
Abstract
Genome-wide association studies (GWAS) play a vital role in identifying important genes those is associated with the phenotypic variations of living organisms. There are several statistical methods for GWAS including the linear mixed model (LMM) which is popular for addressing the challenges of hidden population stratification and polygenic effects. However, most of these methods including LMM are sensitive to phenotypic outliers that may lead the misleading results. To overcome this problem, in this paper, we proposed a way to robustify the LMM approach for reducing the influence of outlying observations using the β-divergence method. The performance of the proposed method was investigated using both synthetic and real data analysis. Simulation results showed that the proposed method performs better than both linear regression model (LRM) and LMM approaches in terms of powers and false discovery rates in presence of phenotypic outliers. On the other hand, the proposed method performed almost similar to LMM approach but much better than LRM approach in absence of outliers. In the case of real data analysis, our proposed method identified 11 SNPs that are significantly associated with the rice flowering time. Among the identified candidate SNPs, some were involved in seed development and flowering time pathways, and some were connected with flower and other developmental processes. These identified candidate SNPs could assist rice breeding programs effectively. Thus, our findings highlighted the importance of robust GWAS in identifying candidate genes.
Similar content being viewed by others

Introduction
One of the major challenges in recent genetics research is to explore the biomarker genes that are linked to complex traits of interests in living organisms. Trait variations in living organisms are related to genetic variations in genes. These variations are observed largely at physiological, developmental, and morphological stages. Identification of important genetic basis such as causal genetic variants for such distinction in phenotypic traits is identifiable at single nucleotide polymorphism (SNP) levels. The techniques to explore the SNP contribution in phenotypic variation are termed as Genome-Wide Association Studies (GWAS). SNPs, however, are usually tested for relationship study through the whole genome with the characters of important trait of interest. The SNPs identified by GWAS can be used for the treatment and prevention of certain complex traits in living organisms. A very large set of SNPs along with a very large number of accessions are simultaneously studied using different GWAS methods to uncover the significant relationship between genomic latent factors and phenotypic variations of interest1.
Linear regression model (LRM) is a popular approach in GWAS. It is implemented through PLINK software for detecting important SNPs associated with quantitative trait2. The PLINK tool focused on parametric or nonparametric-based linear regressions which do not control population stratification. Population stratification is one of the main concerning issues when extensive genome-wide association analysis with numerous subjects is in consideration3,4,5. Some unidentified new population structures are probable to exist due to a large number of subjects that may perhaps liable for regular dissimilarities being selected in SNPs amongst cases and controls4. Due to the higher false discovery rate (FDR), it is imperative to correct the observed population stratification in GWAS4,6. There are, however, several statistical methodologies proposed earlier for GWAS to address the effects of population stratification. The most commonly used statistical methods to avoid the bias of population stratification (PS) or genetic relatedness are genomic control7, structured association8, and principal component analysis9,10. The genomic control (GC) method adjusts the statistical relationship by a common element concerning the whole set of SNPs for precise correction of PS4. If the consequence of population structure increases, the power of GC approach decreases9,11,12,13,14. The structured association (SA) analysis technique suggests locating the samples to separate subpopulation groups for collecting signs of a relationship in each group8. SA method is only useful for small datasets4. Principal Component Analysis (PCA) approach is also used to overcome the influence of population stratification in GWAS by using several topmost principal components (PCs) as covariates4,9. But none of the methods mentioned above can handle the influence of the polygenic effect. To overcome these issues, the linear mixed model (LMM) was proposed which is one of the most popular approaches in GWAS. It is widely using through several computer software such as TASSEL13, EMMA 15, EMMAX16, rrBLUP17, GAPIT18, and GAPIT Version 219.
However, all the methods as early discussed are very much sensitive to phenotypic outliers. So, they can produce misleading results in presence of outlying observations. To overcome these issues, an attempt is made to robustify the LMM based GWAS by using a new type of outlier modification rule based on the minimum β-divergence method20,21. The performance of the proposed approach has been investigated using both simulated and real rice genome datasets related to flowering time.
Results and discussion
We investigated the performance of the proposed method compare to two popular approaches (LMM and LRM) using both synthetic and real data analysis as discussed below:
Results and discussion based on a complete simulation
To investigate the performance of SNPs detections with the synthetic datasets, we considered two original clean simulated datasets that were generated with heritabilities 0.2 and 0.3 respectively, as described in the materials and method section. We contaminated 1%, 2%, 3%, 4%, and 5% phenotypic observations by outliers to generate five contaminated datasets with each clean dataset to investigate the performance against the phenotypic outliers. Before going to the performance comparison, first, we would like to discuss the preprocessing steps for the proposed method as follows.
Outlier detection and modification of phenotypic observations by the proposed method
To analyze these datasets by the proposed method, at first, we identified phenotypic outliers by using the β-weight function for each genotypic group and then replace the outlying phenotypic observations with the corresponding group mean computed by the minimum β-divergence method. To show how β-weight function detects outliers, we plotted β-weight corresponding to each phenotypic observation in the Supplementary Fig. S1 (a-b). Supplementary Fig. S1a consists of two panels, where the left panel plotted the original phenotypic observations and the right panel plotted their β-weights. Similarly, Supplementary Fig. S1b consists of two panels, where the left panel plotted the phenotypic observations including the 5% contaminated observations (red color), and the right panel plotted their β-weights. To select the outlying observations, we used the threshold value τj = pth quantile value of the empirical distribution of \(W_{\beta } (y_{li} |\hat{\theta }_{l,\beta } )\) as introduced in Eq. (7). We observed that the β-weight function correctly identified the outlying observations. Now we would like to discuss the consequence of outliers in the classical and proposed approaches by decomposing phenotypic variations as follows.
The consequence of outliers on the partition of total phenotypic variations
To discuss the consequence outliers on the partition of total phenotypic variations for both classical and proposed approaches, we considered the original clean dataset including two contaminated datasets based on 2% and 5% outlying observations. Table 1 shows the consequence of outliers on the partition of total phenotypic variations for both classical and proposed approaches.
We observed that variance proportions with respect to the genetic effect gradually decrease as increases the rate of outlying observations in the dataset by the classical approach, while the variance proportions with respect to the genetic effects are almost stable for each level of outlying rates in the dataset by the proposed approach. Therefore, the heritability (h2*) in presence of outliers becomes smaller than the heritability (h2) in absence of outliers by the classical approach, while the heritability (\( \, \mathop h\nolimits_{\beta }^{2*} \, \)) in presence of outliers is almost similar to the heritability (h2) in absence of outliers by the proposed approach.
