
Abstract
Lutzomyia umbratilis is the main vector of Leishmania guyanensis in the Brazilian Amazon and in neighboring countries. Previous biological and molecular investigations have revealed significant differences between L. umbratilis populations from the central Brazilian Amazon region. Here, a phylogeographic survey of L. umbratilis populations collected from nine localities in the Brazilian Amazon was conducted using two mitochondrial genes. Statistical analyses focused on population genetics, phylogenetic relationships and species delimitations. COI genetic diversity was very high, whereas Cytb diversity was moderate. COI genealogical haplotypes, population structure and phylogenetic analyses identified a deep genetic differentiation and three main genetic groups. Cytb showed a shallower genetic structure, two main haplogroups and poorly resolved phylogenetic trees. These findings, allied to absence of isolation by distance, support the hypothesis that the Amazon and Negro Rivers and interfluves are the main evolutionary forces driving L. umbratilis diversification. The main three genetic groups observed represent three evolutionary lineages, possibly species. The first lineage occurs north of the Amazon River and east of Negro River, where Le. guyanensis transmission is intense, implying that L. umbratilis is an important vector there. The second lineage is in the interfluve between north of Amazon River and west of Negro River, an area reported to be free of Le. guyanensis transmission. The third lineage, first recorded in this study, is in the interfluve between south of Amazon River and west of Madeira River, and its involvement in the transmission of this parasite remains to be elucidated.
Similar content being viewed by others


Introduction
The diversification patterns (genetically structured populations, evolutionary lineages, complete speciation) in phlebotomine Diptera have been linked to multiple factors, such as climate events, geographic barriers (rivers, mountains), biomes, latitude and altitude, habitat modification and landscape fragmentation caused by anthropogenic actions, among others1. Such factors may reduce the dispersal capacity of sandflies, leading them to become isolated populations, with a resulting loss of genetic diversity within populations and the increase in differentiation between them1,2,3,4,5. The low flight capacity6,7, characteristic of these insects, and the type of soil used for breeding are also factors that may contribute further to population isolation in consequence favor genetic differentiation and, eventually, speciation. In this context, studying the microevolutionary processes that act at the level of sandflies vector populations is key for the identification of these patterns, which are baselines for the epidemiological studies, surveillance and vector control strategies.
It has been proposed that the major rivers of the Amazonian region have acted, and continue to act, as significant dispersal barriers for a variety of taxa, including primates8, lizards9, frogs10, birds11,12 and insects13,14—a scenario referred to as the “river-barrier hypothesis”15. This hypothesis predicts that major Amazonian rivers significantly reduce or prevent gene flow between populations inhabiting opposite banks, so promoting genetic divergence and increasing the opportunity for allopatric speciation. If this is correct, (1) it is expected that sister species will occur on opposite river banks, (2) the genetic similarity between populations will be greater in sites on the same bank that in sites on opposite banks separated by the same distance, and (3) the boundaries of species distributions will coincide with large rivers16,17.
Lutzomyia (Nyssomyia) umbratilis Ward and Fraiha, 1977 is the main vector of the etiologic agent that causes American Cutaneous Leishmaniasis (ACL), Leishmania (Viannia) guyanensis Floch, 1954, in north South America18,19. In the Brazilian Amazon, L. umbratilis is highly anthropophilic and has been identified as the main vector of this parasite in the states of Amazonas, Amapá, Pará and Roraima20,21, an area that together holds > 50% of the cases of human leishmaniasis recorded for the whole Brazilian Amazon21. Lutzomyia umbratilis has also been implicated in the transmission of Le. guyanensis in French Guiana and Suriname18,19,20,22,23.
This phlebotomine species occurs in northern South America, including Bolivia, Brazil, Colombia, French Guiana, Guyana, Peru, Suriname and Venezuela24,25. In Brazil, L. umbratilis has been recorded in all states in the northern region19,20,22,23,24, as well as the states of Mato Grosso and Mato Grosso do Sul (west central and southwest regions)26,27, and the states of Maranhão, Pernambuco and Alagoas, in the northeastern region24,28,29,30. This vast geographic area, with discontinuous distribution and the presence of geographic barriers, along with the low flight capacity of these insects6,7, means that L. umbratilis populations could be highly susceptible to evolve into differentiated populations, evolutionary lineages and distinct species.
Lutzomyia umbratilis was described by Ward and Fraiha31, based on specimens (females) captured in the Monte Dourado area of the Jari River (north of Amazon River), state of Pará, Brazil. Prior to this L. umbratilis had been wrongly identified as Lutzomyia anduzei Rozeboom 194232 due to their great morphological similarity. The species have overlapping geographic distributions across a large portion of northern South America24. Nowadays, they may be distinguished based on the internal and external genitalia morphology of male and female adults and by molecular markers24,33.
In the Brazilian Amazon, adults of L. umbratilis are often found in rainforest, especially in areas of the high humidity and dim light, as consequence, this species has been recognized as ombrophilous, as is expressed in its name – L. umbratilis. Adults of this species have been captured using aspirators in bases of tree trunks during daytime, as well as with CDC (Center for Disease Control) miniature light traps at ground level, and in the forest canopy at night – a behavior termed acrodendrophily34. Lutzomyia umbratilis seems to be abundant in the central Brazilian Amazon region, tending to decline towards the margins of this region35.
Arias and Freitas13,36 reported that the susceptibility of this vector to Leishmania may vary within the central Brazilian Amazon region. These authors reported L. umbratilis populations naturally infected with Le. guyanensis to the north of the Negro and Amazon Rivers; but they did not observe natural infections by Leishmania in this species to the north of the Amazon River and west of the Negro River. Arias and Freitas13 hypothesized that this fluvial system could act as a vicariant barrier to Le. guyanensis transmission. In line with this hypothesis13, a study of experimental infection, performed with two samples of L. umbratilis from opposite banks of these rivers, revealed higher vector competence in a sample from north of Amazon River than in sample from north of the Amazon River and west of the Negro River37, supporting the Arias and Freitas’s hypothesis13. In fact, the presence of cryptic or sibling species complexes in a given region can produce heterogeneous patterns of parasite transmission, because they may differ in vector competence (the intrinsic ability of a vector to transmit a pathogen), host feeding preference, feeding behavior and breeding sites as well as they may either occur sympatrically or have distinct geographical distributions.
A biological study conducted with L. umbratilis populations from Manaus and Manacapuru (north of Amazon River/east of Negro River and north of Amazon River/west of Negro River, respectively) revealed significant biological differences38. A second study that combined morphology and isozymes analyses of four L. umbratilis populations from this fluvial system showed subtle morphological differences in the immature stage and in female genitalia, whereas the isozymes markers did not reveal any differences among populations studied39. However, isozymes are not appropriated markers to separate species that have recently diverged.
Unlike the results with isozymes, a phylogeographic structure study using molecular markers (COI-mtDNA) revealed low and non-significant differentiation between populations situated on the same banks of the Amazon and Negro Rivers, but marked and very significant differences between populations situated on the opposite banks of these rivers33,35,40. This pattern resulted in the absence of isolation by distance (IBD). Although all haplotypes were connected in a unique network, the strong bimodality observed with the mismatch analyses, when all samples were analyzed together, was consistent with long-term isolation, suggesting historical isolation between the analyzed populations40. These groups probably attained complete isolation following the most recent formation of the Negro and Amazon Rivers network (~ 2.4 Mya to present), which was considered as the main evolutionary force. In line with these results, another study, using the Barcode region (COI) of these L. umbratilis two groups, also failed to find evidence for haplotype sharing. However, they could be identified by unique fixed mutation, as well as the genetic distance was rather small33. Later, Freitas and others41, using also a COI fragment, compared Manacapuru and Rio Preto da Eva populations, situated on the opposite banks of the same rivers, with a sample of Recife, in the northeast region from Brazil. Consistent with previous studies33,40, two distinct groups were observed: one group clustered individuals from Rio Preto da Eva (north Amazon River and east Negro River) plus Recife and, another group, clustered only individuals of Manacapuru (interfluve located of the north Amazon River and west of Negro River). This finding indicates that the Recife population may have been derived from populations from the north Amazon River.
However, most of these studies are based on samples from a small number of locations. As a result, additional investigations are needed to fully elucidate the number of species in this complex and the role of each one in the transmission of Le. guyanensis in the Amazonian region. Such information provides an essential baseline for epidemiology, as well as for surveillance and vector control measures. In this context, the current study investigated the molecular variation and phylogeographic structure of L. umbratilis populations collected from nine geographic areas in the Brazilian Amazon region, inferred by sequencing of two mitochondrial fragments (COI and Cytb genes) to test the hypotheses: (1) Are the largest rivers in the Brazilian Amazon acting as barriers on the L. umbratilis populations? (2) Are the populations located on the opposite banks of the Amazon and Negro Rivers and in the interfluves genetically different? (interfluves = beard or the elevated area between two watercourses or two valleys); (3) How many lineages or species exist within L. umbratilis complex in the region studied?
