the docking complexes. JCAT sever ensured cloning efficiency of both vaccine constructs and in-silico cloning into pET30a (+) vector by SnapGene showed successful translation of epitope region. IMMSIM server was identified increased immunological responses. Finally, multi-epitope vaccine candidates were designed and validated their efficiency, it may pave the way for up-coming vaccine and diagnostic kit development.
Similar content being viewed by others
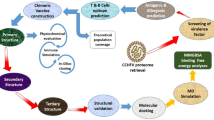

Introduction
Kyasanur Forest Disease (KFD) is a southern Indian endemic zoonotic disease caused by KFDV that belongs to the family Flaviviridae. National institute of Allergy and Infectious Disease has recognized KFDV as category C priority virus. KFDV was first identified in 1957 from isolates of the sick and dying monkeys of black-faced langur (Presbytis entellus) and red-faced bonnet (Macaca radiate) species in Kyasanur Forest, state of Karnataka, India1,2,3,4. Humans are the dead-end host of KFDV life cycle, initial stage KFDV circulates among monkeys, rodents then it transfers into humans by bites of infected ticks, primarily through Haemaphysalis sp5.
The KFDV is an enveloped virus with icosahedral nucleocapsid, spherical in shape and about 40–65 nm in size. It contains 10,774 bases of positive single stranded RNA, encodes 3146 amino acid. Post-translational cleavage of polyprotein into three structural proteins (Capsid, Membrane and Envelope) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5)6,7.
During 1957–2017, totally 9594 KFDV cases were reported in India8. Recently, KFDV outbreak was reported in a new geographic location Sindhudurg district of Maharashtra9. In future, it may spread to other parts of India. But still there is no cure for KFDV, currently formalin inactivated vaccine being used as primary strategy for controlling KFD10, it gives partial effect against to KFDV infections and provides short term immunity to those have received vaccination11. It indicates low efficiency of current vaccine, there is a need for development of new vaccine candidates against KFDV.
There was a study reported the KFDV envelope protein structural variation and different in B-cell epitope antigenicity12. There are no studies on KFDV vaccine development, so we tried to construct the epitope-based subunit vaccine which elite T-cell and B-cell immune response upon KFDV infection. T-cells and B-cells drive the process of adaptive immunity and develop immunological memory by recognizing portions of antigens called epitopes. These epitopes contains short amino acid sequence that can induce more direct and specific immune response, which overcome the disadvantages of live attenuated vaccines13. In-silico based epitope prediction methods have reduced the time consumption and money spent on false epitope candidates and this tools has been employed to develop multi-epitope vaccine against several diseases such as Dengue, SARS-CoV-2, Mycobacterium tuberculosis, Staphylococcus aureus etc14,15,16,17,18,19. Hence, the present investigation has adapted this approach for rational design of epitope-based vaccine against KFDV. Here, first time we have designed vaccine candidates from conserved region of KFDV-envelop protein. And we have successfully designed the multi-immunogenic, non-allergic and high cloning efficient multi-epitope vaccine candidates by utilizing immune-informatics and bioinformatics tools.
Results
Conserved region of KFDV envelope protein
The multiple sequence alignment analysis (Supplementary File 1) reveals that except few amino acids, the entire KFDV envelope protein sequences remain conserve. Phylogenetic tree was constructed from the alignment file, shows separate cluster of 2012 KFDV envelope protein, rest of the sequence were almost the same (Fig. 1A). Based on the conserve score (Supplementary File 2) conservation graph has been drawn, value 1.0 on the Y-axis represents more conserved (Fig. 1B). Amino acids, which got > 0.90 conserve score was considered as a conserved region and taken into further study, conserved amino acid sequence and their position were given in Table 1.

Multiple sequence alignment of KFDV envelop protein by CLC workbench. (A) Phylogenetic tree of KFDV envelope protein. (B) Conservation graph of KFDV envelop protein sequence (value 1.0 is highest conserved).
Conserved regions of KFDV envelope protein containing T-cell and B-cell epitopes
Here we found that the conserved region contains epitopes for T-cell and B-cell targets. Totally 35 epitopes were obtained IEDB-MHC-I tools (Supplementary File 3), whereas the MAPPP tool produced 300 epitopes from 43 human HLA alleles (Supplementary File 4). IEDB-MHC-I top 10 percentile epitopes were also predicted in MAPPP tool, they are listed in Table 2. Combined predictor of proteasomal cleavage, TAP transport, and MHC process has produced 34 epitopes (Supplementary File 5), and top 10 ranked epitopes were listed in Table 3. There are 20 MHC class II epitopes (Supplementary File 6) were identified by IEDB-MHC-II tools, whereas MHC2Pred tool recognized 504 epitopes (Supplementary File 7) on the conserved region of the envelope protein. Based on IEDB = MHC-II percentile > 0.10 (FILT-1) and MHC2Pred epitopes which scored > 0.60 as well having overlap sequence with FILT-1, totally19 epitopes were selected as MHC II targets (Table 4). B-cell liner epitopes prediction tools ABCpred, BCpred and SVMTriP were identified 15, 13 and 4 epitopes respectively (Table 5).
