
Abstract
A symbiosis of bacterial community (sometimes called microbiota) play essential roles in developmental life cycle and health of coral, starting since a larva. For examples, coral bacterial holobionts function nitrogen fixation, carbon supply, sulfur cycling and antibiotic production. Yet, a study of the dynamic of bacteria associated coral larvae development is complicated owning to a vast diversity and culturable difficulty of bacteria; hence this type of study remains unexplored for Acropora humilis larvae in Thai sea. This study represented the first to utilize 16S rRNA gene sequencing to describe the timely bacterial compositions during successfully cultured and reared A. humilis larval transformation in aquaculture (gametes were collected from Sattahip Bay, Chonburi province, Thailand), from gamete spawning (0 h) and fertilization stage (1 h), to embryonic cleavage (8 h), round cell development (28, 39 and 41 h), and planula formation (48 h). The sequencing results as estimated by Good’s coverage at genus level covered 99.65 ± 0.24% of total bacteria. While core phyla of bacteria were observed (Proteobacteria, Actinobacteria, Firmicutes and Bacteroidetes), changes in bacterial population structures and differential predominant core bacterial orders were denoted for each larval developmental stage, from fertilization to embryonic cleavage and subsequently from the embryonic cleavage to round cell development (P = 0.007). For instances, Pseudoalteromonas and Oceanospirillales were found prevalent at 8 h, and Rhizobiales were at 48 h. The bacterial population structures from the round cell stage, particularly at 41 h, showed gradual drift towards those of the planula formation stage, suggesting microbial selection. Overall, this study provides preliminary insights into the dynamics of bacterial community and their potentially functional association (estimated from the bacterial compositions) during the developmental embryonic A. humilis in a cultivation system in Southeast Asia region.
Similar content being viewed by others
Introduction
In the past decades, stresses from climate warming and anthropogenic activities have dramatically affected worldwide coral reef health, including Thailand and Southeast Asia region1,2. Some coral genera, such as Acroporidae, Faviidae, Pocilloporidae and Poritidae, have significantly declined3,4,5. This decline affects ecologic and economic benefits of coral reefs, including damaged and declined marine life habitat and food sources, and disrupted coastal protection6,7,8. Additionally, many new bioactive compounds in human medicine have extracted from coral reef habitat9,10,11. Subsequently, scientists are interested in finding sustainable management for restoring and conserving coral reefs12,13,14.
One of the most recent focus to assist in coral reef restoration and conservation is through the coral-associated microbiome. Microorganisms, mainly bacteria, were reported to colonize various parts of coral tissues (mucus layer, gastrovascular cavity and skeleton) and function in coral growth, health and promoting the higher resistance to stresses that include climate warming15,16. For instances, many coral holobiont bacteria (e.g. Vibrionales, Cyanobacteria, Alteromonas, Rhizobales, Oleibacter and Pseudoalteromonas) recycle nutrients by nitrogen fixation, sulfur cycling and photosynthesis17,18,19, or synthesize bioactive compounds, such as tropodithietic acid (TDA), against pathogenic bacteria20,21,22. Subsequently, abundances of Oceanospirillales and Rhizobiales were reported to be associated with healthy corals of various species23,24,25,26,27. Yet, scientists found that these coral bacteria are dynamics through the coral developmental stages and external factors28,29,30. Damjanovic et al.31 reported the presence of bacteria since Acropora tenuis egg and sperm bundles, and the early coral life stages (0–96 h) were associated with a dynamic and diverse bacterial community; the initial bacteria on gametes could be from a vertical transfer of certain bacteria in the mucus layer surrounding the gametes and a release of bacteria by spawning adults into the water column (a horizontal transfer). Miller et al.32 found that bacterial communities of nursery-reared Acropora cervicornis were relatively consistent among spatial colonies of the same coral host genotype and varied by coral host genotypes. Still, the knowledge about the bacterial population structures during larval development of Acropora humilis in the Southeast Asia has not been deciphered.
The cultivation of corals using sexual reproduction technique is relatively new in the past decade with different levels of success depending on sites33,34,35,36,37, and we are one of the pioneer groups who have actually been successfully cultivating corals using gametes and have successfully been using our cultured baby corals to restore degraded reefs in Thailand. Here, A. humilis can be a model coral species since this species is a predominant species not only in Thailand but also in the tropical regions, particular in the Southeast Asia. However, not many studies had done on this coral species because the hardness of gamete and larval sample collection, and the limitation of the success in coral cultivation from gamete fertilization. At present, it is impossible to collect those wild fertilized coral gametes in the field, and thus, cultured corals in a hatchery represented an excellent alternative choice to study the biology of A. humilis species in larval stages. In addition, even the same genus or species of corals exist in different marine areas, dynamics of the bacterial populations are usually not the same due to different marine biogeography factors, which affect a quality of water, degree and period of sunlight, salinity, oxygen, pH, sedimentation, nutrients, pollutions, etc.16,30,38. In this study, we reported truly the dynamics of the bacterial population structures (and their functional potentials) during the developing A. humilis larvae in aquaculture using metagenomic-derived 16S rRNA gene sequencing. This important knowledge will increase the understanding of the relationship between the bacterial communities and larval development for further apply in coral sexual reproduction cultivation technique.
