
Abstract
An exceptional phenomenon has been observed that SO2 and NOx in flue gas can be effectively adsorbed over activated carbon with a surprising capacity at cold temperatures with the presence of oxygen. In this study, the adsorption characteristics of NO and SO2 over activated carbon at 80, 20, 0, and − 20 is experimentally investigated. Without the presence of oxygen, adsorption of NO is negligible. In the presence of oxygen, NO can be oxidized to NO2 over activated carbon which leads to the co-adsorption of NO/NO2 within the adsorption bed. Catalytic oxidation of NO over activated carbon can be significantly enhanced at cold temperatures, leading to an extraordinary increase of adsorption capacity of NO. With an initial concentration of NO = 200 ppmv and a space velocity of 5000 h−1, the average specific capacity increases from 3.8 to 169.1 mg/g when the temperature decreases from 80 to – 20 ℃. For NO–O2 co-adsorption, the specific capacity increases along the adsorption bed due to the increasing NO2 concentrations. The adsorption capacity of SO2 is also significantly enhanced at cold temperatures. With an initial concentration of SO2 = 1000 ppmv, the specific capacity increases from 12.9 to 123.1 mg/g when the temperature decreases from 80 to – 20 ℃. A novel low-temperature adsorption (LAS) process is developed to simultaneously remove SO2 and NOx from flue gas with a target of near-zero emission. A pilot-scale testing platform with a flue gas flowrate of 3600 Nm3/h is developed and tested. Emission of both SO2 and NOx is less than 1 ppmv, and the predicted energy penalty is about 3% of the net generation.
Similar content being viewed by others


Introduction
SO2 and NOx in flue gas are major air pollutants responsible for acid rain and photochemical smog. SO2 is an acidic gas and can be scrubbed by alkalic solutions such as lime, sodium carbonate, ammonia, etc1,2. Seawater is also considered as a scrubbing agent for desulfurization3,4. NOx is composed of various forms of nitrogen oxides such as NO, NO2, N2O, etc. The dominant species is nitrogen monoxide (NO) which can be either reduced to N2 by selective catalytic reduction or oxidized to NO2 which can be scrubbed by alkalic solutions5. Wet flue gas desulfurization (WFGD) and SCR dinitrification are the dominant technologies in power plants nowadays.
In addition, adsorption technology has also been widely used for gas cleanup. Simultaneous removal of SO2 and NOx by activated carbon or coke has been successfully demonstrated in flue gas treatment6,7. An schematic drawing of the process is shown in Fig. 1.

Schematic drawing of the activated coke desulfurization and denitrification process.
Activated coke technology can remove SO2, NOx, Hg and other adsorbable pollutants simultaneously8. The removal of SO2 over activated carbon in the presence of oxygen and moisture involves a series of reactions that leads to the formation of sulfuric acid. The used carbon is regenerated through heating to recover their adsorbing activity. The desorbed SO2 is recycled as elemental sulfur, sulfuric acid or liquid SO2, as shown in Fig. 1. And the desorbed Hg and other trace contaminant species can be separated and collected during the sulfuric acid or sulfur production process. The typical operating temperature is 80–150 ℃ in the adsorber and 350–450 ℃ in the regenerator. The overall adsorption and desorption reactions are as follows9:
For activated coke technology, NOx is not removed by adsorption. It is removed through catalytic reduction over activated carbon by reacting with injecting NH3, which converts NOx to non-toxic gas N2. The overall reactions are as follows10:
Activated coke technology has been widely used for sintering flue gas treatment in iron and steel industry11, but only a few applications in power plants have been reported12. Power plant flue gas normally has a much higher concentration of SO2 and larger flowrate than sintering flue gas, the specific capacity of SO2 is not sufficient enough to treat the power plant flue gas. To improve the adsorption capacity, researchers have also investigated the possibility of using other adsorbents such as modified activated carbon13,14, activated carbon fibers15, molecular sieve16,17, alumina substrate impregnated with sodium carbonate18, copper oxide19, etc. But none of these adsorbents has been successfully commercialized.
Another defective feature of traditional activated coke technology is that NOx is not able to be removed effectively through adsorption and the injection of ammonia is required to improve the denitrification rate. The removal rare of NOx is less than 20% without NH3 and can be increased to around 70% if sufficient NH3 is injected7,20.
This study proposed a novel low-temperature adsorption (LAS) technology which is able to remove both SO2 and NOx through adsorption with extraordinary adsorption capacity and high efficiency. The development of LAS technology is inspired by an interesting phenomenon observed accidentally that NOx can be adsorbed by activated carbon effectively with an astonishing capacity when flue gas is cooled to cold temperatures. The fundamental behaviors of NO and SO2 adsorption at cold temperatures over activated carbon is investigated in this study. Furthermore, a pilot-scale testing platform with a flue gas treatment capacity of 3600 Nm3/h is designed and built to validate the performance of LAS technology. A brief introduction and first-hand data from the pilot-scale testing facility is also shared in this study. But the detail results from the pilot tests will be discussed in the future.
A comparison between the LAS technology and traditional activated coke technology is given in Table 1. The specific capacity is obtained in this study. The removal rate of traditional activated coke technology is from reported literature7 and the removal rate of LAS technology is obtained from the pilot platform.
Method
Material
Commercial coconut activated carbon (CAC) with granular size between 26 and 30 mesh is used in this study. The surface physical properties of CAC is characterized by BET method (Quantachrome QUDRASORB SI). The specific surface area of CAC is 1314.5 m2/g, the pore size is mainly smaller than 2 nm, as shown in Fig. 2, in which DFT method is used to determine the pore size distribution. CAC is pretreated by heating to 300 ℃ in an vacuum tube for 2 h before each test. Mass of CAC is measured after the pretreatment. The loading density of granular CAC is 0.5 g/cm3.