Performance comparison for SNPs detection
At first, we identified important SNPs by applying each of LRM, LMM, and the proposed methods on each of six datasets with each of two distinct genetic heritabilities 0.2 and 0.3 corresponding to the respective rates 0%, 1%, 2%, 3%, 4% and 5% of outliers. Then we computed statistical power and false discovery rate (FDR) for each of the methods to investigate the performance of the proposed method in a comparison of the classical LRM and LMM approaches. We computed average power and FDR based on 1000 replication of each dataset. Figure 1 showed the effect of outliers on statistical power and FDR with each of two distinct genetic heritabilities 0.2 and 0.3. It is observed from Figs. 1(a, c) that the power of the proposed method slowly decreased compare to the LMR and LMM approaches with the increasing rates of outliers. In absence of outliers, both the proposed and LMM approaches produced almost identical powers but much larger than the power of LRM. For example, in absence of outliers, the power of LRM, LMM and proposed methods were 67.55, 74.55 and 73.00 respectively for the dataset with heritability 0.2 (Fig. 1a), while 72.35, 89.95 and 88.65 respectively for the heritability 0.3 (Fig. 1c). However, with the increasing rates of outliers, the power of LMM approach decreased dramatically faster than the LRM approach. For example, in presence of 5% outliers with the phenotypic observations, the power of LRM, LMM and proposed methods were 58.35, 38.95 and 62.65 respectively for the dataset with heritability 0.2 (Fig. 1a), while 60.15, 49.10 and 83.90 respectively for the heritability 0.3 (Fig. 1c). Thus the, the proposed method produced much higher powers than both LRM and LMM for both scenarios of heritabilities in presence of outliers. The above two examples also indicated that the power of all three methods increase as the increasing of heritabilities in the datasets.

Results computed by LRM, LMM and the proposed methods based on complete simulation (a) plot of statistical power against the rate of phenotypic outliers at heritability h2 = 0.2 and at the cutoff p-value 10–5. (b) Plot of FDR against the rate of phenotypic outliers at h2 = 0.2 and at cutoff = 10–5. (c) plot of statistical power against the rate of phenotypic outliers at h2 = 0.3 and cutoff 10–5 (d) plot of FDR against the rate of phenotypic outliers at h2 = 0.3 and cutoff 10–5.
Figures 1(b,d) showed that FDRs of the proposed and LMM methods were almost same and close to zero in each rate of phenotypic outliers. However FDR for LRM was too high due to the influence of outliers along with the population stratification and polygenic effects22. Thus we may conclude that both LRM and LMM approaches are very much sensitive to phenotypic outliers compare to the proposed method. This results also supported by the decomposition of phenotypic variations described in Table 1, since power of any method decreases as the increasing of computational heritabilities.
Performance comparison with some other robustification techniques
The proposed robustification technique was also compared with other two robustification techniques based on7-sigma rule (\(\overline{y }\pm 7\sigma \)) and inverse-normal transformation (INT)23,24 by the same datasets that were used in Fig. 1. We detected phenotypic outliers by the 7-sigma (7σ) rule and remove them before going to the analysis by LMM model. In the case of INT approach, we performed inverse-normal transformation on the phenotypic observations before going to the analysis by LMM model. In GWAS of quantitative traits/phenotypes, INT is commonly applied when the traits are distributed non-normally24. Figure 2 showed that the proposed method produces slightly higher power compare to 7σ and INT techniques for all cases of heritabilities and outliers. We also observed that the power of INT method is slightly higher compare to 7σ method in presence of outliers, but smaller in absence of outliers. There is one drawback with INT approach for the weak performance compare to the proposed method. The INT approach normalized the traits globally assuming the unimodal trait/phenotypic observations, while proposed method modified outliers corresponding to the genotypic groups assuming the multi-modal trait/phenotypic observations. Actually, phenotypic observations follow 3 modal distribution due to its 3 genotypes. There are two drawbacks with 7σ approach for the weak performance compare to both INT and the proposed methods. This method detected the outlying observations by assuming the unimodal distribution of phenotypic observations like INT approach and reduced the sample size by removing the outlying observation before going to the SNP detection by LMM model. The small sample size decreased the power significantly, which is satisfied by the previous study result25. Thus, the proposed robustification works well compare to 7σ and INT approaches.

Plot of statistical power against the rate of phenotypic outliers based on the same dataset as used in Fig. 1 computed by three methods 7-sigmarule, inverse normal transformation (INT) and the proposed methods.
Performance comparison based on real SNP genotype and simulated phenotype data on rice flowering time
We also investigated the performance of the proposed method in a comparison of LRM and LMM based on partial simulation with the real SNP genotype and simulated phenotype data on rice flowering by considering the same condition like Fig. 1. The top four significant SNPs (id2005919, id2005983, ud2000772 and ud7002027) identified in real data analysis (See Table 2) were considered as main effect with the true effect size (ak). From the rest SNPs, we randomly selected 1996 SNPs as polygenic variants (effects). Finally the phenotypic observation were generated using Eq. (9).
The Fig. 3 shows that the proposed method produces much larger power compare to both LRM and LMM which is supported by the results displayed in Fig. 1. However, with the increasing rate of phenotypic outliers, a slow decreasing rate of power was observed for the LMM method compared to LRM in Fig. 1. Again, with the increasing rate of phenotypic outliers, a decreasing trend of FDR for the LRM method was observed like Fig. 1d. This result could be due to the increase of outlier; the effect of population structure on the phenotype become weak. This result advocating that LMM method can control the confounding due to population stratification but not the outlier, whereas LRM method fail to control both the confounding and outlier. Thus, it may be concluded that the proposed method shows much better performance than the LRM and LMM in presence of phenotypic outliers; otherwise, it keeps the almost equal performance of LMM.

Results computed by LRM, LMM and the proposed methods based on partial simulation with the real dataset (a) plot of statistical power against the rate of phenotypic outliers at heritability h 2= 0.2 and at the cutoff p-value 10–5. (b) Plot of FDR against the rate of phenotypic outliers at h2 = 0.2 and at cutoff = 10–5. (c) plot of statistical power against the rate of phenotypic outliers at h2 = 0.3 and cutoff 10–5 (d) plot of FDR against the rate of phenotypic outliers at h2 = 0.3 and cutoff 10–5.