Selected epitopes display cross-protection against Alkhumra hemorrhagic fever virus
Uniportkb-human BLAST analysis showed similarity with four human proteins such as shish-7, Isoform 2 of Ribonuclease T2, Ribonuclease T2 and Extra-cellular ribonuclease with E-value of 2e−1, 7.7e−1, 2.1e0 and 2.1e0 respectively (Supplementary File 8). These similarity hits can be ignored, as E-value is ≥ 0.1, because as rule of thumb an E-value should be < 10–4 to assure the homology. Whereas UniProtKB BLAST revealed that selected epitopes were sharing the highest similarity with Alkhumra Hemorrhagic Fever Virus (AHFV)20 polyprotein, E-value was 1.7e−43 (Supplementary File 9). So, the proposed vaccine candidates also confer immunity against AHFV.
Multi-epitope vaccine candidate sequences their allergenicity and antigenicity
Totally, two multiple-epitope vaccine candidates were designed from selected high ranked, multi-immunogenic and over lapping epitope sequences. Vaccine construct 1 (VC1) composed with an adjuvant protein β defensin, 6 MHC- I epitope and 11 B-cell epitopes. Vaccine constructs 2 (VC2) composed of VC1 and 4 MHC-II epitopes. Complete information of vaccine construct has given in Table 6. Vaccine candidates VC1 and VC2 were found to be non-allergic in behavior, VC1 got a higher antigenic score (0.6667) and identified as a better antigen, while VC2 antigenic score was 0.5835.
Physiochemical and solubility properties of vaccine construct
The molecular weight (MW) of constructed vaccine candidates VC1 and VC2 were found to 34.6 kDa and 39.4 kDa respectively. Theoretical pI value 9.38(VC1) and 9.25 (VC2) were observed and proteins are expected to alkaline in nature. Estimated half-life for both VC1 and VC2 candidates were 30 h in mammalian reticulocytes in vitro. Instability index were found to be 32.86 and 33.61 for VC1 and VC2 respectively and confirmed as stable proteins. Negative GRAVY values of VC1 (− 0.455) and VC2 (− 0.511) indicate they are hydrophilic in nature and they could interact with water. PROSO II predicted VC1 as soluble protein with score of 0.667, whereas VC2 scored 0.489 and it identified as insoluble protein, obtained physiochemical characteristics are strengthening potency of vaccine candidates.
3D-strucure of designed vaccine candidates
Phyre2 server has built three-dimensional structure of multi-epitope vaccine candidates. The cryo-em structure of tick-borne encephalitis virus complex was used as template for VC1 and VC2 tertiary structure, and both the model got 100% score in terms of model confidence. Initial refinement of vaccine candidates VC1 and VC2 on ModRefiner have produced refine structure with TM-score of 0.9507 and 0.9622 respectively. Further refinement of VC1 and VC2 were done with GalaxyRefine server, based on quality score the best refined structure were chosen and named as VC1R and VC2R respectively. Those were used as ligand molecules in docking study (Fig. 2A, B), quality scores of ligands were given in Table 7.

3D-modeling, refinement and validation of multi epitope vaccine constructs. GalaxyRefine generated Refined structure of (A) VC1 and (B) VC2 construct. RAMPAGE produced of Ramachandran plot analysis of (C) refined VC1 structure and (D) refined VC2 structure. ProSA-web shows Z-score of (E) − 2.74 for VC1 and (F) − 2.66 for VC2.
The Ramachandran plot analysis of modeled vaccine candidates revealed that 97.4% of VC1R protein residues were in favored region and 2.6% of residues in allowed region (Fig. 2C). Similarly, 95.9% of VC2R protein residues were in favored region and 4.1% of the residues in allowed region (Fig. 2D). None of the amino acid residues were fallen in outlier region, which indicates the good quality of protein structure. ProSA-web analysis of yielded Z-score of − 2.74 and − 2.66 for vaccine candidates of VC1R and VC2R respectively. (Fig. 2E, F).
Identification of binding energy between multi-epitope vaccine candidates and TLR2
Docking analysis by Cluspro and PatchDock has revealed that both vaccine candidates (VC1 and VC2) have strong binding capacity with TLR2. Cluspro protein–protein docking result page displayed 10 best-docked confirmations, the top ranked model from balanced coefficient module was downloaded in PDB format and used for LigPlot and iMODS analysis. TLR2 with VC1 exhibited lowest energy value of − 9473.6 kcal/mol, whereas VC2 showed − 876.4 kcal/mol (Fig. 3).

Identification of binding energy by Cluspro docking server (A) VC1 and TLR2 complex (B) VC2 and TLR2 complex.
PatchDock server produced transformation file (posture of 3D-rotational angle) then it was rescored and global energy was obtained by FireDock. The best docking posture was ranked based on the global energy, top ranked docking score, energy contributed by hydrogen bond and Van der waals forces were listed Table 8. The highest global energy of TLR2 with VC1 and VC2 was found to be − 55.71 and − 49.10 respectively.