Material and methods
Sample collections and coral larvae aquaculture
In February 2018, the gametes of five spawning coral colonies of A. humilis were collected from Sattahip Bay, Chonburi Province, Thailand. A. humilis is a hermaphroditic species, which spawns once a year. At Sattahip Bay, the spawning times are usually between 7:30–9:00 pm. For fertilization, collecting gametes from five different coral colonies have been proved to be sufficient for gamete fertilization succession rate at 98–99%. Fertilization rate is determined by random collecting of approximately 100 individuals of eggs after 24 h of encountering with sperms, and counting on fertilized eggs using a dissecting microscope. The average fertilization rates are based on at least 3 replicates. To collect the gametes, the scuba diving technique was used; the divers covered a plankton net in each coral colony before the coral spawning approximately 30 min. Then, after collection, the gametes were immediately transferred to a hatchery facility, mixed, and artificially fertilized. Once gametes were fertilized, they were transferred to 30 × 60 × 30 cm3 tanks filled with 54 L of 0.4 µm filtered seawater at ambient temperature (28 °C), ambient salinity (32 psu), and day-night cycle via natural light intensity of 21 µmol m-2 s-1 for 12 h a day. The density of larvae was < 2000 larvae/L of seawater. The filtered seawater was exchange daily to prevent contaminated growth of other organisms. The fertilization and coral raising techniques were followed Kuanui et al.39,40. Those gametes were fertilized and developed in filtered seawater in the enclosed system. Thus, the variation and the influence of microorganisms from the sea environment should be limited. To exchange the seawater, one-third of the seawater volume were replaced each time to ensure that the fertilized gametes were not disturbed. At specific time points including gamete spawning (0 h) and fertilization stage (1 h), to embryonic cleavage (8 h), round cell development (28 h, 39 h and 41 h), and planula formation (48 h), three replicates of fertilized gamete samples were collected. At 0 h meant the stage of unfertilized eggs prior to be mixed with other colonies for fertilization.
Metagenomic extraction
Protocols for DNA extraction and 16S rRNA gene sequencing were followed https://earthmicrobiome.org/protocols-and-standards/dna-extraction-protocol/41,42. Metagenomic DNA were extracted from 1 g of each ground (consisting of endoderm and ectoderm) larvae sample using Power Soil DNA Isolation Kit (Qiagen Inc., Hilden, Germany). Protocols were as described in the manufacturer’s instructions. The extracted metagenomes were checked for DNA quality and concentration by agarose gel electrophoresis (1% agarose gel) and nanodrop spectrophotometry (A260/A280 and A260)42. Noted that some DNA samples were found degraded and could not successfully amplified for library construction at the next step. For preservation, the extracted DNA were stored at -20 °C.
16S rRNA gene library construction and next generation sequencing
The extracted DNA were used as templates for a preparation of prokaryotic 16S rRNA gene V3–V4 libraries. Amplification was performed using a universal forward primer 515F (5’-GTGCCAGCMGCCGCGGTAA-3’) and a universal reverse primer 806R (5’-GGACTACHVGGGTWTCTAAT-3’), with appended Illumina adapter and Golay barcode sequences42,43. Each biological replicate (n = 3 per sampling time point) was amplified in triplicate PCR reactions to prevent a PCR bias. Each PCR (25 µL) comprised 1 × EmeraldAmp GT PCR Master Mix (TaKaRa, Shiga, Japan), 0.3 µM of each primer, and 50 ng metagenomic DNA template. The amplification parameters included an initial heat activation at 94 °C for 3 min, and 25 cycles of denaturation at 94 °C for 45 s, annealing at 50 °C for 60 s, and extension at 72 °C for 1.5 min, followed by a final extension at 72 °C for 10 min. The PCR products of expected 381 bp in size were purified using Gel Extraction Kit (Bio-Helix Co. Ltd., Keelung, Taiwan). Triplicate PCR products were combined equally and quantified using Qubit 3.0 Fluorometer and Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific Inc., Massachesetts, USA). 150 ng of each sample was sequenced along sequencing primer and index sequence43, on a Miseq platform with 2 × 150 bp paired-end reads (Illumina, California, United States) housed at Omics Science and Bioinformatics Center, Faculty of Science, Chulalongkorn University (Bangkok, Thailand).