Genome-wide association analysis of rice flowering time by the proposed method
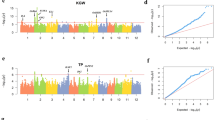
We have analyzed the rice flowering time trait to identify the loci influencing this complex trait. Previous study by Zhao et al.1 on the same trait identified only 2 loci which motivated us to reanalyze the trait using our proposed robust GWAS (rGWAS) method. We have identified 11 significant SNPs using the proposed method (rGWAS) for rice flowering time with the threshold \(p<9.99\times {10}^{-5}\) (Fig. 4). From the Manhattan plot it is observed that among the identified 11 SNPs, six SNPs lie in chromosome 2; one SNP marker belongs to each of the chromosome 6 and 7 and the rest three SNP markers belong to chromosome 8. However, these 11 SNPs did not overlap with the previously identified 2 SNPs. Zhow et al. used the genotype data with missing values as input in EMMA that omit the individuals with missing corresponding to the testing SNP. As a result the sample size was reduced that is responsible to decrease the detection power25,26,27. Moreover, the analyzed trait contained seven phenotypic outliers (Supplementary Fig. S2) that also responsible to reduce the statistical power (Fig. 1–2). A QQ-plot was also constructed using whole genome p-values of the proposed method. QQ-plot showed that the observed values correspond to the expected values are on or near the middle line between the x-axis and the y-axis (Supplementary Fig. S3) indicating that no genomic inflation has occurred in this analysis as well as population stratification is sufficiently controlled.

Manhattan plot using robust GWAS (rGWAS) on the trait of rice flowering time. The x-axis is the genomic position of the SNPs in the genome, and the y- axis is -log10 of the p-values. Each chromosome is colored differently. The grey horizontal line represents the minimal significant level at the cutoff.
Genomic information and characteristics of the candidate genes
To investigate the biological significance of the identified 11 SNPs, we validated these SNPs by using literature review and gold benchmark data. The corresponding chromosome and position of the identified SNPs were used to annotate and find the candidate genes comparing with the reference genome from the Rice Genome Annotation Project (RGAP) (http://rice.plantbiology.msu.edu/) database and collecting necessary genomic information given in Table 2 and Supplementary Table S1.
The SNP id2005644 was identified that located near the gene LOC_Os02g21070 which encodes pentatricopeptide repeats (PPR) domain-containing protein, and is assumed to take part in the biological molecule variation28. Plastid-localized pentatricopeptide repeat protein was reported in a study that is required for both pollen development and plant growth in rice29. A recent study in Arabidopsis showed that PPR containing protein affects flowering time30. Another variant id2005743 located in chromosome 2 and in the gene LOC_Os02g21880 was identified. The gene encodes coiled-coil domain-containing protein. This protein acts as the regulator of protein positioning in the cell during cell division by splitting and organizing signaling paths sequentially and spatially31. A coiled-coil domain containing protein in rice, PAIR1, was reported to express in the early stages of flower development32. Other four candidate genes (LOC_Os02g24770, LOC_Os02g24780 and LOC_Os02g27750, LOC_Os08g01070) were identified those encodes the retrotransposon and transposon transposable elements (TEs). Several studies have reported that these TEs can be induced by heat and cold stress in plants33. For example, Ty1-copia like retrotransposon ONSEN was found to be activated by heat stress in Arabidopsis34. Temperature and photoperiod are also found as two key regulatory factors associated with the flowering time in plants including rice35.
The detected SNPs were also mapped in the region of 100 kb of the genes those involved in the rice flowering time and seed development pathway36 and found five SNPs comprised of four genes involved in the two pathways (Supplementary Table S2). These results are suggesting the potential role of robust GWAS in detecting novel genes.
Functional enrichment analysis of the candidate genes
GO analysis is one of the major bioinformatics techniques for better understanding the underlying biological processes (BP) of the candidate genes along with their molecular functions (MF) and the cellular component (CC) of the genes37,38. Therefore, to more characterize the candidate genes, we have performed GO enrichment analysis and the results are shown in Fig. 5a and Supplementary Fig. S4a-c. The most important pathways or GO terms involving the candidate genes are highlighted with light yellow color in the rectangular boxes (Supplementary Fig. S4a-c). Flower development (GO:0,009,908), abscission (GO:0,009,838), signal transduction (GO:0,007,165), cell death (GO:0,008,219), cellular process (GO:0,009,987), response to stress (GO:0,006,950), and cellular protein modification (GO:0,006,464) were found as the most important pathways activated by the identified candidate genes in rice. Among the identified BPs, flower development (GO:0,009,908) is one of the crucial pathway that play the role for early and healthy grain development in rice39,40 and it is functionally linked to both the reproductive process (GO:0,022,414) and developmental process (GO:0,032,502) of rice plant (Fig. 5a and Supplementary Fig. S4a). Regulation of rice flowering time is delayed by several independent pathways and significantly influenced by prompt vegetative growth and reproductive process41. GO analysis showed that the shoot development (GO:0,048,367) and reproduction stages such as post-embryonic development (GO:0,009,791) are functionally related to rice flower development (Supplementary Fig. S4a).

Expression map of GO and SCL for the 11 candidate genes. (a) Represents the expression map of the functional pathways viz., biological process (BP), molecular function (MF), and cellular component (CC) of the six candidate genes. (b) Represents the predicted subcellular location (SCL) of candidate genes in 10 molecular organs viz., cytosol (cytos), endoplasmic reticulum (ER), extracellular (extra), golgi apparatus (golgi), membrane (membr), mitochondria (mito), nuclear (nucl), peroxisome (pero), plastid (plast) and vacuole (vacu).
Abscission, a part of a multicellular organism (GO:0,007,275), in GO enrichment analysis showed that it is directly related to the developmental process (GO:0,032,502) in rice. Earlier studies showed that seed shattering is controlled by the development of the abscission layer pathways in rice that allows offspring dispersal in the natural environment42. Signaling components via signal transduction (GO: 0,007,165) chain activate the different plant steroid hormones namely Brassinosteroids to regulate various growth and developmental programs, including cell differentiation and elongation, reproductive development, senescence, skotomorphogenesis (seedling development in the dark), and vascular differentiation43,44,45. The hormonal signal transduction pathways are also responsible to control several yield-related traits, including leaf angle, plant height, tiller number, and grain size in rice43,44,46,47. The predicted metabolic routes in this study may execute metabolism activities to convert food to energy to run cellular processes, to build blocks for proteins, lipids, nucleic acids, and some carbohydrates48,49. Some enzyme-catalyzed reactions may allow rice plants to grow and reproduce, maintain their structures, and respond to various stresses. The GO pathway, response to stress (GO:0,006,950) is predicted to play the role for controlling cellular activity in terms of movement, secretion, enzyme production, gene expression for the result of exogenous disturbance, temperature, humidity in rice plants.