Interaction and stability of TLR2 and Vaccine candidates
Interaction of TLR2 and Vaccine candidates were visualized by LigPlot tool. Hydrogen bonds were highlighted in dash line (Fig. 4A), totally seven hydrogen bonds were found between TLR2 with VC1. The receptor’s residue Asn294 showed highest binding affinity with 2.49 Å distance and least binding energy was found towards Phe325 with 3.18 Å distance. VC2 construct also showed identical interaction with TLR2 residues (Fig. 4B).

Ligplot analysis showing intermolecular hydrogen bonds in (A) VC1 and TLR2 complex (B) VC2 and TLR2 complex.
Molecular dynamics simulation study was done by iMODS server to check the stability and physical movement of atoms in docking complex. Simulation were performed in normal mode analysis and obtained results of VC1-TLR2 and VC2-TLR2 docking complexes were depicted in Figs. 5 and 6 respectively. The deformability graph of both complexes was illustrated in Figs. 5B and 6B, hinges are highlighting the deformability regions in the complex. The B-factor (Figs. 5C, 6C) calculates root mean square value and represents the uncertainty of each atom in the docking complex. Eigenvalues of VC1-TLR2 and VC2-TLR2 docking complexes were found to be 1.135892 × 10−5 and 6.807877 × 10−6 respectively (Figs. 5D, 6D). The variance matrix graph of residues displayed in Figs. 5E and 6E. Covariance matrix indicates (Figs. 5F, 6F) coupling between pairs of residue experience correlated (red), uncorrelated (white) and anti-correlated (blue) motions. The elastic of docking complexes was shown in Fig. 5G, it explains the relation between the atoms (darker gray). iMODS simulation results are suggest that vaccine constructs and TLR2 complexes are stable.

Molecular dynamics simulation of VC1 and TLR2 complex by iMODS server. (A) VC1 and TLR2 docking complex. (B) Main-chain deformability. (C) B-factor values. (D) The eigenvalue. (E) Variance. (F) Co-variance map. (G) Elastic network of model.

Molecular dynamics simulation of VC2 and TLR2 complex by iMODS server. (A) VC2 and TLR2 docking complex. (B) Main-chain deformability. (C) B-factor values. (D) The eigenvalue. (E) Variance. (F) Co-variance map. (G) Elastic network of model.
Codon optimization and in silico cloning of KFD-VC1 and KFD-VC2
Java Codon Adaptation Tool was used to check codon optimism of vaccine candidates in Escherichia coli (strain k12) expression system. It revealed that VC1 and VC2 multi-epitope vaccine construct composed of 927 and 1050 nucleotides respectively. The Codon Adaptation Index (CAI) was observed, 1.0 for VC1 and VC2 was 0.98. The GC content of VC1 found to be 51.13% and VC2 was 52.19%. The obtained values indicate that both vaccine candidates were having cloning efficiency. SnapGene tool was used for in-silico insertion of adapted codon sequence of vaccine construct into pET30a (+) expression vector between XhoI and NedI restriction sites and clones were obtained successfully (Fig. 7).

In silico cloning of multiepitope subunit vaccine sequence into pET30a (+) expression vector by SnapGene tool, red color part represents vaccine sequence and black circle represent vector sequence. Restriction sites are heighted in yellow color and translation sequence parts were mention in green color arrows.
Immune response simulation
The immunogenic profile of constructed vaccine candidates was obtained from C-IMMSIM server. It found that our vaccine candidates could able to elicit both humoral and cellular mediated immune responses (Figs. 8, 9). Simulation results showed that secondary and tertiary immune responses is higher than primary response. Antigenic molecules were found to be cleared off after three doses of vaccination, on the other hand B and T memory cell population got increased at the maximum of 500 cells/mm3 and 2000 cells/mm3 respectively, this feature makes our construct VC1 and VC2 as suitable KFDV vaccine candidates. This study also found that elevated level of cytokines such as IFN-γ and IL-2, which important for inhibition of viral replication and T-cell mediated immunity. Increased trend of IgM and IgG antibody titer was observed after third injection, at the same time antigen level were decreased. Above observed immune elicit properties ensured that vaccine constructs will be effective in human subjects.

In silico immune simulation of VC1 by IMMSIM server. (A) Population of B lymphocytes, y2 represents scale of memory B cells. (B) Population of T lymphocytes, y2 represents scale of memory T cells. (C) Concentration of cytokines and interleukins (insect plot shows elevated level of IL-2). (D) Immunoglobulin production upon antigen exposure.

In silico immune simulation of VC2 by IMMSIM server. (A) Population of B lymphocytes, y2 represents scale of memory B cells. (B) Population of T lymphocytes, y2 represents scale of memory T cells. (C) Concentration of cytokines and interleukins (insect plot shows elevated level of IL-2). (D) Immunoglobulin production upon antigen exposure.