Material and methods
Sandfly collection and morphological identification
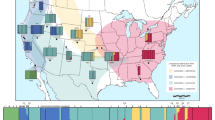
Collection of L. umbratilis adults were performed in nine localities within the Brazilian Amazon, with seven localities in the state of Amazonas (km 43 of BR-174 Federal Highway [BR]; km 65 of AM-010 Highway in the municipality of Rio Preto da Eva [RP]; Manaus [MN]; km 60 of AM-070 Highway in the municipality of Manacapuru [MC]; km 60 and km 70 of AM-352 Highway in the municipality of Novo Airão [NA]; Pitinga [PI]; Ramal do Sampaio in the municipality of Autazes [AU]), one in state of Pará (municipality of Oriximiná, Cachoeira Porteira [CP]), and one in state of Amapá (Porto Grande/Serra do Navio [PG]). Figure 1a shows the locations of all nine study sites: (1) north of Amazon River and east of Negro River: Cachoeira Porteira (CP; purple), km 43 of BR-174 Highway (BR, red), Rio Preto da Eva (RP, orange), Manaus (MN, yellow), Pitinga (PI, pink) and Porto Grande/Serra do Navio (PG, dark green); (2) north of Amazon River and west of Negro River (interfluve 1, between Amazon and Negro Rivers): Manacapuru (MC, dark blue) and Novo Airão (NA, light blue); (3) south of Amazon River and west Madeira River (interfluve 2, between Amazon and Madeira Rivers): Ramal do Sampaio, in the municipality of Autazes (AU, light green). Table 1 provides information on the localities sampled, geographic co-ordinates and sample sizes for each marker.

(a) Collection sites of the nine Lutzomyia umbratilis populations from the Brazilian Amazon and the geographic distribution of three lineages inferred in this study. The colored circles on the map represent the sampled sites. Purple: Cachoeira Porteira (CP); Red: km 43 of BR-174 Highway (BR); Orange: Rio Preto da Eva (RP); Yellow: Manaus (MN); Dark blue: Manacapuru (MC); Light blue: Novo Airão (NA); Pink: Pitinga (PI); Light green: Autazes (AU); Dark green: Porto Grande/Serra do Navio (PG). Map was created using SimpleMappr (https://www.simplemappr.net) (public domain) and adapted for illustrative purposes in Inkscape version 1.0.1 (https://www.inkscape.org). (b) Geographic distribution inferred for the three lineages of this study. Lineage I (Red); Lineage II (Blue), Lineage III (Green). Map was created using SimpleMappr (https://www.simplemappr.net) (public domain) and adapted for illustrative purposes in Inkscape version 1.0.1 (https://www.inkscape.org).
The COI sequences of the individuals from Cachoeira Porteira, km 43 of BR-174 Highway, Rio Preto da Eva, Manaus, Manacapuru and Novo Airão were previously analyzed by Scarpassa and Alencar40, while three new samples (Pitinga, Autazes and Porto Grande/Serra do Navio) were analyzed in this study using the same COI fragment (Table 1). Additionally, in the current study, the sample size of Manaus was expanded, with 18 new specimens sequenced, totaling n=22, and re-evaluated (Table 1). This database was integrated with Scarpassa and Alencar’s data40 and re-evaluated in this study. For the Cytb gene, nine populations were analyzed in the current study (Table 1).
Sandflies were collected with CDC miniature light traps (CDC “miniature”- HAUSHERR MACHINE WORKS, NEW JERSEY, EUA)42, and also with aspirators on the bases of trunks of rainforest trees during the daytime. To collect a random population sample and avoid an excess of offspring from the same females, we followed Scarpassa and Alencar40 and sampled a minimum of five tree trunks within a given study area. Captured specimens were killed at – 20 °C freezer and morphologically identified based on genital morphology, following the taxonomic key of Young and Duncan24 and the characters described in Scarpassa and Alencar33. Following identification, specimens were preserved in 95% ethanol and stored at – 20 °C until genomic DNA was extracted. Sample collections were made under the System of Information on the Biodiversity (SISBIO), using permanent license number 38440-1 awarded to VMS.
Genomic DNA extraction, PCR amplification and sequencing of the COI and Cytb markers
For the COI gene, 176 specimens were analyzed, with 111 from the study of Scarpassa and Alencar40, and 65 analyzed specifically for the current study. For the Cytb gene, 187 specimens from the nine localities were analyzed in this study (Table 1).
Total genomic DNA was extracted individually from whole sandflies using the Phenol–Chloroform protocol43, with the DNA pellet resuspended in 30 μL of 1xTE buffer (10 mM Tris-Cl pH 8.0; 1 mM EDTA pH 8.0) or in sterile water. A small aliquot of this DNA was stored at − 20 °C and used in the amplification by polymerase chain reaction (PCR) of the two analyzed markers. All DNA remaining from the specimens was kept frozen at − 81 °C as voucher DNA in the vector insect collection at the Laboratory of Population Genetics and Evolution of Vector Mosquitoes at the Instituto Nacional de Pesquisas da Amazônia, in Manaus, Brazil. DNA extracted from all individuals was amplified with specific oligonucleotide primers for the two genes, as described below.
A long fragment (1181 bp), representing the variable region of end 3’ of cytochrome c oxidase I gene (COI), was amplified by PCR using the primers described in Zhang and Hewitt44. The PCR reactions was included Platinum Taq DNA Polymerase of High Fidelity (LIFE TECHNOLOGY), and negative control was included for all reactions. The PCR products were electrophoresed in 1% agarose gel stained with Gel Red Nucleic Acid Gel Stain (BIOTIUM INC., HAYWARD, USA) and visualized under UV light to check the size and quality of expected products. The amplified products were purified using PEG precipitation (20% polyethylene glycol 8000 and 2.5 M NaCl). Both DNA strands were sequenced using the Big Dye Terminator Kit, v. 3.1. The reactions were electro-injected in an automated ABI 3130 xl Genetic Analyzer (APPLIED BIOSYSTEMS, THERMO FISHER SCIENTIFIC, WALTHAM, MA, USA).
A fragment of 512 bp from the Cytb gene was amplified by PCR using the primers described in Coutinho-Abreu and others45. The PCR reactions was included Platinum Taq DNA Polymerase of High Fidelity (LIFE TECHNOLOGY), and a negative control was used in all reactions. The PCR products were electrophoresed in 1% agarose gel stained with Gel Red Nucleic Acid Gel Stain (BIOTIUM INC., HAYWARD, USA), and visualized under UV light to verify the size and quality of expected products. Amplified products purification and sequencing reactions were carried out as described above.
Statistical analyses
COI and Cytb sequences were automatically aligned using Clustal W, and manually edited in BioEdit v. 7.0.8.046 with the aid of the electropherogram viewer Chromas Lite47. Consensus sequences of both genes were confirmed with the mitochondrial genome of L. umbratilis48, using the BLAST (BASIC LOCAL ALIGNMENT SEARCH TOOL)49, which resulted in 99 to 100% identity. The sequences of this study were mapped between the positions 1759 and 2939 bp for COI, and between the positions 11,241 and 11,752 bp for Cytb of mitochondrial genome of this species48.
Both COI and Cytb datasets were checked for saturation levels against genetic distances using DAMBE50, which allows testing for the presence of any saturation between transition and transversion rates in relation to genetic distances. Results showed no saturation for either marker, indicating the datasets are appropriate for the applied phylogenetic inferences. Haplotypes were determined using DnaSP v. 5.1051 and TCS v. 1.2152. Identical sequences were considered to represent a single haplotype. To analyze population history and haplotype genealogy, haplotype networks were analyzed for the two markers separately. To calculate the maximum number of mutational connections between pairs of sequences, the 95% parsimony criterion was used, with analyses run on TCS v. 1.2152. Using this criterion, the possibility that all samples analyzed formed a single network or two or more disconnected networks was investigated.
Unfortunately, concatenated analyses with two markers were not possible as the DNAs of the same individuals from Manacapuru and Novo Airão populations did not amplify for both markers used. Thus, the phylogenetic relationships were inferred with all haplotypes for each marker using Neighbor-Joining (NJ) and Maximum Likelihood (ML) in MEGA v. 7.053 and Bayesian Inference (BI) with the Mr.Bayes v. 3.2.554 and *BEAST v. 2.055 algorithms. Kimura2-Parameters (K2P) distances56 were used for the NJ analysis, with 5000 replicates, while the ML and BI analyses were inferred using the General Time Reversible (GTR) + G + I nucleotide substitution model for COI, and the HKY nucleotide substitution model for Cytb, both previously selected with the Akaike Information Criterion (AIC) via jModelTest57. In the ML analyses, a tree was generated with 2000 replicates. In the BI analyses, for each marker two simultaneous independent runs of the Markov Chain Monte Carlo (MCMC) were performed for 100 million generations, while sampling every 1000 generations with a burn-in of 25%. Bayesian Posterior Probabilities (BPP) were used to assess nodal support.