Subcellular location of the candidate genes
The cytosol is the place where the occurrence of the maximum different metabolisms in plants and most of the proteins in the cell are localized50,51. The predicted result of subcellular localization (SCL) of the candidate genes implied that nine genes out of eleven are localized in cytosol (Fig. 5b and Supplementary Data S1). Plastid is an important molecular organ found in plant cells mostly involve in photosynthesis and other gene expressions52. Photosynthesis is the key physiological parameter in rice that relates ultimately in many aspects to increase rice productivity53. Increase photosynthesis rate can utilize the solar radiation properly which leads to creating early flowering time because flowering signals are produced in leaves41,54. This gene expression in plastid likely to enhance the photosynthesis process, which regulates the leaf anatomy for earlier flowering in rice. Four candidate genes LOC_Os02g27750, LOC_Os06g18000, LOC_Os07g45950, and LOC_Os08g25040 were found in the plastid (plast). Out of the four genes the latter two genes were predicted to be located in extracellular (extra) and membrane (membr) of which LOC_Os07g45950 was predicted for vacuole (vacu) (Fig. 5b). It is also observed that three genes named LOC_Os02g21070, LOC_Os02g21880, and LOC_Os08g01070 are predicted to located in nuclear activity. However, no candidate genes were predicted to belong from the cellular locations viz., endoplasmic reticulum (ER), peroxisome (pero), and mitochondria (mito) (Fig. 5b).
Expression profile of the candidate genes
The expression level of the candidate genes in different organs or tissues such as seedling, vascular cell, root, leaves, post-emergence, pre-emergence, seed, endosperm, embryo, shoots, anther, pistil, and panicle were extracted from the database Rice Genome Annotation Project (RGAP)55 (Supplementary Data S2). Heatmap presented in Fig. 6 exhibits the expression levels of the candidate genes through the organs or tissues. From the figure it was observed that the genes LOC_Os02g21880 and LOC_Os02g24134 showed high-level expression in seedling, root, shoot, and panicle in rice while these two genes exhibited only high-level expression in the vascular cell at 14DAP (Fig. 6). Moreover, seedling, vascular cell at 14DAP and shoots specific expressions were maximum for the gene LOC_Os06g18000. The genes LOC_Os02g21880, LOC_Os02g24134, LOC_Os08g01070 and LOC_Os08g25060 were found with high expression in panicle. The earlier study also suggested that leaves, shoot, and panicles have significant roles in regulating flowering time41,56. Our results obtained from real data analysis also consistent with earlier outcomes1,22,31,57,58,59.

Heatmap showing the expression pattern of the identified 11 candidate genes. The heatmap represents the expression in various organs (seedling, vascular cell stage, root, leaves, post, and pre-emergence inflor, seed, endosperm, embryo, shoots, anther, pistil, and panicle) of rice. The color scale bar of the figure represents log2 transformed FPKM values.
Conclusion
The GWAS is a powerful tool to explore the novel biomarker genes at the SNP level. The LMM approach has been considered as the leading statistical procedure to address the two main challenges of embedded population structures and genetic relatedness among individuals when GWAS is performed. It was however observed that this LMM approach is very much sensitive to phenotypic outlier that leads to misleading results. Therefore, in this article, we discussed a way to robustify the LMM procedure for controlling the outlying effect and reducing the puzzling effects of the population stratification and genetic relatedness in GWAS by using the minimum β-divergence method. This method introduced the β-weight function, which played a key role in the robustification procedure. For the convenience of the presentation, we call this method robust GWAS (rGWAS). Simulation results showed that the average power of correct identification of SNPs by LMM and the proposed method is almost the same and greater than 80%, which is much larger than the power of the linear regression model (LRM), in absence of outliers at heritabilities 0.2 and 0.3. The average power of LRM and the proposed method were slowly decreasing as gradually increasing the rate of phenotypic outliers with heritabilities 0.2 and 0.3, while the power of LMM was decreasing sharply and significantly. The false discovery rate (FDR) of LMM and proposed methods are almost the same and much smaller than the FDR of LRM in all cases of our simulation study. Thus, the proposed method outperformed the LRM and LMM in presence of outliers; otherwise, it shows the almost equal performance of LMM which is much better than LRM in absence of outliers. The demonstration of the proposed method with the real genome dataset against rice flowering time identified 11 important SNP makers. To investigate the biological significance of the identified 11 SNPs, we validated these SNPs by using literature review and gold benchmark data. We performed GSEA for the identified 11 SNP makers and SCL analysis to detect more valid SNPs out of 11 that have a significant association with the flowering time and other trait variations in rice. We also studied their expressions in various organs in rice to find the link with the flowering time. From GO analysis, it is observed that the gene LOC_Os06g18000 might play functional roles in flower development and response to stress in rice. Amongst the 11 genes, LOC_Os02g21880, LOC_Os06g18000, LOC_Os02g24134 exhibited larger expression in seedling, vascular cell, root, shoot, and panicle. Also, the gene LOC_Os08g25060 is predicted to provide maximum expression in vascular cell, root, and panicle. SCL results support that the cytosol contains the maximum number of genes. Plastid is an important molecular organ found in plant cells mostly involve in photosynthesis and other gene expressions. In our study, SCL analysis shows that the expression of the gene LOC_Os06g18000 in plastid may act as a flowering promoter. This gene expression in plastid likely to enhance the photosynthesis process which regulates the leaf anatomy for earlier flowering in rice. In GO analysis, it is also observed that this gene expression is associated with flowering in rice. Finally, it can be concluded that phenotypic outliers may significantly affect the analysis results in GWAS. Our proposed robust method outperforms the existing LRM and LMM methods in presence of outliers and the genomic information presented may however provide a basic platform for further biological investigations. To implement the proposed rGWAS method, the R-code and necessary instructions are available at the website.
Materials and methods
Robustification of LMM based GWAS by using the outlier modification rule (proposed)
The linear mixed model (LMM) approaches are extensively applied for genome-wide association studies (GWAS) for observable phenotypic variations in eukaryotic groups. If we consider that, there are m genotypes with n measurements of a phenotype. Efficient mixed-model association (EMMA)15 is such a model generally expressed by the following :
where \({\varvec{y}}\, = \,(y_{1} ,\,y_{2} ,...,y_{n} )^{\prime}\) is the n × 1 vector of phenotypic observations, and X = (xij) is an n × q matrix of fixed effects including mean, SNPs, and other confounding variables.\({\varvec{a}}\) is a q × 1 vector representing coefficients of the fixed effects. Z = (zij) is an n × m incidence (design) matrix mapping each phenotype to one of the m genotypes. b is the vector of random polygenic effects which follows N(0,σg2K), where σg2 is the polygenic variance component, and K = (kjt) is the m × m genomic relationship matrix. The genomic pairwise relationship coefficient between two individuals, j and t, is defined as follows
where \(T_{\varphi }\) is the total number of SNPs, xijand xit measure the numbers (0, 1, 2) of the minor allele(s) for the ith SNP of the jth and tth individuals respectively, and fi is the frequency of the minor allele. ε is the vector of random error which follows N(0,σε2I), where σε2 is the error variance component and where I is the n × n identity matrix. The overall phenotypic variance–covariance matrix can be represented as \( {\mathbf{V}} = \sigma_{g}^{2} {\mathbf{ZKZ^{\prime}}} + \sigma_{\varepsilon }^{2} {\mathbf{I}}\).