Discussion
This genome era enabled with bulk inflow of genomics and proteomics information of almost all clinically important organisms. It facilitates us to choosing the targets for drug design, identifying new strains or drug resistance, diagnosis kit development and personalized medicine etc. Similarity, computational approaches simplified vaccine development process, vaccine informatics is a fast-growing field where in-silico vaccine design ventures are possible. Previous report on KFDV immune response has suggested to target both T cell and B cells mediated immune response for successful KFDV vaccine development21. We have applied few immune informatic tools to identify the epitope which elicit both cellular and humoral immune responses.
In our study, KFDV envelope protein sequence was chosen as target for epitope prediction. The envelope protein of flaviviruses is major protective antigen and consists of three domains EI, EII and EIII. Especially EIII domain contains specific and sub-complex specific neutralizing epitopes, moreover it is easy to express by recombinant techniques22. Another challenge is genetic diversity of virus that resulted in immune escape, always-recombinant vaccines design prefers to target conserved antigenic regions of virus, it might accelerates better immune response23. The KFDV vaccinated individuals were reported for disease occurrence, it may because of variations in circulating virus24. Therefore, we picked the conserved region of KFDV envelope protein sequence, to overcome the genetic diversity of viral strains.
So far, there is no experimental study on identification of T-cell and B-cell epitopes. Here we have identified both of them by using immuno-informatics tools. Epitopes or viral antigens arise the specific immune responses in the body, which induce adaptive immune responses of T-cells mediated cellular immunity and B-cells/antibody mediated humoral immunity25,26. All three MHC-I epitope prediction tools identified KTILTLGDY and KTAEHLPKAW. Also, MHC II epitope KAWQVHRDWFEDLSL overlap with MHC class I epitope KAWQVHRDW, it may serve as target for both CD8 + and CD4 + cells. B-cell epitopes KRPPTDSGHD, TVVLELDKTAEH, DDIHQENPAKTR, AANESHSNRKTASF, TGSKPCRIPVRAVA were identified by any of two-prediction tools out of three.
The high ranked, multi-immunogenic and overlapped epitopes were linked together and multi-epitope construct was obtained. Typical purified proteins are not inherently immunogenic, it requires adjuvant to accelerate the innate immune system and enhance vaccine potency27. We have added β-defensin at N terminal end of the multi-epitope vaccine construct. β-defensin is known to induce lymphokines production which promotes T-cell mediated cellular immunity and antigen-specific Ig production28.
Totally, we made two multi-epitopes vaccines construct against KFDV, 3D structure, and refinement of the vaccine candidates were achieved. The Ramachandran plots of built structures showed that none of the residues were in outlier region, > 95% of the amino acids fallen in favored region. It clearly indicates that modeled structures of vaccine candidates are good in quality.
TLRs plays vital role in innate immunity, especially they detect the virus and activates innate immunity followed by adaptive immunity. Several reports confirmed that TLR2 act as a host sensor to identifying viral envelop proteins and subsequently activates the innate immune system29,30,31. Our docking studies suggest that designed vaccine construct have the binding capacity with TLR2, both Cluspro and PatchDock have identified VC1 as potential binder with TLR2. Hydrogen bonds plays viral role in protein-receptor interaction, and it determines the stability of complex32,33. Intramolecular interactions between vaccine construct and TLR2 was visualized by LigPlot+. Immune simulation study explains the efficiency of vaccine construct to elicit the immune response. Induction of memory B-cells and T-cells is one of the criteria to be a successful vaccine candidate34,35,36. Each injection of VC1 and VC2 increased the level of memory B-cells and T-cells and this population level is sustaining after the third injection (Figs. 7, 8). Also, the elevated level of IFN-γ and IL2 is exhibiting the capacity of vaccine constructs to establish the antiviral state, in general IFN-γ involves in antiviral replication and act as main effector molecule in cell mediated immunity. Escherichia coli is most preferable choice of the host to obtain more quantity of recombinant vaccine protein37. Java Codon Adaptation tool displayed codon adaptability index as > 0.99% and GC content as > 50% for both vaccine candidates, it confirmed the favorable content of vaccine constructs for high-level protein expression in E. coli host.
Conclusion
The present study has made an attempt to design the multi-epitope vaccine against KFDV by using immune-informatics tools. To our knowledge this is first report for identifying T-cell and B-cell targeting epitopes of KFDV. The designed chimeric vaccine peptide could elicit immune response, but still it need to be tested on in-vitro and in-vivo models. Interestingly, our constructed vaccine has cross-protection effect against AHFV. This study has given foresight for development of new prophylaxis for KFDV control in India, and gives the directions in selecting epitopes for KFDV vaccine development.
Material and methods
Retrieval of amino acid sequence and conservation analysis
Totally 13 KFDV envelope protein sequences (GenBank ID: ARJ34245.1, ARJ34247.1, ASF57827.1, ASF57825.1, ASF57832.1, ASF57833.1, ARJ34243.1, ARJ34244.1, ARJ34252.1, ARJ34248.1, ASF57831.1, ARJ34255.1, AJW65394.1) which reported during 1962 to 2016, were retrieved from NCBI database (http://www.ncbi.nlm.nih.gov).