Bioinformatic and statistical analyses
Raw sequences (reads) were quality screened following Mothur’s standard operating procedures (SOP) for MiSeq, which included removal of primer sequences, short read length (< 100 bp), read with ambiguous bases, and chimera sequences44,45. The quality reads were then aligned against GreenGenes 13.8 and SILVA 1.32 databases to remove mitochondria, chloroplast, unknown and eukaryote sequences, and clustered into operational taxonomic units (OTUs) at phylum, class, order, family and genus levels. Nucleic acid sequences in this study were deposited to an open access Sequence Read Archive database of NCBI, accession number SRP275543.
Mothur was used to compute Good’s coverage (an estimate of sequencing depth to the true diversity in a sample) and alpha diversity (Chao richness, Shannon diversity and inverse Simpson indices)44,45. To obtain an equal sequencing depth across samples, the lowest quality read number (7,184 reads) per sample was used for data rarefication and continuing data analyses. Noted that samples with unsuccessful metagenomic extraction or insufficient sequencing depth (< 99% sequencing coverage) were removed from further analyses to prevent bias in representing the true community composition46,47. Mothur was also used to compute beta diversity (thetayc dissimilarity index), a phylogenetic tree (neighbour joining tree), and functional potentials estimated from bacterial community profiles via PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States). The PICRUSt categorizes functions into KEGG (Kyoto Encyclopedia of genes and genomes) pathways48. For statistical analysis, Wilcoxon signed rank test and analysis of molecular variance (AMOVA) were used to test for significant difference between groups (P < 0.05). Data visualization and statistical analyses were conducted using RStudio 1.3.959 (http://www.rstudio.com/)49.
Results
16S rRNA gene sequencing
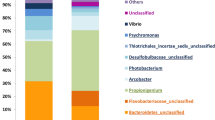
The libraries of our 16S rRNA gene sequences correspoding to samples named 0h, 1h, 8h1, 8h2, 8h3, 28h, 39h1, 39h2, 41h, 48h1, 48h2 and 48h3 passed quality screening and OTU classifications. An average number of quality reads were 16,926 reads per sample. At genus level OTUs, these numbers of quality reads passed > 99% Good’s coverage (Supplemental Table 1). Overall, four phyla were observed (Proteobacteria averagely 57.90%, Actinobacteria 14.23%, Firmicutes 10.11% and Bacteroidetes 8.08%), and each stage of A. humilis larval development carried different bacterial diversity. For alpha diversity, larvae samples at 8 h showed noticably low bacterial diversity compared to the other time points (P = 0.009). Figure 1A showed the consistently least alpha diversity among 8h1, 8h2 and 8h3 (Supplemental Fig. 1A showed an average bacterial composition of the 8 h samples). The 8 h bacteria were dominated by phylum Proteobacteria (89.95%), specifically in order Vibrionales (36.38%) followed by Alteromonadales (18.61%) and an unclassified order in class Gammaproteobacteria (17.76%), in orderly (Supplemental Fig. 1A). Interestingly, these species of genera Pseudoalteromonas and Oleibacter belonged an order Oceanospirillales (Supplemental Fig. 2A). Raw numeric percentages of bacterial genus compositions of all samples were available in Supplemental Table 2.

Relative abundance of bacterial OTU compositions during A. humilis larval development in aquaculture at (A) phylum and (B) order levels. Bacterial phyla and orders with < 1% abundance were represented in “Other (< 1%)”. In (A), pictures corresponding to 1 h and 48 h A. humilis larvae were included.

Relative abundance of bacterial OTU compositions at genus level. For OTU classification where genus could not be identified, the deepest classification was given (abbreviated g_ for genus, f for family, c for class and o for order, respectively). Genus names were color highlighted based on phylum, and in each phylum bacterial genera of < 1% abundance were represented in “p_name_Others (< 1%)”.
Comparative bacterial community profiles during larval development
Figure 1A displayed interesting pattern in the bacterial phyla including the relatively even distributions of four core phyla during 0 and 1 h larvae, the drastic increase of Proteobacteria but almost diminished other phyla in samples 8h1 to 8h3, and the reversion of bacterial population structure in 28 h to be resembling that of 1 h (e.g. increase of Firmicutes and Bacteroidetes) before continuiation of bacterial dynamics such as a transient increase of Actinobacteria at 39h1 and 39h2, and an increasing of Proteobacteria at 48h1 to 48h3. Noted a test of statistical significance between each pairwise time points was impossible because of insufficient sample size. Beta diversity analyses (dissimilarity of bacterial community structures among samples) indicated relatively closer bacterial population structures between stages of gamete spawning and fertilization, and round cell development and planula (P = 0.055). For the latter, increasing relative abundance of genera Magnetospirillum and unclassified Rhizobiales that belong order Rhizobiales were denoted (Supplemental Fig. 2B, averagely 1.98% for 48 h vs. 0.33% for the rests).