To infer the limits between lineages, three models based on single locus data (COI) were used: GMYC (Generalized Mixed Yule Coalescent)58, bGMYC (with implementation Bayesian of GMYC)59 and ABGD (Automatic Barcode Gap Discovery)60. Delimitation with the GMYC approach is based on assigning branching events to two categories, speciation and coalescent within species. With the assumption that species are monophyletic, a set of most recent common ancestor nodes can be specified that determines the type of branching events. bGMYC model identifies uncertainties in the limits of the species as per the changes in the ramification rates in the phylogenetic tree when distinct populations contain several species. ABGD model allows partitioning of the DNA sequence datasets into clusters of similar taxa, establishing a range of maximum values of intraspecific divergence (P), without an a priori species hypothesis. These analyses were conducted using the bGMYC SPLITS package61 and implemented in the R package, version 3.6.061. The ultrametric trees were generated with *BEAUTi and *BEAST v.1.762 and used in these analyses.
The divergence time of the groups (lineages) were also estimated in *BEAST v. 2.055, using a relaxed lognormal clock with a Yule tree prior, assuming a constant lineage birth rate for each tree branch, and with a mutation rate of 2.3% for every million years for the COI gene63.
All phylogenetic analyses also included fragments corresponding to the COI and Cytb regions extracted from mitochondrial genome of L. umbratilis from Saint-Georges de l’Oyapock, French Guiana [Accession numbers: KP702938 and KP702939]48. Saint-Georges de l’Oyapock is situated on the border with the state of Amapá, Brazil, and lies north of the Amazon River (Fig. 1a). Lutzomyia anduzei and Bichromomyia flaviscutellata were used as outgroups in these analyses. Tree topologies were visualized and edited in FigTree v. 1.3.164.
Principal Coordinates Analysis (PCA) was also implemented for the COI using the R package, spider 1.5.0 with the function ordinDNA65. In this analysis, the distance between the clusters is approximately proportional to the genetic distances between species.
Genetic diversity and population structure
The intra-population genetic diversity measures, such as haplotype (h) and nucleotide (π) diversities, the average number of nucleotide differences (K), number of polymorphic sites (NS), transition and transversion rates (Ts/Tv), and the neutrality tests of Tajima’s D66 and Fu’s Fs tests67 for each sample and total were inferred using DnaSP, v. 5.1051 and Arlequin, v. 3.168. Tajima’s D was run to test strict neutrality, while the neutrality test, Fu’s Fs, was estimated to test population size stability. The latter test is more powerful for detecting population expansion and genetic hitchhiking.
Using the FST statistic, genetic differentiation between samples was inferred in Arlequin, v. 3.168. The average number of nucleotide substitutions per site between samples (Dxy), the number of net nucleotide substitutions per site between samples (Da), the number of shared polymorphisms between samples pairs (Ss), and a number of fixed differences between samples pairs (Sf) were calculated in DnaSP, v. 5.1051.
To check the number of genetic groups obtained in the previous analyses, we implemented the Bayesian Analysis of Population Structure (BAPS) with BAPS software69,70 for both markers. In these analyses, all sequences from both datasets were used and 1–9 clusters were employed (the upper corresponding to the total number of sampled localities), and five independent runs were made. The most probable genetic cluster configuration was prepared by comparing the log-likelihood values of the best models.
The genetic groups retrieved in the haplotypes network and phylogenetic analyses were used to estimate the mean intra- and inter-group genetic distances using the uncorrected-p genetic distance. These analyses were calculated in MEGA, v. 7.053, and the standard errors were estimated by bootstrapping with 1000 replicates.
To test the isolation by distance (IBD) hypothesis, the correlation between straight-line geographic distances and FST values among samples was applied using a Mantel test71 in Arlequin, v. 3.168 with 2000 permutations. Straight-line geographic distances among the localities were estimated using Google Earth.
Results
The molecular variation and phylogeographic structure of nine natural populations of L. umbratilis from the Brazilian Amazon were estimated. A total of 176 specimens were analyzed for COI and 187 for Cytb, totaling 363 individuals sequenced (Table 1). Fragment sizes were 1181 bp and 512 bp for COI and Cytb, respectively, totaling 1693 bp. All alignments were unambiguous and no insertions or deletions were detected in either dataset. Transitions (89.68%; 83.33%) were more common than transversions (10.32%; 16.67%), with most occurring at the third codon position (all synonymous) for both markers. COI and Cytb had 110 (9.31%) and 41 (8.01%) variable sites, of which 56 (4.74%) and 14 (2.73%) were parsimoniously informative, respectively. The average nucleotide composition for the COI was 39.9% T, 30.7% A (A + T = 70.6%), 15.2% C, and 14.1% G, while for Cytb these values were 39.5% T, 38.5% A (A + T = 78%), 13.5% C, and 8.5% G.
For COI, the 176 individuals generated 89 haplotypes, of which 52 (H1 to H52) had been described by Scarpassa and Alencar40, and 37 being new haplotypes (H53 to H89) identified in this study. Of these, 79 (88.76%) haplotypes were singletons or exclusive to a single sample. Table S1 shows the haplotype frequencies in the samples studied. The Autazes and Porto Grande/Serra do Navio populations did not share haplotypes with any other locality. The haplotypes genealogy produced two disconnected networks and six haplogroups (Fig. 2). The larger network comprised five haplogroups (H1 to H76; H81 to H89). Haplogroup I clustered 48 haplotypes (H1 to H28; H53 to H61; H73, H74) corresponding to five localities from the north Amazon River and east Negro River: Cachoeira Porteira, BR-174 Highway, Rio Preto da Eva, Manaus and H18 (shared), H73, H74 from Pitinga. Haplogroup II clustered 24 haplotypes (H29 to H52), comprising two samples from north of the Amazon River and west of Negro River (interfluve 1): Manacapuru and Novo Airão. Haplogroup III clustered eight haplotypes (H62 to H66; H68; H75 and H76), corresponding to 24 individuals from Pitinga, north of the Amazon River and east of Negro River. Haplogroup IV clustered all haplotypes (H81 to H89) from the Porto Grande/Serra do Navio, north of the Amazon River and east of Negro River. Haplogroup V clustered five haplotypes (H67; H69 to H72), corresponding to five individuals from Pitinga, which were separated from the remaining haplotypes by at least 12 mutational steps. Haplogroup VI, disconnected from the remaining haplogroups, clustered four haplotypes (H77 to H80) from Autazes. This haplogroup differed from others by 13 fixed sites (eight transitions and five transversions). Here, we defined a fixed site as occurring when a mutation is present in all individuals from one population and absent in all individuals of the other populations.

Parsimony haplotypes network of the 89 haplotypes obtained for COI in the nine Lutzomyia umbratilis populations from the Brazilian Amazon. H1 to H89 = haplotypes. I to VI = Haplogroups. The haplotype circle sizes are proportional to number of individuals observed in each haplotype (see Table S1). Full smaller circles (in black) represent mutational events.
For Cytb, the 187 sequences generated 36 haplotypes, ranging from two (Autazes) to nine (Cachoeira Porteira) haplotypes per sample (Table S1; Fig. 3). Seven (19.44%) haplotypes were shared between localities and 29 (80.56%) were singletons or exclusive to a single sample. The Autazes sample did not share haplotypes with any other locality. All haplotypes were connected in a unique network; however, two star-shaped haplogroups (I and II) were observed; each one represented by a more frequent haplotype (H3 and H22, respectively), positioned more centrally in their respective haplogroups and connected by derived haplotypes, positioned peripherally. In haplogroup I, the H3 haplotype was shared among individuals all localities from north of the Amazon River (Fig. 3). The derived haplotypes from the Cachoeira Porteira, BR-174 Highway, Rio Preto da Eva, Manaus and Porto Grande/Serra do Navio and H8, H24 to H27 haplotypes from Pitinga were separated from H3 by one to four mutations. H28 and H29 haplotypes from Autazes were separated from H3 by seven and eight mutations, respectively.

Parsimony haplotypes network of the 36 haplotypes obtained for Cytb in the nine Lutzomyia umbratilis populations from the Brazilian Amazon. H1 to H36 = haplotypes. I and II = Haplogroups. The haplotype circle sizes are proportional to number of individuals observed in each haplotype (see Table S1). Full smaller circles (in black) represent mutational events.
In haplogroup II, H22 was the most common haplotype (Fig. 3). It was shared by 66 individuals situated on both sides of the Negro River (27 from Manacapuru, 22 from Novo Airão [both north of the Amazon River and west of Negro River, interfluve 1], and 17 from Pitinga [north of the Amazon River and east of Negro River]). H33 to H36 from Manacapuru and Novo Airão, and H23 from Pitinga were derived haplotypes and were connected to H22 by only one mutation. H11 (BR-174 Highway) and H15 (RP and MN) connected the two haplogroups by two mutations. In haplogroup I, H8 haplotype (derived) was shared by individuals of the Pitinga and Cachoeira Porteira (same bank) and could suggest contemporaneous gene flow, whereas in haplogroup II the sharing of H22 haplotype (likely the ancestral form) by individuals from the opposite banks could indicate historical genetic connectivity.