Pore distribution of coconut activated carbon (V: pore volume; dV: reciprocal of volume).
Experimental setup
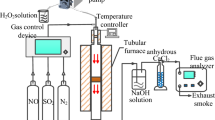
Figure 3 is a schematic drawing of the experimental setup for investigating the adsorption behaviors of SO2 and NO at cold temperatures. The dry flue gas has a volume flow rate of Q = 1 L/min, and has a volume concentration of N2 = 82%, O2 = 6%, CO2 = 12%, SO2 = 1000 ppmv and NO = 200 ppmv. Moisture in flue gas is added by a water injector controlled by a stepper motor. At 80 ℃, 10 vol.% of H2O is added to the flue gas. At 20, 0 and – 20 ℃, the concentration of H2O is reduced to the saturated moisture content at corresponding temperatures, which are 2.3, 0.6 and 0.1 vol.%, respectively. The is because the extra moisture will be removed from the flue gas during cooling process. The flue gas in pre-heated or pre-cooled to adsorption temperature by a coiled copper pipe immersing in a thermostatic bath with a temperature range of – 40 to 100 ℃. The granular activated carbon is loaded in a glass tube with an inner diameter of 5 mm which is also immersed in the thermostatic both. The difference between bath temperature and the gas temperature at the exit of adsorption bed is less than 2 ℃. During each test, 6 g of CAC is loaded and the space velocity of adsorption bed is 5000 h−1. The gas composition of flue gas leaving the adsorption tube is measured by flue gas analyzer testo 350. The gas analyzer is capable of measuring O2, CO2, SO2, NO and NO2. The method, accuracy and resolution of the analyzer for each component is given in Table 2.

Experimental setup for low-temperature adsorption of SO2 and NO.
Pilot platform
A pilot-scale test platform is designed to simultaneously remove SO2, NOx and other adsorbable pollutants based on the novel low-temperature adsorption technology. The designed flue gas flowrate is 3600 Nm3/h. The adsorption temperature is between − 20 to 5 ℃. The pollutants control target is near-zero emission: SO2 and NOx ≦ 1 ppmv.
A schematic flowchart of the pilot-scale test platform is shown in Fig. 4. Flue gas is extracted from the inlet duct before SCR denitrification system. Hot flue gas is cooled to around 120 ℃ by an air preheater and the dust is then removed by a bag-type dust remover. Flue gas is further cooled to around 70 ℃ by a residue heat recovery exchanger (HX1) which can generate usable hot water. Flue gas is cooled to cold temperatures by a direct contact cooling (DCC) tower which has three cooling stages. In the lower stage (stage 1), flue gas is cooled close to room temperature by water scrubbing and the cooling load is provided by cooling water; in the second stage (stage 2), flue gas is cooled to 2–5 ℃ by cold water scrubbing and the cooling load is provided by an industrial chiller; in the upper stage (stage 3), flue gas is cooled to below freezing point by calcium chloride solution scrubbing and the cooling load is provided by an industrial refrigerator. The cold flue gas enters the adsorber in which SO2 and NOx are removed by activated carbon through adsorption. The cold energy of clean flue gas is recovered by cooling the scrubbing water in the DCC cooling tower through HX2. The saturated activated carbon is heated in the regenerator to recover its adsorption activity for repeatable utilization. The desorbed SO2 can be recovered as elemental sulfur or sulfuric acid, and the desorbed NOx can be introduced to the boiler to form a stable thermal balance of NOx–N2–O2, which has been successfully demonstrated in the NOx SO process18. In this pilot study however, the post-treatment of desorbed gas is not considered.