Further diversity analysis at order level in Fig. 1B revealed the orders that correlated with each coral developmental stages. This included the drastic change at 8 h (embryonic cleavage), mainly comprising Vibrionales (36.38%), followed by Alteromonadales (18.61%) and an unclassified order in class Gammaproteobacteria (17.76%). During round cell development, order Actinomycetales, of family Micrococcaceae and genus Kocuria, were prevalent (Figs. 1B and 2). Following at 41 h, these bacteria members were replaced by members of Proteobacteria, such as Novosphingobium and Enhydrobacter (Fig. 2 and Supplemental Fig. 1B). Furthermore, analysing at order and genus levels also revealed differences in relative abundances of bacterial members in phylum Proteobacteria in 8 h vs. 48 h. For instances, genera Novosphingobium, Thalassobius, Enhydrobacter and an unclassified in family Rhodobacteraceae were significantly higher in 48 h (Fig. 2). This difference along the presence of more diversified Bacteroidetes and Firmicutes, in 48 h larval bacterial population structures made the 48h1 to 48h3 cluster closer to the rest of the bacterial population structures than the 8h1 to 8h3 bacterial population structures (Fig. 3A).

Analyses of bacterial community profiles at genus level OTUs into (A) phylogenetic tree and (B) microbial functional potentials. In (B), * denotes a functional category that had a statistical difference in relative abundance across samples.
Moreover, abundances of Oceanospirillales and Rhizobiales were compared. The greater relative abundance of Oceanospirillales in 8 h and Rhizobiales in 48 h, respectively, supported healthy status of our aquacultured corals and suggested their functions be necessary for the coral activities during these points (Supplemental Fig. 2)23,24,25,26,27. Note a somewhat diversity among the 28–41 h bacterial communities in part supported the detail differences in larval morphological development within this time period, such as round vs. tear drop stages.
Functional potentials of bacterial communities
The bacterial communities of our cultured A. humilis larval development demonstrated core microbial functional potentials in membrane transport, carbohydrate and amino acid metabolisms, replication and repair, energy metabolism, cellular processes and signaling, cell motility, and lipid and nucleotide metabolisms, in orderly. In detail differences, functions involved membrane transport, genetic replication and repair, and carbohydrate and amino acid metabolisms were predominated in coral gamete spawning and fertilization; cell motility, cellular processes, and cellular signaling in embryonic cleavage; membrane transport, genetic replication and repair, and carbohydrate and amino acid metabolisms in round cell development; and xenobiotic biodegradation and metabolism in planula (Fig. 3B). Examples of correlation between bacterial compositions and the metabolic potentials are from 39 to 48 h larvae that the increased relative abundance of Rhodobacteraceae and Novosphingobium, which perform role as nitrogen recycler, and nitrogen is essential for coral growth from a transformation of planula into fully developed polyps50,51; thereby relatively high amino acid metabolism were highlighted.
Discussion
Presence of bacteria had been reported in brooded coral larvae52. But, how a marine biogeography factor (i.e. Thai sea) plays role on coral gamete development has never been described and yet neither for the prevalent A. humilis species in the Thai sea, and this study is the first to identify the bacterial compositions associated with the early coral development stages from gamete fertilization to the formation of planula in aquacultured A. humilis. Little information were available on bacteria associated with corals particular at the young stages and larval stages, and none for the A. humilis gamete, due to the limitation and availability of sample collection, sexual reproduction and culture techniques, and advanced sequencing technique. Scientists reported that periods of day and night lights, tides and temperature have effects on A. humilis spawning; hence successful culturing of Thai sea A. humilis since gametes likely involves particular microbial association, and how the developing corals select certain bacteria from the surroundings (Thai sea reef ecosystem, in this case) remain unknown53,54,55.
Our findings of four core phyla through cultured A. humilis larval development were similar to those reported in Acropora digitifera at 48 h and 4 day larval stages, and that Bernasconi et al.30 also reported the relatively higher abundance of Bacteroidetes than Firmicutes. Nonetheless, this latter report was not found consistent in our results. This finding highlights the differences by different coral species, different hour stages and that the surrounding Thai sea could also provide a differently bacterial population involvement16,56,57, so the bacterial associations found in our study were not completely same to the previous studies. Our findings also highlight the essential to study the specific coral species at specific marine site to help understand the bacterial involvement and bacterial manipulation in enhancing A. humilis larval growth in the Thai sea. In 0 and 1 h bacteria, relative abundances of unclassified Alphaproteobacteria, Gammaproteobacteria and order Actinomycetales were consistent with previous findings29.
The relatively lowest alpha diversity at 8 h might support the dynamic point of coral bacteria from fertilization to cell rounding stages following Zhou et al.29 who reported an increasing bacterial diversity during larvae to juvenile. In addition, the 8 h bacterial population structures were uniquely distinct from the other samples (Figs. 1 and 2), providing bacteria to support functions in cell motility and cellular processes and signaling (Fig. 3B).