Multiple sequence alignment (MSA) of retrieved sequences were performed in CLCworkbench (https://www.qiagenbioinformatics.com/products/clc-main-workbench/), alignment file (.aln) was generated from all retrieved envelop protein sequences, neighbor-joining method was used to the phylogenetic tree and conservation score of each amino acids were obtained.
Prediction of T-cell and B-cell epitopes
Eight different immunoinformatic tools were used to predict T-cell (MHC class I and MHC class II) and B-cell epitope regions on KFDV envelope protein. Three different tools (IEDB-MHC-I, IEDB- combined and MAPPP) were used to obtain MHC class I epitope, proteasomal cleavage score, Transporter of Antigenic Peptide (TAP) and MHC scores. MHC class II epitope was prediction by IEDB- MHC-II and MHC2Pred tools. B-cell epitopes were identified by ABCpred, BCpred, and SVMTriP tools, the detailed information on tools algorithm, URL site and threshold values were in Table 9.
Self-antigen and cross-protection analysis
Uniport-BLAST tool (https://www.uniprot.org/blast/) was used to checked all top ranked T-cell and B-cell epitope sequences for self-antigen and cross-protection analysis. Self–antigen or similarity of human proteins with predicted epitopes were identified by Uniport- BLAST search against uniportkb_human databases. And UniProtKB database BLAST search was used to identify the cross protection of selected epitopes with other pathogenic organisms.
Construction of multi-epitope vaccine candidate
Based on high score, multi-immunogenic and overlapping sequence of T-cell and B-cell epitopes were conjugated to construct the multi-epitope vaccine candidates. Construct of vaccine has started with adjuvant β defensin (ACK99045.1) peptide sequence, in order to augment the immunogenicity of the vaccine candidate. Adjuvant was connected to MHC-I epitopes with EAAAK linker, every individual epitopes of MHC-I were linked by GGGS and all MHC-II epitopes assembled with GPGPG linker. Whereas B-cell epitopes were combined by KK linker. Usage of the linker between two epitopes will increase the immunogenicity and function of the vaccine constructs54.
Allergenicity and antigenicity of the vaccine candidate
Algpred (https://webs.iiitd.edu.in/raghava/algpred/submission.html) used to predict the allergenicity of vaccine candidate based on the similarity of known epitope. The vaccine candidate sequence was uploaded into the server, the IgE epitope and PID-BLAST search on allergen representative peptide algorithms were chosen for allergen prediction55. Antigenicity capacity of the vaccine candidate were evaluate by VaxiJen 2.0 server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html)56.
Physiochemical properties and solubility prediction
Physiochemical parameters of vaccine candidate such as molecular weight, theoretical pI, instability index, aliphatic index, estimated half-life and grand average of hydropathicity (GRAVY) were identified by ProtParam (https://web.expasy.org/protparam/) tool57. PROSO II server (http://mbiljj45.bio.med.uni-muenchen.de:8888/prosoII/prosoII.seam) was used to evaluate the solubility of vaccine candidates58.
3D structure prediction, refinement and validation
Phyre2 server (http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index)59 used to build the 3D-structure of the designed vaccine candidates. The server is build the 3D model based on homologues with known protein structure, and produce accurate model of about 70% of the domains in a typical genome. The obtained 3D structures of designed multi-epitope vaccine candidates were subjected into two-step refinement. Initial refinement was performed in ModRefiner (https://zhanglab.ccmb.med.umich.edu/ModRefiner/)60, an atomic-level protein structure refinement tool. This program makes protein into full-atomic relaxation, where initial model or reference model does not restrain the refined model. This improves both global and local structures with more accurate side chain positions, better hydrogen-bonding networks, and fewer atomic overlaps. Then second refinement was done with GalaxyRefine server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE)61,62, it performs repeated structure perturbation to side-chains and for secondary structure elements and loops are also applied followed by overall structural relaxation by molecular dynamics simulation. The obtained 3D-structures of vaccine candidates were validated in RAMPAGE server (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php)63 and ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php)64,65. ProSA recognize the errors in experimental and theoretical model of protein structure and z-core will tell about the quality of the protein structure.
Molecular docking of designed vaccine candidates with TLR2
TLR plays viral role in innate immune response upon the viral infections, especially TLR2 were reported for recognition of viral envelope protein. Hence, TLR2 (PDB ID: 2Z7X) was selected as target protein against to constructed vaccine candidates. ClusPro 2.0 server (https://cluspro.org)66 and PatchDock (https://bioinfo3d.cs.tau.ac.il/PatchDock/php.php)67 were used for this purpose.
ClusPro 2.0 server was used to predict the binding energy between TLR-2 and vaccine construct, experiment was performed by uploading PDB files of receptors and ligands into server and submitted with default parameters. PatchDock was also performed at default settings, top 1000 transformed docking position generated from PatchDock server was re-scored by FireDock server (http://bioinfo3d.cs.tau.ac.il/FireDock/php.php)68,69 and global energy of each vaccine candidate with TLR2 was obtained.