Schematic flowchart of pilot-scale test platform.
Results and discussion
Phenomenon
This section is to introduce an interesting observation on the adsorption of SO2 and NO at cold temperatures, which to our knowledge has not yet been reported in open literature. This phenomenon is the origin of this study as well as the development of LAS technology.
The adsorption characteristics of SO2 and NO at 80, 20 and − 20 ℃ is investigated by the experimental setup shown in Fig. 3. The simulated flue gas has a composition described in section “Experimental setup”. During each test, 6 g of CAC is loaded in the tube and the space velocity is 5000 h−1. The concentration of SO2 and NO is 1000 and 200 ppmv, respectively. The dry flue gas has a volume flow rate of 1 L/min. The water content is 10 vol.% at 80 ℃, 2.3 vol.% (saturated) at 20 ℃, 0.1 vol.% (saturated) at – 20 ℃, respectively.
Figure 5 is the breakthrough curve of SO2 (blue) and NO (red) of the three experiments. The concentrations of SO2 and NO leaving the adsorption bed (Cout) are measured and plotted. Since the gas analyzer has a resolution of 1 ppmv for both SO2 and NO, the breakthrough time (tb, min) is defined as the time when Cout(SO2) = 1 ppmv and Cout(NO) = 1 ppmv. The breakthrough adsorption capacity (Ab, mg/g) is defined as the adsorption capacity which can maintain near-zero emission (less than 1 ppmv). The saturated adsorption capacity (As, mg/g) is defined as the maximum adsorption capacity when the bed stops adsorption. These two specific capacities are key parameters which determine the loading space velocity of adsorption bed and the frequency of bed regeneration.

Breakthrough curves of SO2 (blue) and NO (red) adsorption at 80, 20 and − 20℃ (Simulated flue gas: SO2 = 1000 ppmv, NO = 200 ppmv, O2 = 6 vol.%, CO2 = 12 vol.%, space velocity = 5000 h−1).
The breakthrough and saturated adsorption capacity are calculated by the following equations:
where Ab and As are the breakthrough and saturated adsorption capacity (mg/g), respectively. tb(min) is the breakthrough time. Q (L/min) is the flow rate of simulated flue gas, which is 1 L/min. M(g/mol) is the molecular weight of SO2 or NO. m(g) is the mass of loaded CAC, which is 6 g when the space velocity is 5000 h−1. Cin (ppmv) is the inlet concentration, which is 1000 ppmv for SO2 and 200 ppmv for NO. Cout (t) is the outlet concentration of SO2 or NO at time t(min). In the time integral of Eq. (6), t0 is the starting time and te is ending time when the bed stops adsorption (Cout = Cin).
As shown in Fig. 5, NO breaks through the adsorption bed almost instantaneously at an adsorption temperature of 80 ℃ which is close to the operating temperature of traditional activated coke technology. The breakthrough time of NO is about 5 min at 20 ℃ and 225 min at – 20 ℃. The breakthrough time of SO2 is 20, 68 and 295 min at 80, 20 and – 20 ℃, respectively. The specific capacity of SO2 and NO calculated by Eqs. (5) and (6) is given in Table 3.
Based on the observation from Fig. 5 and Table 3, it is found that the specific capacity of both NO and SO2 increases substantially with decreasing temperature. At above room temperature, the specific capacity of NO is less than 1 mg/g, which is insufficient for adsorption removal. At – 20 ℃, the specific capacity of NO increases to 13.24 mg/g, and the removal of NOx from flue gas through adsorption becomes possible. At above room temperatures, the breakthrough time of NO is much shorter than that of SO2. At – 20 ℃, although the specific capacity of NO is far smaller than that of SO2, the breakthrough time of NO is close to that of SO2. This is because the concentration of SO2 is much higher than NO in the flue gas. Therefore, it is technically feasible to remove SO2 and NOx by adsorption at cold temperatures.
The above experiments reveal the phenomenon of simultaneous adsorption of NO and SO2 in simulated flue gas. To understand the mechanism of NO and SO2 adsorption at cold temperatures, the adsorption of NO and SO2 are studied and discussed separately in sections “Adsorption of NO” and “Adsorption of SO2”.
Adsorption of NO
Figure 6 shows the breakthrough curves of the NO adsorption over CAC at 80, 20, 0 and − 20 ℃. 6 g of pretreated CAC is loaded in the adsorption tube and the space velocity is 5000 h−1. The inlet gas has flow rate of 1 L/min, and is composed of NO and N2, with an concentration of Cin (NO) = 200 ppmv. The concentration of NO at the outlet of adsorption bed is measured and plotted in Fig. 6. The breakthrough time and adsorption capacity of NO adsorption is given in Table 4. The physisorption (Van der Waals adsorption) of NO over activated carbon is enhanced by decreasing the adsorption temperature since physisorption is an exothermic process21. Without the presence of oxygen, the specific capacity of NO physisorption is less than 0.5 mg/g even at – 20 ℃.