where I is the n × n identity matrix. The variance components for polygenic effects and errors were estimated by restricted maximum likelihood (REML) using spectraldecomposition instead of the iterative expectation–maximization algorithm (EM algorithm)15. The full-likelihood function is maximized when \({\hat{a} = (X^{\prime}H}^{{{ - 1}}} {\mathbf{X}})^{ - 1} {\mathbf{X^{\prime}H}}^{ - 1} {\mathbf{y}}\) and the optimal variance component is \( \hat{\sigma }_{F}^{2} = R/n\) for full-likelihood and \(\hat{\sigma }_{R}^{2} = R/(n - q)\) for restricted likelihood, where \(R = ({\varvec{y}} - {\varvec{Xa}})^{\prime}H^{ - 1} ({\varvec{y}} - {\varvec{Xa}})\) is a function of δ as well and \(H = \sigma^{ - 1} V = {\varvec{Z}}K{\varvec{Z}} + \delta I\) is a function of δ, defined by \(\delta = \sigma_{\varepsilon }^{2} /\sigma_{g}^{2} ,\,\,\,\sigma = \sigma_{g}\). When the maximum likelihood (ML) or restricted maximum likelihood (REML) variance component \({\hat{\mathbf{V}}} = \hat{\sigma }_{g}^{2} {\mathbf{ZKZ^{\prime}}} + \hat{\sigma }_{\varepsilon }^{2} {\mathbf{I}}\) is estimated, the classical F-statistic for testing the null hypothesis Ma = 0 for an arbitrary full-rank p × q matrix M13,60.
with p numerator degrees of freedom and n-q denominator degrees of freedom. The Satterthwaite degrees of freedom are calculated to avoid computationally intensive matrix operations. Both ML and REML estimators are very much sensitive if one or more phenotypic observations in y are outliers. Thus, LMM based F-statistic in Eq. (3) produces misleading results to test the null hypothesis in presence of outliers. In this study, we, therefore, consider the robustification of LMM based F-statistic in Eq. (3) by using the outlier identification and modification rule. There are some procedures for the identification of outliers in the literature; those are suitable when usual/clean data follows the unimodal distribution. However, in the current problem, phenotypic observations follow the multimodal distribution. So, conventional procedures cannot be used to identify the outlying observations properly. Therefore, in this paper, an attempt is made to propose a new outlier identification and modification rule by using the minimum β-divergence methods20,61 as follows:
(i) Select the top-ranking significant SNP associated with the phenotypic variations by using the minimum β-divergence based robust ANOVA62.
(ii) Divide the phenotypic data into m groups corresponding to the m genotypic labels of the selected most significant SNP. For example, let
be the partition of phenotypic observations corresponding to the selected SNP, where, n = n1 + n2 + …….. + nm.
(iii) Detect the outlying observations from the lth (l = 1,2,….,k) group using the β-weight function defined by
where i = 1, 2, ... , nl
The minimum β-divergence estimators \(\hat{\theta }_{l,\beta } = (\hat{\mu }_{l,\beta } ,\,\hat{\sigma }_{l,\beta }^{2} )\) of the parameters \(\theta_{l,\beta } = (\mu_{l,\beta } , \,\sigma_{l,\beta }^{2} )\) are calculated iteratively by using the following formulas:
and
The notation θt+1 denotes the update of \(\theta_{t}\) in the (t + 1)th iteration. The robustness of these estimators has been discussed in the background of influence function20 and their reliability61. It is noteworthy that the minimum β-divergence estimators \(\hat{\theta }_{l,\beta } = (\hat{\mu }_{l,\beta } ,\hat{\sigma }_{l,\beta }^{2} )\) reduce to the classical maximum likelihood estimators (MLEs) \(\hat{\theta }_{l} = (\hat{\mu }_{l} ,\hat{\sigma }_{l}^{2} )\) when β = 0.
It is considered that the MLEs of a Gaussian distribution are consistent and asymptotically efficient in absence of outlying objects63. Therefore, in this article, an effort has been provided to develop a robust linear mixed model (LMM) method in which the classical MLEs \(\hat{\theta }_{l}\) are used in absence of outlying objects and minimum β-divergence estimators \(\hat{\theta }_{l,\beta }\) stated in Eq. (5) and (6) are used in presence of outliers for estimation of θl in the mixed model. The minimum β-divergence method suggests two approaches for combining the robustness and efficiency of estimation in LMM. The tuning parameter β is selected through the cross-validation (CV) technique20. CV process produces β = 0 for the minimum β-divergence method estimators and is then equivalent to the classical estimators. When there are outlying subjects in the phenotypic traits, the technique generates β > 0 for the minimum β-divergence estimators. To overcome the challenges of outlying observations in GWAS, an alternative approach that is the β-weight function mentioned in (4) has been proposed with β = 0.2 for outlier detection. This weight function imposes smaller weights (≥ 0) to outlying observations and larger weights (≤ 1) to uncontaminated/usual objects.
An outlying phenotypic observation yli in the lth group is defined based on the β-weight function mentioned below:
where the threshold value τl is the pth quantile value of the empirical distribution of \(W_{\beta } (y_{li} |\hat{\theta }_{l,\beta } )\).
(iv) Then replace the outlying phenotypic observations of lth group by its robust mean \(\mu_{l,\beta }\)(l = 1, 2,…,k), where m is the number of genotype in the selected SNP.
(v) After that apply an efficient mixed-model association (EMMA) to the modified dataset discussed in the previous step.
Simulated data generation
To investigate the performance of the proposed algorithm in a comparison of the conventional algorithms, we generated both the genotypic and phenotypic data as follows:
Genotype simulation
To explore how the proposed method performs, a set of synthetic genotype and phenotype data were generated. A synthetic genotype dataset was simulated that reflects population structure. For this purpose, m* = 2000 SNPs were generated for n = 1000 individuals, and these individuals were taken from k = 3 distinct population by considering different minor allele frequencies (MAFs). To do this, first, a set of latent vectors {v1, v2, …..vm*} was generated from a multivariate normal distribution with mean zero and variance–covariance matrix Cov(vj,vk) = ρ|j-k|64,65. In our simulation, we considered ρ = 0.5 to avoid the linkage disequilibrium (LD) between the SNPs. Finally, two cutoff values s1 and s2 were used to convert the design matrix V = [v1, v2, …, vm* ] = [vij] of latent vectors to the genotypic score matrix \( x_{ij} (i = 1,2,...,n,j = 1,2,...,m_{1} )\,{\text{and }}\,z_{ij} (i = 1,2,...,n;j = 1,2,...,m_{2} )\) as follows:
where s1 and s2 determine the minor allele frequency.