Ligplot analysis and molecular dynamic simulation of Docking complex
Interaction of TLR2 with vaccine candidates were assessed by Ligplot, docking complexes resulted from cluspro analysis were saved as PDB format and uploaded into Ligplot tool70 and intermolecular interactions such as hydrogen bonds, hydrophobic contact was obtained in the form of 2D representation.
The molecular dynamics simulation was performed to check the stability of docking complexes. The iMODS (http://imods.chaconlab.org/)71,72,73 web-server was used to calculate B-factor (disorder of atoms), structural deformability and eigenvalue.
In silico cloning optimization of KFD multi-epitope vaccine candidates
The Java Codon Adaptation Tool (http://www.prodoric.de/JCat) was used for reverse translation and codon optimization of designed vaccine candidates74. Codon optimization was executed in order to express the KFD multi epitope vaccine construct in E. coli (strain K12). The output files were checked for Codon Adaptation Index (CAI) (> 0.8–1.0), and GC content (30–70%), to ensure transcription and translational efficiency of designed vaccine construct. Resulted optimized codon sequence of vaccine candidates were introduced with XhoI and NdeI restriction sties at N-terminal and C-terminal ends respectively. SnapGene (www.snapgene.com) was utilized to insert the vaccine sequence in expression vector pET-30a(+), between XhoI and NdeI cloning site, final clones of vaccine candidates VC1 and VC2 were obtained.
Immune response simulation
Immune response profile of the constructed vaccine candidates was evaluated by C-IMMSIM server75. Experiment was performed by uploading PDB files of vaccine constructs, random speed, simulation volume and simulation steps were set as 12,345, 50 and 1100 respectively. Three insilco injections were given at the time steps of 1, 84 and 168 respectively (1-time step is equal to 8 h in real life), with no LPS and maintained minimum 30 days of time interval between two injections.
References
Work, T. H., A new virus disease in India. Summary of preliminary report on investigations of the Virus Research Center on an epidemic disease affecting forest villagers and wild monkeys of Shimoga Districk, Mysore. Indian J. Med. Sci. 11, 341–342 (1957).
Sreenivasanh, M. A., Bhat, R. & Rajagopalan, P. K. The epizootics of kyasanur forest disease in wild monkeys during 1964 to 1973. Trans. R. Soc. Trop. Med. Hyg. 80, 810–814 (1986).
Sreejith, K. A. B. K. N. K. Kyasanur forest disease virus breaking the endemic barrier: An investigation into ecological effects on disease emergence and future outlook. 1–8. https://doi.org/10.1111/zph.12349 (2017).
Yadav, P. D. et al. Phylogeography of Kyasanur Forest Disease virus in India (1957–2017) reveals evolution and spread in the Western Ghats region. Sci. Rep. 1–12. https://doi.org/10.1038/s41598-020-58242-w (2020).
Naren Babu, N. et al. Spatial distribution of Haemaphysalis species ticks and human Kyasanur Forest Disease cases along the Western Ghats of India, 2017–2018. Exp. Appl. Acarol. 77, 435–447 (2019).
Dodd, K. A. et al. Ancient ancestry of KFDV and AHFV revealed by complete genome analyses of viruses isolated from ticks and Mammalian hosts. PLoS Negl. Trop. Dis. 5, 1–7 (2011).
Gritsun, D. J., Jones, I. M., Gould, E. A. & Gritsun, T. S. Molecular archaeology of Flaviviridae untranslated regions: Duplicated RNA structures in the replication enhancer of flaviviruses and pestiviruses emerged via convergent evolution. PLoS One 9, 1–11 (2014).
Chakraborty, S., Andrade, F. C. D., Ghosh, S., Uelmen, J. & Ruiz, M. O. Historical expansion of Kyasanur Forest Disease in India from 1957 to 2017: A retrospective analysis. GeoHealth 3, 44–55 (2019).
Gurav, Y. K. et al. Kyasanur Forest Disease prevalence in Western Ghats proven and confirmed by recent outbreak in Maharashtra, India, 2016. Vector-Borne Zoonotic Dis. 18, 164–172 (2018).
Mehendale, S. et al. Kyasanur Forest Disease outbreak and vaccination strategy, Shimoga District, India 2013–2014. Emerg. Infect. Dis. 21, 2013–2014 (2019).
Kasabi, G. S., Murhekar, M. V., Sandhya, V. K. & Raghunandan, R. Coverage and effectiveness of Kyasanur Forest Disease (KFD) vaccine in Karnataka, South India, 2005–10. PLoS Negl. Trop. Dis. 7, 13–16 (2013).
Shil, P., Yadav, P. D., Patil, A. A., Balasubramanian, R. & Mourya, D. T. Bioinformatics characterization of envelope glycoprotein from Kyasanur Forest disease virus. Indian J. Med. Res. https://doi.org/10.4103/ijmr.IJMR (2018).
de Sousa, C. B. P., da Soares, I. S. & Rosa, D. S. Editorial: Epitope discovery and synthetic vaccine design. Front. Immunol. 9, 9–11 (2018).