During round cell development, prevalent species in family Micrococcaceae and genus Kocuria that we observed had been described58 and might support the observed larval bacterial biofilm and function in quarum sensing to induce specific bacterial and/or substrate attachment essential for larval development. In many marine invertebrates whose the development of larvae involves a settlement on specific substrates to initiate metamorphosis to juvenile form, such as Hydractinia symbiolongicarpus (a cnidaria hydroid that grows on a gastropod shell), the bacterial isolates Microbacterium and Kocuria could synthesize and secrete quorum sensing signaling molecule, for example in their bacterial biofilms on coral larvae. This signal molecule could participate in the coral substrate selection by inducing larval chemotaxis and substrate attachment58. Further, species in Micrococcaceae, such as Micrococcus MCCB104, had been described to produce antimicrobial compounds against pathogenic bacteria (e.g. Vibrio spp.) and prevent disease in prawns and other marine organisms59.
Recently, there have been evidences that during the coral oocytes’ stages, some symbiotic microorganisms, such as algae (photosynthetic eukaryotic microbes), were found inside, and could function as an additional nutrition and energy source for metabolism during the oocyte development60. Subsequently, there were studies that investigated original sources of symbiotic algae and the other microorganisms, and how they could enter to the oocyte membranes61. Moreover, on an ectoderm, symbiotic bacteria could help prevent gamete and embryo from pathogenic microbe inhabitation and infection62,63.
In conclusion, our bacterial community and metabolic potential analyses among 0–1 h, 8 h, 28–41 h and 48 h suggested coral-bacteria association to correlate with nutrient metabolism and genetic information processing in gamete spawning and fertilization, cellular processes and microbial selection in embryonic cleavage, environmental information processing in round cell development and a greater nitrogen supply in planula formation. Nonetheless, bacterial transcriptome study should be future performed to confirm our speculation. Pseudoalteromonas in 8 h and Pseudomonas in 41–48 h were reported to inhibit coral pathogens64,65, and Rhodobacteraceae involve reproduction and ontogeny of various coral species, i.e. Acropora tenuis, Acropora digitifera, Pocillopora damicornis and Pocillopora acuta30,38,53,66,67. In addition, orders Oceanospirillales and Rhizobiales were essential coral holobionts worldwide. Subsequently, as Oceanospirillales function in degradation of dimethylsulfoniopropionate (DMSP) into usable carbon and sulfur forms and also produces antimicrobial compounds, so their predominance at 8 h might support coral growth and maintianance of healthy coral status by preventing pathogenic bacteria21,22. During the latter larval development, in particular planula, Rhizobiales function in nitrogen fixation, so their predominance might support amino acid (protein) synthesis and hence the coral growth28. Neave et al.68 also reported functional specificity and diversification in developing A. humilis associated Oceanospirilllales genomes to be more enriched for functions in carbohydrate and protein cyclings. Note the large variation between 39h1 and 39h2 independent replicates perhaps suggested an effect of transitioning bacterial habitation in shaping the holobionts69, or an effect from natural variation among corals. Similarly, 48h1 and 48h2 (but 48h3) replicates showed a more community similarity to the 41 h, supportive of ongoing transitioning stage; the shortcoming of our preliminary study is a limited number of sequencing replicates to allow consensus evaluation of relative abundances of bacteria during this coral transitioning time points. Our findings of bacterial association and their dynamics strengthen the assertion of bacterial involvement in coral development and the perspective in using bacterial manipulation in enhancing coral growth. The knowledge from this study can be a baseline data and helped improve an intervention in cultivation of corals through sexual reproduction to enhance the coral growth and juvenile settlement in a hatchery for mass culture to support sustainable coral management.
References
Chavanich, S., Viyakarn, V., Loyjiw, T., Pattaratamrong, P. & Chankong, A. Mass bleaching of soft coral, Sarcophyton spp. in Thailand and the role of temperature and salinity stress. ICES J. Mar. Sci. 66, 1515–1519 (2009).
Phongsuwan, N. et al. Status and changing patterns on coral reefs in Thailand during the last two decades. Deep Sea Res. Pt. II Top. Stud. Oceanogr. 96, 19–24 (2013).
Gardner, T. A., Côté, I. M., Gill, J. A., Grant, A. & Watkinson, A. R. Long-term region-wide declines in Caribbean corals. Science 301, 958–960 (2003).
Bruno, J. F. & Selig, E. R. Regional decline of coral cover in the Indo-Pacific: Timing, extent, and subregional comparisons. PLoS ONE 2, e711. https://doi.org/10.1371/journal.pone.0000711 (2007).
De´ath, G., Fabricius, K. E., Sweatman, H. & Puotinen, M. The 27-year decline of coral cover on the Great Barrier Reef and its causes. PNAS 109, 17995–17999 (2012).
Moberg, F. & Folke, C. Ecological goods and services of coral reef ecosystems. Ecol. Econ. 29, 215–233 (1999).