Phenotype simulation
Phenotypic datasets were produced by considering several factors including genetic variation, error variation, and population stratification. To generate phenotype data, two distinct situations were considered in terms of two heritability rates 0.2 and 0.3. In every situation, m1 = 4 SNPs were considered as causal variants and the remaining m2 = m*-m1 = 1996 SNPs were allocated as polygenic variants (effects). The quantitative trait/phenotypic values were simulated using Eq. (1) which can be re-written as
The total phenotypic variation was decomposed by assuming all three sources of variations in Eq. (8) are independent each other as
Then the contribution of main genetic effect in the total phenotypic variation (known as heritability) was defined by the ratio of main genetic variance over the phenotypic variance and it is written as
The genomic outcomes of the SNPs were simulated from a normal distribution such that it satisfies a certain proportion of genetic variance for different genetic effects as given in Table 3.
To check the performance of the proposed method in a comparison of LRM and LMM approaches in presence of different rate of outliers, we contaminated 1%, 2%, 3%, 4%, and 5% of phenotypic data randomly by using theoutlying observations to generate five different contaminated datasets. We replicated these five contaminated datasets including original clean dataset 1000 times. Tthe outlying phenotypic observations was generated \(\left( {\mathop y\nolimits_{j}^{*} } \right)\) satisfying \( {\text{2}} \times {\mathbf{max}}({\mathbf{y}}){\mkern 1mu} < {\text{y}}_{j}^{*} < {\text{5}} \times \max ({\mathbf{y}}) \).
Consequence of phenotypic outliers on the partition of total variations
Let y* be the modified phenotypic response variable which is generated by replacing 5% observation of y by the outlying observations. Then var(y*) > var(y) obviously, which implies
where c > 0. Now we can write the following equation similar to the Eq. (8) as
by assuming the same genetic effects but changed to the error variations. Then
Combining Eq. (9), (11) and (13), we get
Thus, error variance in presence of outliers must be larger than the error variance in absence of outliers. Conversely, the heritability (h2*) in presence of outliers must be smaller than the heritability (h2) in absence of outliers as follows.
Therefore, both LMM and LRM approach losses the SNPs identification power in presence of phenotypic outliers. However, in the case of the proposed method, the error variance in presence of outliers must be almost same to the error variance in absence of outliers. Similarly, the heritability \( \, \mathop h\nolimits_{\beta }^{*} \, \) in presence of outliers must be almost same to the heritability (h2) in absence of outliers, since
where \({\text{var}} \left( {\mathop y\nolimits_{\beta }^{*} } \right)\) is the variance of total phenotypic variations after the preprocessing of phenotypic observations by the proposed method. Therefore, the power of the proposed method must be larger than the power of both LMM and LRM methods in presence of phenotypic outliers.
Performance measures
To investigate the performance, the statistical power and FDR of 3 methods were calculated by using the formula, Power = (PT/PC) × 100 and FDR = [PF/(PT + PF)] × 100, respectively, where PT measures the truly detected SNPs and PC measures total causal variants and PF is the number of falsely detected SNPs. For each situation, 1000 replications were performed to account for the average value of the power and FDR for comparison.
Real genotype and phenotype data on rice flowering time
We applied the proposed rGWAS method to explore the potential SNPs influencing rice flowering time. The genotypic and phenotypic data used to carry out the analysis for investigation were collected from the rice diversity database (www.ricediversity.org). The data set contain 413 accessions along with 36,901 SNPs of Oryza Sativa1. All selected SNPs were taken into consideration in the analysis with call rate > 70% and minor allele frequency (MAF) > 0.051. Missing genotypes were imputed with weighted k-Nearest-Neighbors method66 based on the five weighted nearest varieties. To compute the kinship matrix (using Eq. (2)), LD-pruned set of variants was used with an R2 greater than 0.9 in a 200 variant sliding window of size 1000. The individual with missing observation in the phenotypic dataset was not considered in this study. Experimental data on flowering time were obtained as the number of days until the inflorescence was 50% emerged from the flag leaf calculated from the day of planting. The phenotypic data used in this analysis for the flowering time were recorded at Faridpur district in Bangladesh.
SNP annotation and candidate gene identification
11 SNPs were identified through whole genome association analysis by using rGWAS. Rice Genome Annotation Project (RGAP) Release 7 (http://rice.plantbiology.msu.edu/) database was used to annotate the identified SNPs. Among the identified genes, six were annotated with protein-coding genes and the rest five were non-coding (Supplementary Table S1). For the non-coding SNPs, the nearest genes were considered as candidate gene and used for further functional characterization. We also aligned the significant SNP tags against the genes those involved in rice flowering time and seed development pathway36. Region within 100 kb were searched for the pathway genes (Supplementary Table S2). The QQ-plot were generated using qqmath function in the R package lattice67.
Gene-set enrichment analysis
To characterized the candidate genes/SNPs that may have a significant association with the phenotypic variations, we performed gene ontology (GO) enrichment analysis of the candidate genes in terms of biological process (BP), molecular function (MF), and cellular component (CC) were performed using the on-line tool QuickGO (https://www.ebi.ac.uk/QuickGO). A gene set was considered as significantly enriched for GO terms if \(p<0.05\). The heatmap of the GO corresponding to the candidate genes was plotted using R package ComplexHeatmap68.
Prediction of the subcellular location
An online-based tool called Plant Subcellular Localization Integrative (PSI) predictor69 was used for predicting the subcellular locations of the candidate genes in the plant cell. The predicted subcellular location corresponding to the candidate genes were visualize by using the R package corrplot70.
Tissue-specific expression
We explored the expression profile of the candidate genes in different tissues. Tissue-specific expression of the genes were obtained from the database Rice Genome Annotation Project (RGAP)55. The heatmap of the expression level of the genes was created via R package ComplexHeatmap68.
Data availability
To implement the proposed method, the necessary codes in R can be downloaded from the repository:http://www.ru.ac.bd/biorgru/software/r-code-robustgwas-zip/
Change history
10 November 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41598-021-00399-z
References
Zhao, K. et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. https://doi.org/10.1038/ncomms1467 (2011).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. https://doi.org/10.1086/519795 (2007).
Li, Q. & Yu, K. Improved correction for population stratification in genome-wide association studies by identifying hidden population structures. Genet. Epidemiol. https://doi.org/10.1002/gepi.20296 (2008).