Neighbor-Joining (NJ) tree topology inferred with the 89 haplotypes from COI retrieved five main groups (Fig. 4); however, most these showed weak support. Group 1, weakly supported, clustered the haplotypes from the Cachoeira Porteira (purple), BR-174 Highway (red), Rio Preto da Eva (orange), Manaus (yellow), the most haplotypes from Pitinga (pink) and the sequence of Saint Georges l’Oyapock, French Guiana. Group 2, moderately supported (66%), clustered all haplotypes from Manacapuru (dark blue) and Novo Airão (light blue). Group 3, weakly supported (55%), clustered all haplotypes from Porto Grande/Serra do Navio (dark green). Group 4, strongly supported (99%), clustered five haplotypes from Pitinga (pink). Group 5, the most basal and strongly supported (100%), clustered all haplotypes from Autazes (light green). Maximum Likelihood (ML) tree generated a very similar topology to Fig. 4 (Fig. S1).

Neighbor-Joining (NJ) tree generated based on the 89 haplotypes of COI. Tree inferred under the Kimura 2-Parameters model and 5000 replications. The support values are indicated above of the branches. The colors on the terminal branches of the tree represent the haplotypes observed in each locality, following the same color pattern of the Figs. 1, 2 and 3. The black color on the terminal branches of tree represent the shared haplotypes between the localities. See Table S1 for identification of the haplotypes. Saint Georges l’ Oyapock, French Guiana. Lutzomyia anduzei and Bichromomyia flaviscutellata were used as outgroups.
Bayesian Inference (BI) tree based on COI (Fig. 5) retrieved two major groups from L. umbratilis. The first group, the most basal, clustered all haplotypes from Autazes (BPP: 1.0). The second group consisted of two large subgroups: A) the most apical subgroup consisted three clusters: (A1) all haplotypes from the Cachoeira Porteira, BR-174 Highway, Rio Preto da Eva, Manaus plus H18 (shared), H73 and H74 from Pitinga and the Saint Georges l’Oyapock sequence (BPP: 0.96); (A2) all haplotypes from Porto Grande/Serra do Navio (BPP: 0.99); (A3) haplotypes (H67, H69-H72) from Pitinga, which formed sister cluster with Porto Grande/Serra do Navio (BPP: 1.0); (B) the most basal subgroup consisted two clusters (BPP: 0.93): (B1) all haplotypes from Manacapuru and Novo Airão (BPP: 0.98), and B2) haplotypes (H62-H66, H68, H75, H76) from Pitinga (BPP: 0.99), which formed sister cluster with Manacapuru and Novo Airão populations. In this analysis L. anduzei and B. flaviscutellata were used as outgroups.

Bayesian Inference (BI) tree (maximum clade of credibility) based on the 89 haplotypes of COI and generated in BEAST. Tree inferred under the Time Reversible (GTR) + G + I nucleotide substitution model. The support values, in BPP (Bayesian Posterior Probability), are indicated above of the branches. See Table S1 for identification of the haplotypes. Saint: Saint Georges l' Oyapock, French Guiana. Lutzomyia anduzei and Bichromomyia flaviscutellata were used as outgroups. The species-genetic lineages delimited by the GMYC, bGMYC and ABGD models are represented by colored boxes. The 9, 8 and 6 indicate the lineage numbers recognized by the GMYC, bGMYC and ABGD models, respectively. Each color represents one lineage. The colors of the lineages delimitated by the three models followed the same pattern of the Fig. 2.
The three GMYC, bGMYC and ABGD delimiters employed identified seven, six and four lineages for L. umbratilis, respectively (Fig. 5). The ABGD model was more conservative. In this model, the populations from north group (except Porto Grande/ Serra do Navio and lineage 5 from Pitinga) and Manacapuru and Novo Airão were included in the same lineage. However, the bGMYC model identified a lineages number (six) compatible with others analyses.
Neighbor-Joining (data not shown) and BI trees inferred with the 36 haplotypes from Cytb generated identical topologies. The groups were not well resolved, with polytomies and most branches having weak support. Figure 6 shows the BI tree topology. The most basal and the best resolved group was from Autazes (BPP: 1.0). The remaining haplotypes did not form defined groups. Therefore, it was not possible to establish evolutionary relationships between these groups. The absence of resolution may be due to the few informative sites (2.73%) recorded for this marker. In this analysis only L. anduzei was used as an outgroup due to the absence of Cytb sequences for B. flaviscutellata.

Bayesian Inference (BI) tree generated based on the 36 haplotypes of Cytb. Tree inferred under the HKY nucleotide substitution model. The support values, BPP (Bayesian Posterior Probability), are indicated above of the branches. The colors on the terminal branches of the tree represent the haplotypes observed in each locality, following the same color pattern of the Figs. 1, 2 and 3. The black color on the terminal branches of tree represent the shared haplotypes between the localities. See Table S1 for identification of the haplotypes. Saint Georges l’ Oyapock. French Guiana (square shape in black color). Lutzomyia anduzei was used as outgroup.
The BI tree generated in *BEAST v. 2.055 (Fig. S2) shows the divergence times estimated between groups retrieved. The most recent cladogenesis event occurred between groups Cachoeira Porteira/BR-174 Highway/Rio Preto da Eva/Manaus/Pitinga (some haplotypes)/Porto Grande/Serra do Navio and Manacapuru/Novo Airão and was dated to ~ 180,000–230,000 years ago (Holocene). The node that indicates the oldest event occurred between Autazes and remaining groups and was dated to ~ 400,000–500,000 years ago (middle Pleistocene) (Fig. S2).
PCA analysis clearly separated six genetic groups for the COI (Fig. 7), with the sample from Autazes (Lineage 6) being the most distant one.

Principal Coordinates Analysis (PCA) generated based on the 89 haplotypes of COI. The colors of the lineages followed the same pattern of the Fig. 5.
Table S2 shows the intra-population genetic diversity measures and neutrality tests for COI and Cytb. Remarkably, genetic variation was higher in COI than Cytb for all samples, especially in the samples from Manacapuru and Novo Airão.
For COI, Tajima’s D test was negative, but not significant (P > 0.05), in the samples from Manaus, Pitinga, Autazes, Porto Grande/Serra do Navio (Table S2); therefore, the neutral model cannot be rejected. Fu’s Fs test was negative and significant in samples from Manaus (P < 0.0001) and Porto Grande/Serra do Navio (P < 0.05), consistent with recent population expansion or positive selection. For Cytb, Tajima’s D and Fu’s Fs tests were negatives and significant (P < 0.05) in samples from Cachoeira Porteira, BR-174 Highway, Rio Preto da Eva and Manaus, favoring the interpretation of recent population expansion or selective sweeps. This finding is compatible with the observed star-shaped haplotype network (Fig. 3).
Table S3 shows the pairwise genetic distances values, based on the FST statistic, among nine populations for the COI and Cytb genes. For COI, when the samples from Pitinga, Autazes and Porto Grande/Serra do Navio were integrated into the Scarpassa and Alencar dataset40, most pairwise comparisons had high and significant FST values, regardless whether the compared populations were situated on the same or opposite sides of the rivers and between interfluves. This result is due to the genetic subdivision of the Pitinga sample, and the highly divergent haplotypes of Porto Grande/Serra do Navio. Nonetheless, the highest and most significant FST values were observed in the comparisons involving the Autazes sample, that ranged from 0.7171 (Pitinga versus Autazes) to 0.9333 (Rio Preto da Eva versus Autazes).
For Cytb, the results obtained for the six samples (Cachoeira Porteira, BR-174 Highway, Rio Preto da Eva, Manaus, Manacapuru and Novo Airão) were similar those described for COI by Scarpassa and Alencar40, where low genetic distances were observed between localities situated on the same side of the rivers and within interfluves, whereas high and significant genetic distance values were detected between localities from opposite sides and between interfluves. But, when the Pitinga, Autazes and Porto Grande/Serra do Navio samples were included in the comparisons, the genetic distance values varied depending on the sample. Higher FST values were observed (0.4870) between Manaus versus Pitinga (same side) than between Manacapuru versus Pitinga samples (0.1559) and Novo Airão versus Pitinga (0.1395), situated on opposite sides of the rivers.
Table S3 also provides the average number of nucleotide substitutions per site between samples (Dxy), the number of net nucleotide substitutions per site between samples (Da), the number of shared polymorphisms between pairs of samples (Ss), and the number of fixed differences between pairs of samples (Sf) for the two genes. For COI, similarly FST statistic, the highest Dxy, Da and Sf values were found in the comparisons between Autazes versus Cachoeira Porteira and between Autazes versus Manacapuru (both with Sf = 15), and between Autazes versus Manaus and between Autazes versus Porto Grande/Serra do Navio (both with Sf = 18). For Cytb, the highest values were between Autazes versus Manacapuru and between Autazes versus Novo Airão (both with Sf = 8).