Breakthrough curve of NO adsorption at 80, 20, 0 and − 20℃ (NO = 200 ppmv, space velocity = 5000 h−1).
Figure 7 shows the breakthrough curves of the NO-O2 co-adsorption over CAC at 80, 20, 0 and − 20 ℃. 6 g of pretreated CAC is loaded in the adsorption tube and the space velocity is 5000 h−1. The inlet gas has flow rate of 1 L/min, and is composed of NO (200 ppmv), O2 (6 vol.%) and N2. With the presence of oxygen, NO can be oxidized to NO2 over activated carbon22,23, a steady NO–NO2 equilibrium will be formed at the exit of activated carbon bed24,25. Therefore, the concentration of both NO and NO2 at the outlet of adsorption bed is measured and plotted in Fig. 7. At 80 ℃, NO (red) breakthrough the adsorption bed within a few minutes and reaches a steady concentration after about 400 min, and about 2% of NO is oxidized to NO2. At 20 ℃, NO (red) breakthrough the bed after about 56 min, and NO2 (blue) is detected after about 1600 min. After about 3500 min, both NO and NO2 reaches a steady concentration and the oxidation rate of NO is about 45%. When adsorption temperature is lowered to 0 ℃ and − 20℃, the breakthrough time of NO increases to 392 and 1591 min, respectively. Meanwhile, the oxidation rate of NO increases to 69% and 91.5%, respectively. For each test, the sum of NO and NO2 concentration is 200 ppmv when the steady state is reached.

Breakthrough curves of NO (red) and NO2 (blue) during NO–O2 co-adsorption at 80, 20, 0 and – 20 ℃ (NO = 200 ppmv, O2 = 6 vol.%, space velocity = 5000 h−1).
The adsorption capacity of NO–O2 co-adsorption at various temperatures is given in Table 4. Both NO and NO2 has a breakthrough time during NO–O2 co-adsorption, as shown in Fig. 7. The breakthrough time for NO–O2 co-adsorption is defined as the breakthrough of NO which always occurs first. The breakthrough adsorption capacity Ab is calculated by Eq. (5) and the saturated adsorption capacity As is calculated by Eq. (6), where Cout (t) = Cout (NOx) = Cout (NO) + Cout (NO2) in the case of NO–O2 co-adsorption.
As shown in Table 4, the presence of oxygen can significantly increase the breakthrough time and specific capacity of NO. With presence of oxygen, NO can be oxidized to NO2 which is a much more adsorbable species over activated carbon26,27. The oxidation rate η(NO2) is calculated by the following equation:
where Cout (NO2) is the concentration of NO2 when it reaches steady state. Cin (NO) is the inlet NO concentration. The oxidation rate of NO at 80, 20, 0 and – 20 ℃ is given in Table 4 as well. With the presence of oxygen, the catalytic oxidation of NO is significantly enhanced by at sub-zero temperatures. The mechanism of NO oxidation over activated carbon is complicated, involving both surface reactions and gaseous reactions. The oxidation of gaseous NO by adsorbed oxygen over the activate surface site is believed to be the dominant pathway26.
where C( ) represents the activated carbon with active surface site. Based on reaction (9), the oxidation reaction rate can be calculated by the following equation:
Temperature can impact the oxidation reactions in many ways. First of all, the physisorption of oxygen over activated carbon is enhanced and the concentration of C(O) is increased by reducing adsorbing temperature28. Secondly, the rate constant k(T) of NO oxidation increases with decreasing temperature29.
As shown in Table 4, temperature has a significant impact on the breakthrough time and specific capacity of NO–O2 co-adsorption. When the adsorption temperature decreases from 80 to – 20 ℃, the breakthrough time increases from 3.5 to 1591 min, and the saturated capacity increases from 3.8 to 169 mg/g. This extraordinary increase of specific capacity is due to the NO2 adsorption.
Figure 8 shows the breakthrough curves of the NO–O2 co-adsorption over CAC with various loading space velocity. In Fig. 8a, 0.25 g CAC is loaded and the space velocity is 120,000 h−1; in Fig. 8b, 0.5 g CAC is loaded and the space velocity is 60,000 h−1; in Fig. 8c, 1 g CAC is loaded and the space velocity is 30,000 h−1; in Fig. 8d, 2 g CAC is loaded and the space velocity is 15,000 h−1; in Fig. 8e, 4 g CAC is loaded and the space velocity is 7500 h−1; in Fig. 8f, 6 g CAC is loaded and the space velocity is 5000 h−1. All six experiments are conducted at – 20 ℃. The inlet gas has flow rate of 1 L/min, and is composed of NO (200 ppmv), O2 (6 vol.%) and N2.