Sheppard, C. et al. The Gulf: A young sea in decline. Mar. Pollut. Bull. 60, 13–38 (2010).
Cruz-Trinidad, A., Aliño, P. M., Geronimo, R. C. & Cabral, R. B. Linking food security with coral reefs and fisheries in the coral triangle. Coast Manag. 42, 160–182 (2014).
Chavanich, S. et al. A tunicate from a Thai coral reef: A potential source of new anticancer compounds. Coral Reefs 24, 621. https://doi.org/10.1007/s00338-005-0036-y (2005).
Rocha, J., Peixe, L., Gomes, N. & Calado, R. Cnidarians as a source of new marine bioactive compounds-an overview of the last decade and future steps for bioprospecting. Mar. Drugs 9, 1860–1886 (2011).
Cooper, E. L., Hirabayashi, K., Strychar, K. B. & Sammarco, P. W. Corals and their potential applications to integrative medicine. Evid. Based Complement. Alternat. Med. 2014, 184959. https://doi.org/10.1155/2014/184959 (2014).
Petersen, D. et al. The application of sexual coral recruits for the sustainable management of ex situ populations in public aquariums to promote coral reef conservation-SECORE Project. Aquat. Conserv. 16, 167–179 (2006).
Chavanich, S. & Viyakarn, V. Conservation and restoration of coral reefs under climate change: Strategies and practice. in The Cnidaria, Past, Present and Future. 787–792. (Springer, 2016).
Boström-Einarsson, L. et al. Coral restoration–A systematic review of current methods, successes, failures and future directions. PLoS ONE 15, e0226631. https://doi.org/10.1371/journal.pone.0226631 (2020).
Webster, N. S. & Reusch, T. B. Microbial contributions to the persistence of coral reefs. ISME J. 11, 2167–2174 (2017).
van Oppen, M. J. & Blackall, L. L. Coral microbiome dynamics, functions and design in a changing world. Nat. Rev. Microbiol. 17, 557–567 (2019).
Lesser, M. P., Mazel, C. H., Gorbunov, M. Y. & Falkowski, P. G. Discovery of symbiotic nitrogen-fixing cyanobacteria in corals. Science 305, 997–1000 (2004).
Chimetto, L. A. et al. Vibrios dominate as culturable nitrogen-fixing bacteria of the Brazilian coral Mussismilia hispida. Syst. Appl. Microbiol. 31, 312–319 (2008).
Ceh, J. et al. Nutrient cycling in early coral life stages: Pocillopora damicornis larvae provide their algal symbiont (Symbiodinium) with nitrogen acquired from bacterial associates. Ecol. Evol. 3, 2393–2400 (2013).
Gochfeld, D. J. & Aeby, G. S. Antibacterial chemical defenses in Hawaiian corals provide possible protection from disease. Mar. Ecol. Prog. Ser. 362, 119–128 (2008).
Kirkwood, M., Todd, J. D., Rypien, K. L. & Johnston, A. W. The opportunistic coral pathogen Aspergillus sydowii contains dddP and makes dimethyl sulfide from dimethylsulfoniopropionate. ISME J. 4, 147–150 (2010).
Raina, J.-B. et al. Isolation of an antimicrobial compound produced by bacteria associated with reef-building corals. PeerJ 4, e2275. https://doi.org/10.7717/peerj.2275 (2016).
Lodwig, E. M. et al. Amino-acid cycling drives nitrogen fixation in the legume—Rhizobium symbiosis. Nature 422, 722–726 (2003).
Bourne, D., Iida, Y., Uthicke, S. & Smith-Keune, C. Changes in coral-associated microbial communities during a bleaching event. ISME J. 2, 350–363 (2008).
Mouchka, M. E., Hewson, I. & Harvell, C. D. Coral-associated bacterial assemblages: Current knowledge and the potential for climate-driven impacts. Integr. Comp. Biol. 50, 662–674 (2010).
Lema, K. A., Willis, B. L. & Bourne, D. G. Corals form characteristic associations with symbiotic nitrogen-fixing bacteria. Appl. Environ. Microbiol. 78, 3136–3144 (2012).
Bourne, D. G., Morrow, K. M. & Webster, N. S. Insights into the coral microbiome: Underpinning the health and resilience of reef ecosystems. Ann. Rev. Microbiol. 70, 317–340 (2016).
Lema, K. A., Bourne, D. G. & Willis, B. L. Onset and establishment of diazotrophs and other bacterial associates in the early life history stages of the coral Acropora millepora. Mol. Ecol. 23, 4682–4695 (2014).
Zhou, G. et al. Microbiome dynamics in early life stages of the scleractinian coral Acropora gemmifera in response to elevated pCO2. Environ. Microbiol. 19, 3342–3352 (2017).