As in the previous analyses, the BAPS analyses identified six genetic groups for COI, and three for Cytb (log ML = −2028.6632 and −564.0626; posterior probability = 0.9999 and 0.9784, respectively) (Figs. S3 and S4).
Genetic distance (uncorrected-p distance) was also calculated between lineages retrieved from the haplotype networks (Figs. 2 and 3). Table 2 shows COI genetic distances. These ranged from 0.7% (between lineages I and III and between lineages II and III) to 2.3% (between lineages IV and VI). Between lineages and L. anduzei and B. flaviscutellata s.s. the mean genetic distances were 5.7% and 11.9%, respectively. Table 3 gives the Cytb genetic distances. These ranged from 0.05% (between lineages I and II) to 1.7% (between lineages II and III). Between the three lineages and L. anduzei genetic distances varied from 4.4% to 4.5%.
A Mantel test was used to analyze the correlation between genetic and geographic distances among samples for each marker. These analyses showed that there was no correlation between genetic and geographic distances for either marker (COI: r = 0.128849, P = 0.2730; Cytb: r = -0.110475, P = 0.4825).
Discussion
Population genetics through access to population structure analyses, demographic history and evolutionary relationships has become an indispensable discipline in evolutionary biology, allowing the discovery of evolutionary lineages and hidden species.
The findings of this study on population structure (Figs. 2, 3; Table S3) revealed a deep and significant genetic split for L. umbratilis from the Brazilian Amazon, confirming the previous biological and molecular studies33,35,38,40,41. On the other hand, phylogenetic analyses with COI identified up to six groups, but only the groups represented by five (H67; H69 to H72) haplotypes from Pitinga (haplogroup V; Fig. 2) and the sample from Autazes were strongly supported in most of the analyses (Figs. 4, 5). For Cytb, the BAPs analysis indicated three genetic groups, whereas in the phylogenetic analyses, the tree topologies showed little resolution preventing to establish the evolutionary relationships among the groups, except for the Autazes population, that as was found in COI, formed a group strongly supported (Fig. 6). Thus, the weak support observed for most groups and the absence of reciprocally monophyletic clades suggest an incomplete lineage sorting caused by retention of ancestral polymorphism due to the recent evolutionary timing of the L. umbratilis diversification process72.
Populations situated north of the Amazon River and east of Negro River (Cachoeira Porteira, BR-174 Highway, Rio Preto da Eva, Manaus, H18, H74 and H75 haplotypes of Pitinga) showed shared haplotypes and low genetic differentiation, with no or few fixed sites, and having most haplotypes clustered within the same haplogroup, as well as in phylogenetic analyses (Figs. 2, 3 and 5; Table S3) reflecting ongoing gene flow or a recent common history for these populations. This outcome indicates a degree of structuring consistent with populations within a single species73, consequently, they represent the same lineage (Lineage I; Fig. 1B). Nonetheless, the sample of Porto Grande/Serra do Navio, also situated north of the Amazon River and east of Negro River, revealed some degree of genetic structure (Figs. 2, 5; Table S3) and it was recognized as a distinct lineage with three models of delimitation used (Fig. 5), as well as in the PCA analysis (Fig. 7). However, the BAPS results for Cytb (Fig. S4) identified this population genetically similar to those of the group northern as well as the results of genetic structure (Table S3) provided no evidence for historic isolation between this and the remaining populations from group north. Thus, it also was included in the Lineage I (Fig. 1b). It is possible that additional analysis of the Porto Grande/Serra do Navio population using a larger sample size, would reduce these differences. Based on the phylogenetic analyses, L. umbratilis from Saint-Georges l’Oyapock48 also belongs to the same lineage, as does the population from the state of Pernambuco41.
Like COI40, the Cytb analyses also found that populations from Manacapuru and Novo Airão, situated in the interfluve of the north Amazon River and west of Negro River, were genetically similar to each one (Figs. 3, 6; Table S3); accordingly, they represent the same lineage or species. On the other hand, well-marked genetic differences, with Nm values < 1, absence of shared sites and presence of fixed sites, were observed between populations from Manacapuru/Novo Airão and those from areas north (except Pitinga) of the Amazon River and east of Negro River (Ss = 0 and Sf = 6–7), and between Manacapuru/Novo Airão and Autazes (interfluves 1 versus 2; Ss = 0 and Sf = 8). These findings reinforce the evidences that the Manacapuru and Novo Airão populations could represent a distinct evolutionary lineage (Lineage II; Fig. 1b), although the genetic differentiation was low (from 0.7% to 2.1%; Table 2) and for the Cytb, they shared haplotypes with the Pitinga population (Fig. 3). The differences in levels of diversity and genetic structure between COI and Cytb genes observed here indicate that they evolve at different rates, with Cytb seeming to evolve more slowly, consequently it shows an older evolutionary history for these populations compared with the highly variable COI gene.
The specimens from Autazes generated COI haplotypes very different (from 13 to 18 fixed sites) from the remaining specimens studied here. This was shown by a disconnected haplotype network (Fig. 2), the highest genetic distance values and an absence of shared sites (Ss) in the most comparisons (Tables 2 and S3), clearly indicating the genetic discontinuity of this population. In the phylogenetic analyses with both markers, haplotypes from Autazes were clustered in a strongly supported basal group, demonstrating an independent evolutionary trajectory for this lineage. This evidence, allied to a large number of fixed sites (Table S3), suggests historic fragmentation between the Autazes population and the other populations analyzed, and indicates that its divergence process may have started earlier. The municipality of Autazes is situated in the interfluve between Amazon and Madeira Rivers (named here interfluve 2). Taken together, the Amazon River may act as significant barrier, preventing the gene flow between Autazes populations and the remaining populations from the opposite banks, so promoting genetic differentiation. Based on this evidence, we suggested this population, first reported in this study, could represent a new lineage within L. umbratilis (Lineage III; Fig. 1b).
The IBD-based hypothesis was rejected for both markers, suggesting that most of the genetic subdivision observed in L. umbratilis populations cannot be explained by geographic distance, without physical barrier. As previously observed40, when sandfly populations from the same side of the rivers and those within interfluves are compared, they exhibited very low genetic differentiation, even when separated by large geographic distances, that may range from ~ 354 to 369 km (Table S3). Otherwise, when comparisons are made between populations from the opposite sides of rivers and between interfluves, very high and significant genetic differentiation were observed, even when these populations are separated by short geographical distances, ranging from ~ 59 to 96 km10. However, the exceptions were observed between Pitinga and the remaining locations from northern Amazon and east of Negro Rivers and between Pitinga and Manacapuru/Novo Airão.
In recent decades, the Amazon River and its tributaries have been the focus of many phylogeographical and biogeographical studies on their potential role in population isolation10,11,74,75,76,77,78,79,80,81,82,83,84,85. Studies have suggested that this complex river system most likely originated in the Miocene, ~ 11 Mya, and took its present shape in the late Pliocene, ~ 2.4 Mya86. Rossetti and others87 proposed that the Amazon River reached its current flow during the late Pleistocene. In this study, the separation time estimated between groups falls from middle Pleistocene to Holocene (Fig. S2), coinciding with the drainage system of Amazon River86,88. In this context, our findings provide support for the proposal that the Amazon and Negro Rivers and their tributaries are the main evolutionary forces driving the diversification process by allopatry in L. umbratilis40. Besides this vicariant event, the low dispersal rate of these sandflies, which seldom move more than 1 km from their birth place6,7, along with the amenable environmental conditions for adaptation and also drift may have contributed to the emergence and marked differentiation of the three main genetic lineages observed in this study.
Curiously, molecular studies conducted with other sandflies from the Brazilian Amazon, such as L. anduzei5, B. flaviscutellata and Bichromomyia nociva89,90, which we presumed to have similar dispersion patterns, did not support this fluvial system acting as physical barriers between populations of these species. Such differences may suggest either lack of a common evolutionary history affecting these species or distinct dispersion rates for these species. Further studies will be required to support or refute these hypotheses.
Two or three species?