Breakthrough curves of NO (red) and NO2 (blue) during NO–O2 co-adsorption at various cross sections of adsorption bed: (a) 120,000 h−1, (b) 60,000 h−1, (c) 30,000 h−1, (d) 15,000 h−1, (e) 7500 h−1 and (f) 5,000 h−1 (NO = 200 ppmv, O2 = 6 vol.%, T = − 0℃).
The purpose of conducting these six independent experiments is to mimic the experiment of simultaneously motoring NO and NO2 at six different cross sections of a single adsorption bed which is difficult to achieve in our experimental setup.
The average adsorption capacity and oxidation rate along the axial direction of the CAC bed are given in Fig. 9 and Table 5. As shown in Fig. 9, the NO2 concentration increases along the bed, indicating that the oxidation rate increases along the axial direction of the adsorption bed. At each cross section of the adsorption bed, a stable NO–NO2 equilibrium is formed both in the gas phase and the adsorption surface. Therefore the NO–O2 co-adsorption mechanism involves the adsorption of both NO and NO2 over activated carbon. Since NO2 is a much more adsorbable species than NO30, the adsorption capacity increases with the increasing NO2 concentration along the axial direction of activated carbon bed, as shown in Fig. 9. This is quite different with the adsorption of SO2 which has an uniform adsorption capacity along the adsorption bed. Due to this distinct characteristics, it should be noted that the adsorption capacity given in Fig. 9 and Table 5 should be defined as average adsorption capacity of a specific CAC bed. The real adsorption capacity at each cross section should be larger than the average value.

Oxidation and adsorption characteristics of NO–O2 along activated carbon bed (NO = 200 ppmv, O2 = 6 vol.%, T = − 20 ℃, space velocity = 5000 h−1 when thickness is 60 cm).
Based on the above analysis, it is now quite clear why the breakthrough time and adsorption capacity of NO–O2 co-adsorption increases dramatically when the adsorption temperature is lowered to below room temperatures. At cold temperatures, the catalytic oxidation of NO is fastened, therefore the breakthrough time and breakthrough adsorption capacity are largely increased. In addition, the adsorption capacity of NO2 increases significantly at cold temperatures, leading to prominent increase of saturated adsorption capacity.
Adsorption of SO2
Figure 10 shows the breakthrough curves of the SO2 adsorption over CAC at 80, 20, 0 and – 20 ℃. 6 g of pretreated CAC is loaded in the adsorption tube and the space velocity is 5000 h−1. The inlet gas has flow rate of 1 L/min, and is composed of SO2 (1000 ppmv), O2 (6 vol.%) and N2. The concentration of SO2 at the outlet of adsorption bed is measured and plotted in Fig. 10. Results indicate that the breakthrough time and specific capacity of SO2 adsorption increases with decreasing temperature. When the temperature decreases from 80 to – 20 ℃, the breakthrough time and adsorption capacity increase by about 13 and 10 times, respectively. SO2 adsorption over activated carbon is fast and the breakthrough adsorption capacity is quite close to the saturated adsorption capacity. The breakthrough curve has a sharp slope which is quite different with that of NO adsorption.