Bernasconi, R. et al. Establishment of coral-bacteria symbioses reveal changes in the core bacterial community with host ontogeny. Front. Microbiol. 10, 1529. https://doi.org/10.3389/fmicb.2019.01529 (2019).
Damjanovic, K., Menéndez, P., Blackall, L. L. & van Oppen, M. J. H. Early life stages of a common broadcast spawning coral associate with specific bacterial communities despite lack of internalized bacteria. Microb. Ecol. 79, 706–719 (2020).
Miller, N., Maneval, P., Manfrino, C., Frazer, T. K. & Meyer, J. L. Spatial distribution of microbial communities among colonies and genotypes in nursery-reared Acropora cervicornis. PeerJ 8, e9635. https://doi.org/10.7717/peerj.9635 (2020).
Chamberland, V. F. et al. Four-year-old Caribbean Acropora colonies reared from field-collected gametes are sexually mature. Bull. Mar. Sci. 92, 263–264 (2016).
Baria-Rodriguez, M. V., dela Cruz, D. W., Dizon, R. M., Yap, H. T. & Villanueva, R. D. Performance and cost-effectiveness of sexually produced Acropora granulosa juveniles compared with asexually generated coral fragments in restoring degraded reef areas. Aquat. Conserv. Mar. Freshwater Ecosyst. 29, 891–900 (2019).
Henry, J. A., O’Neil, K. L. & Patterson, J. T. Native herbivores improve sexual propagation of threatened staghorn coral Acropora cervicornis. Front. Mar. Sci. 6, 713. https://doi.org/10.3389/fmars.2019.00713 (2019).
Ligson, C. A., Tabalanza, T. D., Villanueva, R. D. & Cabaitan, P. C. Feasibility of early outplanting of sexually propagated Acropora verweyi for coral reef restoration demonstrated in the Philippines. Restor. Ecol. 28, 244–251 (2019).
Tabalanza, T. D. et al. Successfully cultured and reared coral embryos from wild caught spawn slick in the Philippines. Aquaculture 525, 735354. https://doi.org/10.1016/j.aquaculture.2020.735354 (2020).
Apprill, A., Marlow, H. Q., Martindale, M. Q. & Rappé, M. S. Specificity of associations between bacteria and the coral Pocillopora meandrina during early development. Appl. Environ. Microbiol. 78, 7467–7475 (2012).
Kuanui, P., Chavanich, S., Viyakarn, V., Omori, M. & Lin, C. Effects of temperature and salinity on survival rate of cultured corals and photosynthetic efficiency of the zooxanthellae in coral tissues. Ocean Sci. J. 50, 263–268 (2015).
Kuanui, P. et al. Effect of light intensity on survival and photosynthetic efficiency of cultured corals of different ages. Estuar. Coast Shelf Sci. 235, 106515. https://doi.org/10.1016/j.ecss.2019.106515 (2020).
Marotz, C. et al. DNA extraction for streamlined metagenomics of diverse environmental samples. Biotechniques 62, 290–293 (2017).
Bulan, D. E. et al. Spatial and seasonal variability of reef bacterial communities in the upper Gulf of Thailand. Front Mar. Sci. 5, 441. https://doi.org/10.3389/fmars.2018.00441 (2018).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624 (2012).
Schloss, P. D. et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120 (2013).
Pollock, J., Glendinning, L., Wisedchanwet, T. & Watson, M. The madness of microbiome: attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microbiol. 84. https://doi.org/10.1128/AEM.02627-17 (2018).
Bharti, R. & Grimm, D. G. Current challenges and best-practice protocols for microbiome analysis. Brief Bioinform. 22, 178–193. https://doi.org/10.1093/bib/bbz155 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
RStudio Team. RStudio: Integrated Development for R. (RStudio, PBC, 2020).
Olson, N., Ainsworth, T., Gates, R. & Takabayashi, M. Diazotrophic bacteria associated with Hawaiian Montipora corals: Diversity and abundance in correlation with symbiotic dinoflagellates. J. Exp. Mar. Biol. Ecol. 371, 140–146 (2009).
Sharp, K. H., Sneed, J., Ritchie, K., Mcdaniel, L. & Paul, V. J. Induction of larval settlement in the reef coral Porites astreoides by a cultivated marine Roseobacter strain. Biol. Bull. 228, 98–107 (2015).
Sharp, K. H., Distel, D. & Paul, V. J. Diversity and dynamics of bacterial communities in early life stages of the Caribbean coral Porites astreoides. ISME J. 6, 790–801 (2012).
Apprill, A., Marlow, H. Q., Martindale, M. Q. & Rappé, M. S. The onset of microbial associations in the coral Pocillopora meandrina. ISME J. 3, 685–699 (2009).
Boch, C. A., Ananthasubramaniam, B., Sweeney, A. M., Doyle, F. J. III. & Morse, D. E. Effects of light dynamics on coral spawning synchrony. Biol. Bull. 220, 161–173 (2011).