Lineage delimitation based on the bGMYC model identified six lineages for L. umbratilis of this study (Fig. 5), a result compatible with the haplotype network, phylogenetic analyses, BAPS and PCA analyses for COI. For both markers, the haplotypes from Pitinga population segregated into different haplogroups in the networks (Figs. 2, 3) and in the phylogenetic analyses (Figs. 4, 5, 6). Two (GMYC and bGMYC) of the three models used recognized three distinct lineages within Pitinga (Fig. 5) indicating admixture or an expressive genetic subdivision for this population. Based on this, could the Pitinga population be a polytypic lineage or species? PCA analysis (Fig. 7) indicated that the Lineage 3 from Pitinga is more closely related to remaining populations from group north (Lineage 1), as well as the genetic distance was low between them (1.3%; Table 2), supporting close relationship. PCA analysis (Fig. 7) also showed that the Lineage 5 from Pitinga is more genetically closely related to Lineage 4 from Manacapuru and Novo Airão (0.7%; Table 2) and for the Cytb, the Pitinga population shared haplotypes with the Manacapuru and Novo Airão populations. It is possible that in recent past the individuals from Pitinga had contact with individuals from Novo Airão and/or neighboring areas, via the Anavilhanas archipelago situated on Negro River (Fig. 1a). Anavilhanas archipelago is second largest group of freshwater islands in the world and comprises an intricate pattern of islands, channels, lagoons, swamps, and partially submerged sandbanks that are refuge for a diversified fauna and flora. This archipelago is located on the lower course of the Negro River, where the widest part of the river may reach approximately 20 km91. Thus, the dispersal of the individuals would have occurred step by step across of these islands, especially in the dry season when the land span of the islands may be greater. Hence, this region from Negro River would be acting (or acted) as a porous barrier to gene flow between populations of the opposite sides in this stretch of River. Further investigations, focusing on a broader sampling and the use of genomic approaches, such as RADseq that provide higher resolution to detect genetic structure and heterozygous individuals, will be needed to clarify these findings recorded at Pitinga.
On the other hand, the Lineage 6 (Autazes) was the most isolated from remaining lineages (Fig. 7); a result congruent with haplotype network, genetic structure and phylogenetic analyses. The population of Autazes is situated to south Amazon River and did not share haplotypes with any other population analyzed. Amazon River is considered the widest in the world and in the rainy season it may reach up ~ 50 km wide. Thus, the genetic isolation observed for Autazes population indicates that, unlike the Negro River, the Amazon River acts as effective barrier to the dispersal between individuals from opposites banks, as consequence the Autazes population was the most divergent.
The values for uncorrected-p distances were low between lineages (COI: from 0.7 to 2.3%; Cytb: from 0.05% to 1.7%), contrasting with very high FST values between samples (Table S3). Studies carried out with other Diptera have also reported low genetic distance values between cryptic species, such as Anopheles albitarsis (1.7–3.5%)92, Anopheles triannulatus (1.7–2.3%)93 and Anopheles nuneztovari (1.6–2.8%)94 complexes, which characterize these species as of recent evolutionary origin. Thus, the differentiation level observed here is indicative of recent divergence between lineages, supporting our conclusions as reported above. This occurs due to the fact that, in young species, fixed differences may be observed only in genes involved in the speciation process95.
Previous studies conducted with L. umbratilis populations north of Amazon/east of Negro Rivers and north of the Amazon/west of Negro Rivers revealed appreciable and significant biological differences between them38. Justiniano and others38 compared the first laboratory bred generations of L. umbratilis populations obtained of the opposite banks of the Negro River (Manaus and Manacapuru). Interestingly, these samples showed remarkable differences in their life cycle, fecundity, fertility and adult longevity, with the population from Manaus being more productive and living longer than the population from Manacapuru. Such differences could be due to intrinsic biological features resulting to distinct evolutionary adaptations due to their geographical isolation by the Negro River. On the other hand, Justiniano39 observed discrete morphological differences in the immature stage, and in the number and size of the spines of armature of the female genital atrium between populations of these locations. The results of this study support the previous biological38 and molecular studies33,35,40,41 and, when taken together, it is possible to infer that the populations north and south of the Amazon River and interfluves represent distinct “species”, which could have diverged more biological and molecular levels than morphologic39 level, with implications distinct epidemiologic13,37. Genomics approach using loci involved in the different biologic aspects of L. umbratilis may clarify this issue. For example, some of these efforts may be focused on the mechanisms that determine the efficiency as vector, as well as to identify specific loci (outliers) that are acting in the most variable adaptability processes of these lineages. In this study, we also recorded a new independently evolving lineage in Autazes. Hence, our findings provide convincing arguments in favor of the hypothesis that what is currently termed L. umbratilis represents, in fact, at least three evolutionary lineages, possibly species, in the studied area.
Overall, the first lineage occurs north of the Amazon and east of Negro Rivers (Lineage I; Fig. 1b), where Le. guyanensis transmission is intense, implying that L. umbratilis is an important vector there. The second lineage occurs in the interfluve between north of the Amazon and west of the Negro Rivers (Lineage II; Fig. 1b), previously reported to be an area free of Le. guyanensis transmission. The third lineage, first recorded by this study, occurs in the interfluve between south of the Amazon and west of the Madeira Rivers (Lineage III; Fig. 1b) and was the most divergent lineage and could be the most ancestral. The involvement of this lineage in Leishmania transmission remains to be elucidated and should be investigated further. The first lineage is most likely the type form (L. umbratilis s.s.), because it occurs in the same bank as the type locality (Monte Dourado area of the Jari River, state of Pará, north of the Amazon and east of the Negro Rivers)31, so that the other two taxa therefore represent unrecognized species.
In conclusion, our findings revealed L. umbratilis three lineages in the Brazilian Amazon. The origin of these lineages is most likely to be associated with the formation of the Amazon drainage system and its tributaries. The first and second lineages are on the north of the Amazon/east of the Negro Rivers and north of the Amazon/west of the Negro Rivers, respectively; the third lineage is known from the interfluve between south of the Amazon River and west of the Madeira River. The three lineages may represent three species. However, their confirmation will require detailed examination morphological of immature and adult stage combined with molecular studies to provide diagnostic characters that can allow them to be recognized as valid species. Given the strong evidence of genetic subdivision within the Pitinga population, we do not rule out the possibility that more lineages or species exist in the Brazilian Amazon, as well as in other locations unsampled in northern South America. These findings will aid epidemiological studies, surveillance and vector control programs in these regions, especially north of the Amazon River, where the transmission by Le. guyanensis can be very high.
Change history
19 August 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41598-021-96387-4
References
Ready, P. D. et al. Phylogenetic species and domesticity of Lutzomyia whitmani at the southeast boundary of Amazonian Brazil. Trans. Royal Soc. Trop. Med. Hyg. 92, 159–160 (1998).
Khalid, N. M. et al. Evidence for genetic differentiation at the microgeographic scale in Phlebotomus papatasi populations from Sudan. Parasit. Vectors 5, 249 (2012).
Péch-May, A. et al. Genetic structure and divergence in populations of Lutzomyia cruciata, a phlebotomine sand fly (Diptera: Psychodidae) vector of Leishmania mexicana in southeastern Mexico. Infect. Genet. Evol. 16, 254–262 (2013).
Kato, H. et al. Genetic divergence in populations of Lutzomyia ayacuchensis, a vector of Andean-type cutaneous leishmaniasis in Ecuador and Peru. Acta Trop. 141, 79–87 (2015).
Scarpassa, V. M., Figueiredo, A. S. & Alencar, R. B. Genetic diversity and population structure in the Leishmania guyanensis vector Lutzomyia anduzei (Diptera, Psychodidae) from the Brazilian Amazon. Infect. Genet. Evol. 31, 312–320. https://doi.org/10.1016/j.meegid.2015.02.007 (2015).
Dye, C. et al. Communication among phlebotomine sandflies: A field study of domesticated Lutzomyia longipalpis populations in Amazonian Brazil. Animal Behav. 42, 183–192 (1991).
Morrison, A. C. et al. Dispersal of the sandfly Lutzomyia longipalpis (Diptera: Psychodidae) at an endemic focus of visceral leishmaniasis in Colombia. J. Med. Entomol. 30, 427–435 (1993).
Carneiro, J. et al. Phylogenetic relationships in the genus Cheracebus (Callicebinae, Pitheciidae). Am. J. Primatol. 82(9), e23167. https://doi.org/10.1002/ajp.23167 (2020).
Garda, A. A. et al. Microhabitat variation explains local-scale distribution of terrestrial Amazonian lizards in Rondônia, western Brazil. Biotropica 45, 245–252. https://doi.org/10.1111/j.1744-7429.2012.00906.x (2013).
Gascon, C. et al. Patterns of genetic population differentiation in four species of Amazonian frogs: A test of the riverine barrier hypothesis. Biotropica 30, 104–119 (1998).
Ribas, C. C. et al. A palaeobiogeographic model for biotic diversification within Amazonia over the past three million years. Proc. R. Soc. B Biol. Sci. 279, 681–689 (2012).
Ferreira, M. et al. Biogeography of the Neotropical genus Malacoptila (Aves: Bucconidae): The influence of the Andean orogeny, Amazonian drainage evolution and palaeoclimate. J. Biogeogr. 44, 748–759 (2017).
Arias, J. R. & Freitas, R. A. Sobre os vectores da leishmaniose cutânea na Amazônia central do Brasil. 2. Incidência de flagelados em flebotomíneos selváticos. Acta Amaz. 8, 387–396 (1978).