Ali, M., Pandey, R. K., Khatoon, N., Narula, A. & Mishra, A. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. https://doi.org/10.1038/s41598-017-09199-w (2017).
Can, H., Köseoğlu, A. E., Alak, S. E., Güvendi, M. & Döşkaya, M. In silico discovery of antigenic proteins and epitopes of SARS-CoV-2 for the development of a vaccine or a diagnostic approach for COVID-19. Sci. Rep. https://doi.org/10.1038/s41598-020-79645-9 (2020).
Kar, T. et al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. https://doi.org/10.1038/s41598-020-67749-1 (2020).
Behmard, E., Soleymani, B., Najafi, A. & Barzegari, E. Immunoinformatic design of a COVID-19 subunit vaccine using entire structural immunogenic epitopes of SARS-CoV-2. Sci. Rep. https://doi.org/10.1038/s41598-020-77547-4 (2020).
Bibi, S., Ullah, I., Zhu, B., Adnan, M. & Liaqat, R. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Sci. Rep. https://doi.org/10.1038/s41598-020-80899-6 (2021).
Klimka, A. et al. Epitope-specific immunity against Staphylococcus aureus coproporphyrinogen III oxidase. NPJ Vaccines https://doi.org/10.1038/s41541-020-00268-2 (2021).
Palanisamy, N., Akaberi, D., Lennerstrand, J. & Lundkvist, Å. Comparative genome analysis of Alkhumra hemorrhagic fever virus with Kyasanur forest disease and tick-borne encephalitis viruses by the in silico approach. Pathog. Glob. Health 112, 1–17 (2018).
Devadiga, S., McElroy, A. K., Prabhu, S. G. & Arunkumar, G. Dynamics of human B and T cell adaptive immune responses to Kyasanur forest disease virus infection. Sci. Rep. 10, 1–9 (2020).
Liu, G. et al. Immunogenicity and efficacy of flagellin-envelope fusion dengue. Clin. Vaccine Immunol. 22, 516–525 (2015).
Bauer, A. et al. Preferential targeting of conserved gag regions after vaccination with a heterologous DNA prime-modified vaccinia virus Ankara boost HIV-1 vaccine regimen. J. Virol. 91, e00730-17 (2017).
Rajaiah, P. Kyasanur Forest Disease in India: Innovative options for intervention. Hum. Vaccines Immunother. 15, 2243–2248 (2019).
Sette, A. et al. Definition of epitopes and antigens recognized by vaccinia specific immune responses: Their conservation in variola virus sequences, and use as a model system to study complex pathogens. Vaccine 27(Suppl 6), G21–G26 (2009).
Mackay, L. K. et al. T Cell Detection of a B-Cell Tropic Virus Infection: Newly-Synthesised Versus Mature Viral Proteins as Antigen Sources for CD4 and CD8 Epitope Display. 5, (2009).
De Gregorio, E., Caproni, E. & Ulmer, J. B. Vaccine adjuvants: Mode of action. Front. Immunol. 4, 1–6 (2013).
Tani, K. et al. Defensins act as potent adjuvant taht promote cellular and humoral immune response in mice to a lymphhoma idiotype and carrier antigents. Int. Immunol. 12, 691–700 (2000).
Barton, G. M. Viral recognition by Toll-like receptors. Semin Immunol. 19, 33–40 (2007).
Zheng, M. et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. https://doi.org/10.1038/s41590-021-00937-x (2021).
Xagorari, A. & Chlichlia, K. Toll-like receptors and viruses: Induction of innate antiviral immune responses. Open Microbiol. J. 2, 49–59 (2008).
Pauling, L., Corey, R. B. & Branson, H. R. The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. 37, 205–211 (1951).
Pauling, L. & Corey, R. B. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. U. S. A. 37, 251–256 (1951).
Carsetti, R. The role of memory B cells in immunity after vaccination. Paediatr. Child Health 19, S160–S162 (2009).
Palm, A. E. & Henry, C. Remembrance of things past: Long-term B cell memory after infection and vaccination. Front. Immunol. 10, 1–13 (2019).
Cox, R. J. & Brokstad, K. A. Not just antibodies: B cells and T cells mediate immunity to COVID-19. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-020-00436-4 (2020).
Rosano, G. L. & Ceccarelli, E. A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 5, 1–17 (2014).
Kim, Y., Sidney, J., Pinilla, C., Sette, A. & Peters, B. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinform. 10, 1–11 (2009).
Nielsen, M. et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 12, 1007–1017 (2003).
Lundegaard, C., Lund, O. & Nielsen, M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics 24, 1397–1398 (2008).
Peters, B. & Sette, A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform. 6, 1–9 (2005).
Sidney, J. et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 4, 1–14 (2008).
Tenzer, S. et al. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell. Mol. Life Sci. 62, 1025–1037 (2005).
Peters, B., Bulik, S., Tampe, R., van Endert, P. M. & Holzhütter, H.-G. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J. Immunol. 171, 1741–1749 (2003).