Liu, L., Zhang, D., Liu, H. & Arendt, C. Robust methods for population stratification in genome wide association studies. BMC Bioinformatics https://doi.org/10.1186/1471-2105-14-132 (2013).
Xu, H., Sarkar, B. & George, V. A new measure of population structure using multiple single nucleotide polymorphisms and its relationship with FST. BMC. Res. Notes https://doi.org/10.1186/1756-0500-2-21 (2009).
Campbell, C. D. et al. Demonstrating stratification in a European American population. Nat. Genet. https://doi.org/10.1038/ng1607 (2005).
Devlin, B. & Roeder, K. Genomic control for association studies. Biometrics https://doi.org/10.1111/j.0006-341X.1999.00997.x (1999).
Pritchard, J. K., Stephens, M., Rosenberg, N. A. & Donnelly, P. Association mapping in structured populations. Am. J. Hum. Genet. https://doi.org/10.1086/302959 (2002).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. https://doi.org/10.1038/ng1847 (2006).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. https://doi.org/10.1371/journal.pgen.0020190 (2006).
Aranzana, M. J. et al. Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet. https://doi.org/10.1371/journal.pgen.0010060 (2005).
Devlin, B., Roeder, K. & Wasserman, L. Genomic control, a new approach to genetic-based association studies. Theor. Popul. Biol. https://doi.org/10.1006/tpbi.2001.1542 (2001).
Yu, J. et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. https://doi.org/10.1038/ng1702 (2006).
Zhao, K. et al. An Arabidopsis example of association mapping in structured samples. PLoS Genet. https://doi.org/10.1371/journal.pgen.0030004 (2007).
Hyun, M. K. et al. Efficient control of population structure in model organism association mapping. Genetics https://doi.org/10.1534/genetics.107.080101 (2008).
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. https://doi.org/10.1038/ng.548 (2010).
Endelman, J. B. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome J. https://doi.org/10.3835/plantgenome2011.08.0024 (2011).
Lipka, A. E. et al. GAPIT: Genome association and prediction integrated tool. Bioinformatics https://doi.org/10.1093/bioinformatics/bts444 (2012).
Tang, Y. et al. GAPIT Version 2: an enhanced integrated tool for genomic association and prediction. Plant Genome https://doi.org/10.3835/plantgenome2015.11.0120 (2016).
Mollah, M. N. H., Eguchi, S. & Minami, M. Robust prewhitening for ICA by minimizing β-divergence and its application to FastICA. Neural Process. Lett. https://doi.org/10.1007/s11063-006-9023-8 (2007).
Mollah, M. N. H. & Eguchi, S. Robust QTL analysis by minimum β-divergence method. Int. J. Data Min. Bioinform. https://doi.org/10.1504/ijdmb.2010.034199 (2010).
Ahsan, A. et al. Identification epistasis loci underlying rice flowering time by controlling population stratification and polygenic effect. DNA Res. https://doi.org/10.1093/dnares/dsy043 (2018).
Beasley, T. M., Erickson, S. & Allison, D. B. Rank-based inverse normal transformations are increasingly used, but are they merited?. Behav. Genet. https://doi.org/10.1007/s10519-009-9281-0 (2009).
McCaw, Z. R., Lane, J. M., Saxena, R., Redline, S. & Lin, X. Operating characteristics of the rank-based inverse normal transformation for quantitative trait analysis in genome-wide association studies. Biometrics https://doi.org/10.1111/biom.13214 (2020).
Jiang, W. & Yu, W. Power estimation and sample size determination for replication studies of genome-wide association studies. BMC Genom. https://doi.org/10.1186/s12864-015-2296-4 (2016).
Wang, M. & Xu, S. Statistical power in genome-wide association studies and quantitative trait locus mapping. Heredity https://doi.org/10.1038/s41437-019-0205-3 (2019).
Hong, E. P. & Park, J. W. Sample size and statistical power calculation in genetic association studies. Genom. Inf. https://doi.org/10.5808/gi.2012.10.2.117 (2012).
Sharma, M. & Pandey, G. K. Expansion and function of repeat domain proteins during stress and development in plants. Front. Plant Sci. https://doi.org/10.3389/fpls.2015.01218 (2016).
Liu, Y. J. et al. A plastid-localized pentatricopeptide repeat protein is required for both pollen development and plant growth in rice. Sci. Rep. https://doi.org/10.1038/s41598-017-10727-x (2017).
Emami, H. & Kempken, F. PRECOCIOUS1 (POCO1), a mitochondrial pentatricopeptide repeat protein affects flowering time in Arabidopsis thaliana. Plant J. https://doi.org/10.1111/tpj.14441 (2019).
Rose, A. Genome-wide identification of arabidopsis coiled-coil proteins and establishment of the ARABI-COIL database. Plant Physiol. https://doi.org/10.1104/pp.103.035626 (2004).
Nonomura, K. I. et al. The novel gene homologous pairing aberration in rice Meiosis1 of rice encodes a putative coiled-coil protein required for homologous chromosome pairing in meiosis. Plant Cell https://doi.org/10.1105/tpc.020701 (2004).
Ito, H. et al. A stress-activated transposon in arabidopsis induces transgenerational abscisic acid insensitivity. Sci. Rep. https://doi.org/10.1038/srep23181 (2016).
Ito, H. et al. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature https://doi.org/10.1038/nature09861 (2011).
Craufurd, P. Q. & Wheeler, T. R. Climate change and the flowering time of annual crops. J. Exp. Bot. https://doi.org/10.1093/jxb/erp196 (2009).
Hanumappa, M. et al. WikiPathways for plants: a community pathway curation portal and a case study in rice and arabidopsis seed development networks. Rice https://doi.org/10.1186/1939-8433-6-14 (2013).
Harris, M. A. et al. The gene ontology project in 2008. Nucleic Acids Res. https://doi.org/10.1093/nar/gkm883 (2008).
Harris, M. A. et al. The gene oncology (GO) database and informatics resource. Nucleic Acids Res. https://doi.org/10.1093/nar/gkh036 (2004).
Huang, X. et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. https://doi.org/10.1038/ng.1018 (2012).
Cho, L. H., Yoon, J. & An, G. The control of flowering time by environmental factors. Plant J. https://doi.org/10.1111/tpj.13461 (2017).
Lee, Y. S. & An, G. Regulation of flowering time in rice. J. Plant Biol. https://doi.org/10.1007/s12374-015-0425-x (2015).
Zhou, Y. et al. Genetic control of seed shattering in rice by the APETALA2 transcription factor Shattering Abortion1. Plant Cell https://doi.org/10.1105/tpc.111.094383 (2012).