Baquiran, J. I. P. et al. The prokaryotic microbiome of Acropora digitifera is stable under short-term artificial light pollution. Microorganisms 8, 1566. https://doi.org/10.3390/microorganisms8101566 (2020).
Rohwer, F., Seguritan, V., Azam, F. & Knowlton, N. Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 243, 1–10 (2002).
Pootakham, W. et al. High resolution profiling of coral-associated bacterial communities using full-length 16S rRNA sequence data from PacBio SMRT sequencing system. Sci. Rep. 7, 2774. https://doi.org/10.1038/s41598-017-03139-4 (2017).
Franco, Á. G., Cadavid, L. F. & Arévalo-Ferro, C. Biofilms and extracts from bacteria producing “quorum sensing” signaling molecules protomote chemotaxis and settlement behaviors in Hydractinia symbiolongicarpus (Cnidaria: Hydrozoa) larvae. Acta Biol. Colomb. 24, 150–162 (2019).
Jayaprakash, N. et al. A marine bacterium, Micrococcus MCCB 104, antagonistic to vibrios in prawn larval rearing systems. Dis. Aquat. Org. 68, 39–45 (2005).
Tsai, S., Chang, W.-C., Chavanich, S., Viyakarn, V. & Lin, C. Ultrastructural observation of oocytes in six types of stony corals. Tissue Cell 48, 349–355 (2016).
Lin, C., Kup, F.-W., Chavanich, S. & Viyakarn, V. Membrane lipid phase transition behavior of oocytes from three gorgonian corals in relation to chilling injury. PLoS ONE 9, e92812. https://doi.org/10.1371/journal.pone.0092812 (2014).
Shnit-Orland, M. & Kushmaro, A. Coral mucus-associated bacteria: A possible first line of defense. FEMS Microbiol. Ecol. 67, 371–380 (2009).
Leite, D. C., Salles, J. F., Calderon, E. N., van Elsas, J. D. & Peixoto, R. S. Specific plasmid patterns and high rates of bacterial co-occurrence within the coral holobiont. Ecol. Evol. 8, 1818–1832 (2018).
Rypien, K. L., Ward, J. R. & Azam, F. Antagonistic interactions among coral-associated bacteria. Environ. Microbiol. 12, 28–39 (2010).
ElAhwany, A. M., Ghozlan, H. A., ElSharif, H. A. & Sabry, S. A. Phylogenetic diversity and antimicrobial activity of marine bacteria associated with the soft coral Sarcophyton glaucum. J. Basic Microbiol. 55, 2–10 (2015).
Damjanovic, K., van Oppen, M. J., Menéndez, P. & Blackall, L. L. Experimental inoculation of coral recruits with marine bacteria indicates scope for microbiome manipulation in Acropora tenuis and Platygyra daedalea. Front. Microbiol. 10, 1702. https://doi.org/10.3389/fmicb.2019.01702 (2019).
Damjanovic, K., Blackall, L. L., Menéndez, P. & van Oppen, M. J. H. Bacterial and algal symbiont dynamics in early recruits exposed to two adult coral species. Coral Reefs 39, 189–202 (2020).
Neave, M. J., Michell, C. T., Apprill, A. & Voolstra, C. R. Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci. Rep. 7, 40579. https://doi.org/10.1038/srep40579 (2017).
Hernandez-Agreda, A., Leggat, W., Bongaerts, P., Herrera, C. & Ainsworth, T. D. Rethinking the coral microbiome: Simplicity exists within a diverse microbial biosphere. MBio 9, e00812. https://doi.org/10.1128/mBio.00812-18 (2018).
Acknowledgements
This research was supported by Thailand Research Fund (RSA6180046), Thailand Research Fund through the Royal Golden Jubilee Ph.D. Program (PHD/0126/2558), NRCT-JSPS Core-to-Core Program, CREPSUM Project, Mubadala Petroleum (Thailand) Limited, Microbiome Research Unit for Probiotics in Food and Cosmetics, and the Second Century Fund (C2F) of Chulalongkorn University. The authors acknowledged the National Center for Genetic Engineering and Biotechnology (BIOTEC) and the National Science and Technology Development Agency (NSTDA) for allowing us to compute the data on the server, and thanked D. Sripan and P. Chinklang for general lab help.
Author information
Authors and Affiliations
Contributions
C.K. helped data analysis and drafted the manuscript. S.J. collected samples and performed experiments. M.P. performed experiments and analyzed data. V.V. conceived of the study. S.C. provided samples and conceived of the study. N.S. conceived of the study, coordinated the experiments and data analysis, and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kullapanich, C., Jandang, S., Palasuk, M. et al. First dynamics of bacterial community during development of Acropora humilis larvae in aquaculture. Sci Rep 11, 11762 (2021). https://doi.org/10.1038/s41598-021-91379-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-91379-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