Hakenberg, J. et al. MAPPP: MHC class I antigenic peptide processing prediction. Appl. Bioinform. 2, 155–158 (2003).
Lin, H. H., Ray, S., Tongchusak, S., Reinherz, E. L. & Brusic, V. Evaluation of MHC class I peptide binding prediction servers: Applications for vaccine research. BMC Immunol. 9, 1–13 (2008).
Wang, P. et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 11, 568 (2010).
Wang, P. et al. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 4, e1000048 (2008).
Lata, S., Bhasin, M. & Raghava, G. P. S. Application of machine learning techniques in predicting MHC binders. In Immunoinformatics: Predicting Immunogenicity In Silico (ed. Flower, D. R.) 201–215 (Humana Press, 2007). https://doi.org/10.1007/978-1-60327-118-9_14.
Saha, S. & Raghava, G. P. S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65, 40–48 (2006).
Chen, J., Liu, H., Yang, J. & Chou, K. C. Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids 33, 423–428 (2007).
El-Manzalawy, Y., Dobbs, D. & Honavar, V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. 21, 243–255 (2008).
Yao, B., Zhang, L., Liang, S. & Zhang, C. SVMTriP: A method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PLoS One 7, 5–9 (2012).
Nezafat, N., Ghasemi, Y., Javadi, G., Khoshnoud, M. J. & Omidinia, E. A novel multi-epitope peptide vaccine against cancer: An in silico approach. J. Theor. Biol. 349, 121–134 (2014).
Saha, S. & Raghava, G. P. S. AlgPred: Prediction of allergenic proteins and mapping of IgE epitopes. Nucl. Acids Res. 34, 202–209 (2006).
Doytchinova, I. A. & Flower, D. R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 8, 1–7 (2007).
Gasteiger, E. et al. Protein identification and analysis tools in the ExPASy server (ed. Walker, J. M.) 571–607 (Humana Press Inc, 2005).
Smialowski, P., Doose, G., Torkler, P., Kaufmann, S. & Frishman, D. PROSO II—A new method for protein solubility prediction. FEBS J. 279, 2192–2200 (2012).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Xu, D. & Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 101, 2525–2534 (2011).
Shin, W.-H., Lee, G. R., Heo, L., Lee, H. & Seok, C. Prediction of protein structure and interaction by GALAXY protein modeling programs. Bio Des. 2, 1–11 (2014).
Ko, J., Park, H., Heo, L. & Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucl. Acids Res. 40, 294–297 (2012).
Carugo, O. & Djinović-Carugo, K. A proteomic Ramachandran plot (PRplot). Amino Acids 44, 781–790 (2013).
Wiederstein, M. & Sippl, M. J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucl. Acids Res. 35, 407–410 (2007).
Sippl, M. J. Recognition of errors in three-dimensional structures of proteins. Proteins Struct. Funct. Bioinform. 17, 355–362 (1993).
Vajda, S. et al. New additions to the ClusPro server motivated by CAPRI. Proteins Struct. Funct. Bioinform. 85, 435–444 (2017).
Schneidman-duhovny, D., Inbar, Y., Nussinov, R. & Wolfson, H. J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucl. Acids Res. 33, 363–367 (2005).
Mashiach, E., Schneidman-Duhovny, D., Andrusier, N., Nussinov, R. & Wolfson, H. J. FireDock: A web server for fast interaction refinement in molecular docking. Nucl. Acids Res. 36, W229–W232 (2008).
Andrusier, N., Nussinov, R. & Wolfson, H. J. FireDock: Fast interaction refinement in molecular docking. Proteins 69, 139–159 (2007).
Wallace, A. C., Laskowski, R. A. & Thornton, J. M. LIGPLOT: A program to generate schematic diagrams of protein–ligand interactions. Protein Eng. 8, 127–134 (1995).
Lopéz-blanco, J. R., Garzón, J. I. & Chacón, P. iMod: Multipurpose normal mode analysis in internal coordinates. Bioinformatics 27, 2843–2850 (2011).
Aliaga, I., Quintana-ort, E. S. & Chac, P. iMODS: Internal coordinates normal mode analysis server. Nucl. Acids Res. 42, 271–276 (2014).
Kovacs, J. A., Chaco, P. & Abagyan, R. Predictions of protein flexibility: First-order measures. Proteins Struct. Funct. Bioinform. 668, 661–668 (2004).
Grote, A. et al. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucl. Acids Res. 33, 526–531 (2005).
Rapin, N., Lund, O., Bernaschi, M. & Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS One 5, e9862 (2010).
Author information
Authors and Affiliations
Contributions
S.A.—Concept making, In-silico experiments, result interpretations. P.V.—contributed insilico experiments and writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arumugam, S., Varamballi, P. In-silico design of envelope based multi-epitope vaccine candidate against Kyasanur forest disease virus. Sci Rep 11, 17118 (2021). https://doi.org/10.1038/s41598-021-94488-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-94488-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