Zhang, C., Bai, M. & Chong, K. Brassinosteroid-mediated regulation of agronomic traits in rice. Plant Cell Rep. https://doi.org/10.1007/s00299-014-1578-7 (2014).
Mori, M. et al. Isolation and characterization of a rice dwarf mutant with a defect in brassinosteroid biosynthesis. Plant Physiol. https://doi.org/10.1104/pp.007179 (2002).
Clouse, S. D., Langford, M. & McMorris, T. C. A brassinosteroid-lnsensitive mutant in arabidopsis thaliana exhibits multiple defects in growth and development. Plant Physiol. https://doi.org/10.1104/pp.111.3.671 (1996).
Divi, U. K. & Krishna, P. Brassinosteroid: a biotechnological target for enhancing crop yield and stress tolerance. New Biotechnol. https://doi.org/10.1016/j.nbt.2009.07.006 (2009).
Yamamuro, C. et al. Loss of function of a rice brassinosteroid insensitive1 homolog prevents internode elongation and bending of the lamina joint. Plant Cell https://doi.org/10.1105/tpc.12.9.1591 (2000).
Smith, E. & Morowitz, H. J. Universality in intermediary metabolism. Proc. Natl. Acad. Sci. U.S.A. https://doi.org/10.1073/pnas.0404922101 (2004).
Smith, R. L., Soeters, M. R., Wüst, R. C. I. & Houtkooper, R. H. Metabolic flexibility as an adaptation to energy resources and requirements in health and disease. Endocr. Rev. https://doi.org/10.1210/er.2017-00211 (2018).
Kholodenko, B. N. Four-dimensional organization of protein kinase signaling cascades: the roles of diffusion, endocytosis and molecular motors. J. Exp. Biol. https://doi.org/10.1242/jeb.00298 (2003).
Ohlrogge, J. B., Kuhn, D. N. & Stumpf, P. K. Subcellular localization of acyl carrier protein in leaf protoplasts of Spinacia oleracea. Proc. Natl. Acad. Sci. U.S.A. https://doi.org/10.1073/pnas.76.3.1194 (1979).
Jansen, R. K. et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. https://doi.org/10.1016/S0076-6879(05)95020-9 (2005).
Hidayati, N., Triadiati & Anas, I. Photosynthesis and transpiration rates of rice cultivated under the system of rice intensification and the effects on growth and yield. HAYATI J. Biosci. (2016). doi:https://doi.org/10.1016/j.hjb.2016.06.002
Karki, S., Rizal, G. & Quick, W. P. Improvement of photosynthesis in rice (Oryza sativa L.) by inserting the C4 pathway. Rice (2013). doi:https://doi.org/10.1186/1939-8433-6-28
Kawahara, Y. et al. Improvement of the oryza sativa nipponbare reference genome using next generation sequence and optical map data. Rice https://doi.org/10.1186/1939-8433-6-4 (2013).
Weng, X. et al. Grain number, plant height, and heading date7 is a central regulator of growth, development, and stress response. Plant Physiol. https://doi.org/10.1104/pp.113.231308 (2014).
Shivani et al. Genome-wide analysis of transcription factors during somatic embryogenesis in banana (Musa spp.) cv. Grand Naine. PLoS ONE (2017). doi:https://doi.org/10.1371/journal.pone.0182242
Pasam, R. K. et al. Genome-wide association studies for agronomical traits in a world wide spring barley collection. BMC Plant Biol. https://doi.org/10.1186/1471-2229-12-16 (2012).
Assaad, F. F., Huet, Y., Mayer, U. & Jürgens, G. The cytokinesis gene KEULE encodes a Sec1 protein that binds the syntaxin KNOLLE. Journal of Cell Biology (2001).
Kennedy, B. W., Quinton, M. & van Arendonk, J. A. Estimation of effects of single genes on quantitative traits. J. Anim. Sci. https://doi.org/10.2527/1992.7072000x (1992).
Nurul Haque Mollah, M., Sultana, N., Minami, M. & Eguchi, S. Robust extraction of local structures by the minimum β-divergence method. Neural Networks (2010). doi:https://doi.org/10.1016/j.neunet.2009.11.011
Mollah, M. M. H., Jamal, R., Mokhtar, N. M., Harun, R. & Mollah, M. N. H. A hybrid one-way ANOVA approach for the robust and efficient estimation of differential gene expression with multiple patterns. PLoS ONE https://doi.org/10.1371/journal.pone.0138810 (2015).
Zhang, B., Kirov, S. & Snoddy, J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. https://doi.org/10.1093/nar/gki475 (2005).
Wang, K. & Abbott, D. A principal components regression approach to multilocus genetic association studies. Genet. Epidemiol. https://doi.org/10.1002/gepi.20266 (2008).
Li, J., Zhong, W., Li, R. & Wu, R. A fast algorithm for detecting gene-gene interactions in genome-wide association studies. Ann. Appl. Stat. https://doi.org/10.1214/14-AOAS771 (2014).
Schwender, H. Imputing missing genotypes with weighted k nearest neighbors. J. Toxicol. Environ. Health Part A Curr. Issues https://doi.org/10.1080/15287394.2012.674910 (2012).
Sarkar, D. Package ‘lattice’: Trellis Graphics for R. (2017). http://lattice.r-forge.r-project.org/
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics https://doi.org/10.1093/bioinformatics/btw313 (2016).
Liu, L., Zhang, Z., Mei, Q. & Chen, M. PSI: a comprehensive and integrative approach for accurate plant subcellular localization prediction. PLoS ONE https://doi.org/10.1371/journal.pone.0075826 (2013).
Wei, T. et al. R package ‘corrplot’: Visualization of a Correlation Matrix. (2017). https://github.com/taiyun/corrplot
Acknowledgements
We are very grateful to Bangladesh Agricultural Research Institute and Bioinformatics Lab, Department of Statistics, University of Rajshahi to provide us the opportunity to conduct this research using lab facilities. Munirul Alam of icddr,b thanks to the Governments of Bangladesh, Canada, Sweden, and the United Kingdom for core/unrestricted support.
Author information
Authors and Affiliations
Contributions
N.H.M. Conceptualize the proposed method. Z.A., M.A.A. and N.H.M. worked together to develop the algorithm. Z.A. and M.A.A. analyzed the data and drafted the manuscript. N.H.M. and M.A. critically revised this manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained errors in the Equations. Modifications have been made to Table 1, Table 3 and the Materials and methods section. Full information regarding the corrections made can be found in the correction for this Article.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Akond, Z., Ahsan, M.A., Alam, M. et al. Robustification of GWAS to explore effective SNPs addressing the challenges of hidden population stratification and polygenic effects. Sci Rep 11, 13060 (2021). https://doi.org/10.1038/s41598-021-90774-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90774-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