Foley, D. H. et al. Geographic distribution, evolution, and disease importance of species within the Neotropical Anopheles albitarsis Group (Diptera, Culicidae). J. Vector Ecol. 39, 168–181 (2014).
Wallace, A. R. On the monkeys of the Amazon. Proc. Zool. Soc. Lond. 20, 107–110 (1852).
Nazareno, A. G. et al. Wide but not impermeable: Testing the riverine barrier hypothesis for an Amazonian plant species. Mol. Ecol. 00, 1–13. https://doi.org/10.1111/mec.14142 (2017).
Leite, R. N. & Rogers, D. S. Revisiting Amazonian phylogeography: Insights into diversification hypotheses and novel perspectives. Org. Divers. Evol. 13, 639–664. https://doi.org/10.1007/s13127-013-0140-8 (2013).
Ready, P. D. et al. A pilot study to control Lutzomyia umbratilis (Diptera: Psychodidae), the major vector of Leishmania braziliensis guyanensis, in a peri-urban rainforest of Manaus, Amazonas state, Brazil. Mem. Inst. Oswaldo Cruz 80, 27–36 (1985).
Lainson, R. The Neotropical Leishmania species: A brief historical review of their discovery, ecology and taxonomy. Rev. Pan-Amaz. Saúde 1, 13–32 (2010).
Lainson, R. et al. Leishmaniasis in Brazil: XIII. Isolation of Leishmania from armadillos (Dasypus novemcinctus), and observation on the epidemiology of cutaneous leishmaniasis in north Pará State. Trans. R. Soc. Trop. Med. Hyg. 73, 239–242 (1979).
Figueira, E. A. G. et al. Phlebotomine sandflies (Diptera: Psychodidae) from Lábrea, state of Amazonas, Brazil, with a description of Evandromyia (Aldamyia) apurinan Shimabukuro, Figueira & Silva, sp. nov.. Mem. Inst. Oswaldo Cruz 108, 280–287. https://doi.org/10.1590/S0074-02762013000300004 (2013).
Lainson, R. et al. Cutaneous leishmaniasis in North Brazil: Lutzomyia anduzei as a major vector. Trans. R. Soc. Trop. Med. Hyg. 70, 171–172 (1976).
Lainson, R. et al. Leishmaniasis in Brazil: XVI. Isolation and identification of Leishmania species from sandflies, wild mammals and man in north Pará State, with particular reference to L. brasiliensis guyanensis causative agent of “pian-bois”. Trans. R. Soc. Trop. Med. Hyg. 75, 530–536 (1981).
Young, D. G. & Duncan, N. A. Guide to the identification and geographic distribution of Lutzomyia sandflyies in Mexico, the West Indies, Central and South America (Diptera: Psychodidade). Mem. Am. Entomol. Inst. 54, 1–881 (1994).
Azevedo, A. C. R. et al. Comparative studies of populations of Lutzomyia umbratilis (Diptera: Psychodidae) in Brazil and Venezuela. J. Med. Entomol. 39, 587–600 (2002).
Azevedo, A. C. R. et al. Studies on the sandfly fauna (Diptera: Psychodidae: Phlebotominae) from transmission areas of American Cutaneous Leishmaniasis in state of Acre, Brazil. Mem. Inst. Oswaldo Cruz 103, 760–767 (2008).
Gil, L. H. S. et al. Species structure of sand fly (Diptera: Psychodidae) fauna in the Brazilian western Amazon. Mem. Inst. Oswaldo Cruz 104, 955–959 (2009).
Forattini, O. P. Notas sobre Phlebotomus do Território do Amapá, Brasil (Diptera, Psychodidae). Studia Entomol. 11, 467–480 (1960).
Balbino, V. Q. et al. First report of Lutzomyia (Nyssomyia) umbratilis Ward & Frahia, 1977 outside of Amazonian region, in Recife, State of Pernambuco, Brazil (Diptera: Psychodidae: Phlebotominae). Mem. Inst. Oswaldo Cruz 96, 315–317 (2001).
Freitas, M. T. S. et al. New records of phlebotomine sandflies (Diptera: Psychodidae) from the state of Alagoas, northeast of Brazil. J. Med. Entomol. 20, 1–6. https://doi.org/10.1093/jme/tjx175 (2017).
Ward, R. D. & Fraiha, H. Lutzomyia umbratilis, a new species of sandfly from Brazil (Diptera: Psychodidae). J. Med. Entomol. 14, 313–317 (1977).
Almeida, F. B. Flebótomos da Amazônia. 1—Sobre a presença de Lutzomyia anduzei (Rozeboom, 1942) no Brasil (Diptera: Psychodidae). Boletim do Instituto Nacional de Pesquisas da Amazônia: Série Patologia Tropical 3, 1–16 (1970).
Scarpassa, V. M. & Alencar, R. B. Molecular taxonomy of the two Leishmania vectors Lutzomyia umbratilis and Lutzomyia anduzei (Diptera: Psychodidae) from the Brazilian Amazon. Parasit. Vectors 6, 258 (2013).
Ready, P. D. et al. The ecology of Lutzomyia umbratilis Ward & Fraiha (Diptera: Psychodidae) the major vector to man of Leishmania braziliensis guyanensis in north-eastern Amazonian Brazil. Bull. Entomol. Resour. 76, 21–40 (1986).
Scarpassa, V. M.; Alencar, R. B. Speciation in the Leishmania guyanensis Vector Lutzomyia umbratilis (Diptera: Psychodidae) from Northern Brazil—Implications for Epidemiology and Vector Control. An Overview of Tropical Diseases, Dr. Amidou Samie (Ed.), InTech. https://doi.org/10.5772/60921 (2015).
Arias, J. R. & Freitas, R. A. On the vectors of cutaneous leishmaniasis in the central Amazon of Brazil I. Preliminary findings. Acta Amaz. 7, 293–294 (1977).
Soares, R. P. et al. Lutzomyia umbratilis from an area South of the Negro river is refractory to in vitro interaction with Leishmania guayanensis. Mem. Inst. Oswaldo Cruz 113, 202–205 (2018).
Justiniano, S. C. B. et al. Comparative biology of two populations of Lutzomyia umbratilis (Diptera: Psychodidae) of central Amazonia, Brazil, under laboratory conditions. Braz. J. Biol. 64, 227–235 (2004).
Justiniano, S. C. B. Biologia comparada de populações de Lutzomyia umbratilis (Diptera: Psychodidae) da Amazônia Central Brasileira. Doctoral Thesis. 151pp. Instituto Nacional de Pesquisas da Amazônia, Manaus, AM. (2004).
Scarpassa, V. M. & Alencar, R. B. Lutzomyia umbratilis, the main vector of Leishmania guyanensis, represents a novel species complex?. PLoS ONE 7, e37341 (2012).
Freitas, M. T. S. et al. Phenotypic and genotypic variations among three allopatric populations of Lutzomyia umbratilis, main vector of Leishmania guyanensis. Parasit. Vectors 8, 448. https://doi.org/10.1186/s13071-015-1051-7 (2015).
Sudia, W. D. & Chamberlain, R. W. Battery operated light trap, an improved model. Mosq. News 22, 126–129 (1962).
Sambrook, J. F. Russell, D. W. Molecular Cloning: A laboratory manual, 3rd edn. Vol. 1–3, p. 2100 (Cold Spring Harbor Laboratory Press, New York, UK, 2001).
Zhang, D. X. & Hewitt, G. M. Assessment of the universality and utility of a set of conserved mitochondrial COI primers in insects. Insect Mol. Biol. 6, 143–150 (1996).
Coutinho-Abreu, I. V. et al. Lutzomyia longipalpis sl in Brazil and the impact of the São Francisco River in the speciation of this sand fly vector. Parasit. Vectors 1, 16 (2008).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 41, 95–98 (1999).
Chromas Lite®Software; Technelysium Pty. Ltd, Tewantin, Queensland, Australia. http://www.technelysium.com.au. 1998.
Kocher, A. et al. Complete mitochondrial genome of Lutzomyia (Nyssomyia) umbratilis (Diptera: Psychodidae), the main vector of Leishmania guyanensis. Mitochondrial DNA https://doi.org/10.3109/19401736.2015.1022748 (2015).
Altschul, S. F. et al. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Xia, X. & Xie, Z. DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 92, 371–373 (2001).
Librado, P. & Rozas, J. DnaSP vol 5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. https://doi.org/10.1093/bioinformatics/btp187 (2009).
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1660 (2000).
Tamura, K. et al. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. https://doi.org/10.1093/molbev/mst197 (2013).
Ronquist, F. et al. Mr. Bayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. https://doi.org/10.1093/sysbio/sys029 (2012).
Bouckaert, R. et al. BEAST2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10, E1003537 (2014).
Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. https://doi.org/10.1007/BF01731581 (1980).
Darriba, D. et al. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 9, 772. https://doi.org/10.1038/nmeth.2109 (2012).
Fujisawa, T. & Barraclough, T. G. Delimiting species using single-locus data and the generalized mixed yule coalescent approach: A revised method and evaluation on simulated data sets. Syst. Biol. 62, 707–724 (2013).
Reid, N. M. & Carstens, B. C. Phylogenetic estimation error can decrease the accuracy of species delimitation: A Bayesian implementation of the general mixed Yule-coalescent model. BMC Evol. Biol. 12, 196. https://doi.org/10.1186/1471-2148-12-196 (2012).
Puillandre, N. et al. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877. https://doi.org/10.1111/j.1365-294X.2011.05239.x (2012).
Ezard, T. et al. SPLITS: Species’ Limits by threshold statistics. R package version 1.0–18/r45. http://R-Forge.R-project.org/projects/splits/ (2009).
Drummond, A. J. et al. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. https://doi.org/10.1093/molbev/mss075 (2012).
Brower, A. V. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc. Natl. Acad. Sci. 91, 6491–6495. https://doi.org/10.1073/pnas.91.14.6491 (1994).
Rambaut, A. FigTree. Tree figure drawing tool version 1.3.1. Institute of Evolutionary biology, University of Edinburgh. http://tree.bio.ed.ac.uk/software/figtree/ (2006).
Brown, S. D. J. et al. Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol. Ecol. Resour. 12, 562–565 (2012).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphisms. Genetics 123, 585–595 (1989).
Fu, Y. X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925 (1997).
Excoffier, L.; Laval, G.; Schneider, S. An integrated software package for population genetics data analysis, Version 3.01. Computational and Molecular Population Genetics Lab. Institute of Zoology, University of Berne, Switzerland. http://www.cmpg.unibe.ch/software/arlequin3 (2006).
Corander, J. & Tang, J. Bayesian analysis of population structure based on linked molecular information. Math. Biosci. 205, 19–31. https://doi.org/10.1016/j.mbs.2006.09.015 (2007).
Corander, J. et al. Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinform. 9, 539. https://doi.org/10.1186/1471-2105-9-539 (2008).
Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220 (1967).
Krzywinski, J. & Besansky, N. J. Molecular systematics of Anopheles: From subgenera to subpopulations. Annu. Rev. Entomol. 48, 111–139. https://doi.org/10.1146/annurev.ento.48.091801.112647 (2003).
Wright, S. Evolution and the genetics of populations, vol. 4, variability within and among natural populations. University of Chicago Press, Chicago, IL. (1978).
Salo, J. et al. River dynamics and the diversity of Amazon lowland forest. Nature 322, 254–258 (1986).
Ayres, J. M. & Clutton-Brock, T. H. River boundaries and species range size in Amazonian primates. Am. Nat. 140, 531–537 (1992).
Lougheed, S. C. et al. Ridges and rivers: A test of competing hypotheses of Amazonian diversification using a dart-poison frog (Epipedobates femoralis). Proc. R. Soc. Lond. Ser. B Biol. Sci. 266, 1829–1835 (1999).
Patton, J. L. et al. Mammals of the Rio Jurua and the evolutionary and ecological diversification of Amazonia. Bull. Am. Mus. Nat. Hist. 244, 1–306 (2000).
Hall, J. P. W. & Harvey, D. J. The phylogeography of Amazonia revisited: New evidence from riodinid butterflies. Evolution 56, 1489–1497 (2002).
Bates, J. M. et al. Avian mitochondrial DNA sequence divergence across a headwater stream of the Rio Tapajos, a major Amazonian river. J. Ornithology 145, 199–205 (2004).
Boubli, J. P. et al. Spatial and temporal patterns of diversification on the Amazon: A test of the riverine hypothesis for all diurnal primates of Rio Negro and Rio Branco in Brazil. Mol. Phylog. Evol. 82, 400–412 (2015).
Collevatti, R. G. et al. Contrasting patterns of genetic structure in Caryocar (Caryocaraceae) congeners from flooded and upland Amazonian forests. Biol. J. Linnean Soc. 98, 278–290 (2009).
Maldonato-Coelho, M. et al. Rivers, refuges and population divergence of fire-eye antbirds (Pyriglena) in the Amazon Basin. J. Evol. Biol. 26, 1090–1107 (2013).
Fernandes, A. M. et al. Phylogeography of the chestnut-tailed antbird (Myrmeciza hemimelaena) clarifies the role of rivers in Amazonian biogeography. J. Biogeog. 39, 1524–1535 (2012).
Smith, B. T. et al. The drivers of tropical speciation. Nature 515, 406–409 (2014).
Moraes, L. J. C. L. et al. The combined influence of riverine barriers and flooding gradients on biogeographical patterns for amphibians and squamates in south-eastern Amazonia. J. Biogeog. 43, 2113–2124 (2016).
Baker, P. A. et al. The emerging field of geogenomics: Constraining geological problems with genetic data. Earth Sci. Rev. 135, 38–47 (2014).
Rossetti, D. F. et al. New geological framework for western Amazonia (Brazil) and implications for biogeography and evolution. Quatern. Res. 63, 78–89 (2005).
Figueiredo, J. et al. Late Miocene onset of the Amazon river and the Amazon deep-sea fan: Evidence from the Foz do Amazonas Basin. Geology 37, 619–622 (2009).
Melo, L. B., Alencar, R. B. & Scarpassa, V. M. Molecular taxonomy and phylogenetic inferences of Bichromomyia flaviscutellata complex based on the COI gene DNA barcode region. Infect. Genet. Evol. 81, 104256. https://doi.org/10.1016/j.meegid.2020.104256 (2020).
Melo, L. B. Taxonomia molecular do complexo Bichromomyia flaviscutellata e estrutura genética de Bichromomyia flaviscutellata sensu stricto (Diptera: Psychodidae) principal vetor da Leishmania amazonensis (Kinetoplastea: Trypanosomastidae). Master Thesis 59 pp., Instituto Nacional de Pesquisas da Amazônia, Manaus, AM (2019).
Almeida-Filho, R., Shimabukuro, Y. E. & Beisl, C. H. Assessment of landscape changes in the Anavilhanas archipelago during the flood peak and drought events in the Rio Negro, central Amazônia, Brazil. Rev. Brasil. Cartografia 68, 957–964 (2016).
Lehr, M. A. et al. Cryptic species in the Anopheles (Nyssorhynchus) albitarsis (Diptera: Culicidae) complex: Incongruence between random amplified polymorphic DNA-polymerase chain reaction identification and analysis of mitochondrial DNA COI gene sequences. Ann. Entomol. Soc. Am. 98, 908–917 (2005).
Moreno, M. et al. Phylogeography of the neotropical Anopheles triannulatus complex (Diptera: Culicidae) supports deep structure and complex patterns. Parasit. Vectors 6, 47. https://doi.org/10.1186/1756-3305-6-47 (2013).
Scarpassa, V. M., Cunha-Machado, S. & Saraiva, J. F. Evidence of new species for malaria vector Anopheles nuneztovari sensu lato in the Brazilian Amazon region. Malaria J. 15, 205. https://doi.org/10.1186/s12936-016-1217-6.ISBN:1475-2875 (2016).
Moreno, M. et al. Complete mtDNA genome of Anopheles darlingi and an approach to anopheline divergence time. Malaria J. 9, 127. https://doi.org/10.1186/1475-2875-9-127.
Funding
This study was funded by National Council of Scientific and Technological Development (CNPq) grants, process numbers 409058/2006-8 to Dr. Toby Vincent Barrett and 575482/2008-7 to V. M. Scarpassa, by the Foundation of Research Support of the State of Amazonas (FAPEAM), process number 1501/2008 to V. M. Scarpassa, by the INCT/CENBAM/CNPq/FAPEAM/INPA, process number 722069/2009, granted to Dr. William Ernest Magnusson and by the PROEQUIPAMENT-CAPES, Brazil. The authors also thank Dr. Raimundo Nonato Picanço Souto (Universidade Federal do Amapá) by logistic support during the collections in Porto Grande/Serra do Navio, in Amapá and to MSc. João Rafael Oliveira (Entomology at INPA) who kindly prepared the figures of this article. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
V.M.S.: Designed the experiment. R.B.A.: Collected and identified the specimens. V.M.S.: Generated the data. VMS: Processed the statistical analyzes. A.S.C.M.: Contributed with phylogenetic analyses. R.B.A.: Prepared the figures. V.M.S.: Wrote the manuscript. V.M.S. and R.B.A.: Revised the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained errors. Modifications have been made to the Abstract and Figure 5. Full information regarding the corrections made can be found in the correction for this Article.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scarpassa, V.M., Cunha-Machado, A.S. & Alencar, R.B. Multiple evolutionary lineages for the main vector of Leishmania guyanensis, Lutzomyia umbratilis (Diptera: Psychodidae), in the Brazilian Amazon. Sci Rep 11, 15323 (2021). https://doi.org/10.1038/s41598-021-93072-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93072-4
This article is cited by
-
Trichophoromyia auraensis: evidence for cryptic species and first record in the state of Maranhão, Brazil
Parasitology Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