Breakthrough curve of SO2 adsorption over CAC at various temperatures (SO2 = 1000 ppmv, O2 = 6 vol.%, flow rate = 1 L/min, space velocity = 5000 h−1).
The impact of O2, CO2 and H2O on the adsorption of SO2 at various temperatures are also investigated. As shown in Table 6, the presence of oxygen has a slight improvement of SO2 adsorption due to the catalytic oxidation over activated carbon. The presence of CO2 in the opposite has a negative impact on the SO2 adsorption due to the occupation of active carbon surface. The presence of H2O and O2 can enhance the adsorption of SO2 through H2SO4 adsorption9.
The impact of space velocity of CAC load on the adsorption of SO2 is shown in Fig. 11. The breakthrough time is doubled when the CAC load is doubled. This indicates that the adsorption capacity (mg/g CAC) is irrelevant with space velocity and is a constant value at certain temperature and partial pressure of SO2. This is also quite different with the adsorption capacity of NO (with the presence of O2) which increases along the adsorption bed as shown in Fig. 9.

Breakthrough curve of SO2 adsorption over CAC at various space velocity (SO2 = 1000 ppmv, O2 = 6 vol.%, T = − 20 ℃, flow rate = 1 L/min).
The adsorption process of SO2 and NO with the presence of oxygen over activated carbon is illustrated in Fig. 12. There are three regions during the SO2 adsorption process (upleft). In the saturated region, an equilibrium of [SO2(g), SO2(a)] is established. Where (g) present gaseous phase and (a) represent adsorbed phase. In the adsorption region, SO2(g) is being adsorbed and converted to SO2(a). Since the adsorption of SO2 is fast, the adsorption region is within a narrow region. In the fresh carbon region, both SO2(g) and SO2(a) are zero. When the adsorption bed reaches the saturated status (upright), a homogeneous equilibrium of SO2 (g) and SO2 (a) is established cross the entire adsorption bed:
For NO + O2 adsorption (lower left), due to the catalytic oxidation, equilibriums of [NO(g), NO(a)] and [NO2(g), NO2(a)] co-exist, with increasing NO2 and decreasing NO along the bed. Since the adsorption of NO is almost neglectable compared with NO2, the total adsorption capacity increases along the adsorption bed in the saturated region. In the adsorption region, the remaining NO is further oxidized to NO2 and adsorbed. Since the adsorption rate is limited by oxidation rate, the adsorption of NO + O2 is much slower than SO2 and the adsorption region is much wider. When it is reaches saturated status, equilibrium of [NO(g), NO(a)] with decreasing partial pressure and equilibrium of [NO2(g), NO2(a)] with increasing partial pressure are established along the adsorption bed. At each cross section of the bed, the following equilibriums co-exist:

Adsorption process of SO2 (red) and NO (blue) with presence of oxygen over activated carbon.
Pilot test
The pilot test platform is built in Huaneng Yueyang Power Plant and is accomplished in September 2020. A detail description of the process is given in section “Pilot platform”. A picture of the pilot test platform is shown in Fig. 13. A preliminary low-temperature adsorption test is conducted in October. The flue gas flow rate is 3600 Nm3/h and the operating temperature is − 15 to − 20 ℃. Figure 14 is simplified process diagram of the pilot system. Figure 15 shows the inlet and outlet flue gas composition monitored by online CEMS system. Figures 16 and 17 shows some of the operating data of a successive 72 h test. The inlet flue gas has an SO2 concentration of around 500–1000 ppmv and NOx concentration of around 70–100 ppmv. The concentrations of both SO2 and NOx are reduced to below 1 ppmv when leaving the adsorber. More tests are undergoing and will be shared as soon as the data are unclassified. In addition, the performance and energy penalty are evaluated by conducting Aspen Plus modeling, and the energy penalty is about 2–3% of the total net power generation depending on the ambient temperatures. The detail data of the pilot test and the modeling work will be presented and discussed shortly.

3D model (left) and a photo (right) of the pilot test platform.

Process diagram of the low-temperature adsorption system.

Inlet and outlet flue gas composition monitored by online CEMS system.

SO2 (red) and NOx (blue) concentration of inlet flue gas during 72-h operation.

SO2 (red) and NOx (blue) concentration of outlet flue gas during 72-h operation.
Conclusion
In this study, the oxidation and adsorption characteristics of NO over activated carbon at cold temperatures is investigated. With the presence of oxygen, both the oxidation rate and adsorption of NO over activated carbon is enhanced significantly at cold temperatures. The breakthrough time of NO increases from 3.45 to 1591.75 min when the adsorption temperature decreases from 80 to −20 ℃. At each cross sections along the adsorption bed, NO and NO2 equilibrium is established with decreasing NO and increasing NO2 along the bed, which leads to a increasing adsorption capacity along the bed. The adsorption of SO2 also increases significantly at cold temperatures. The adsorption capacity increases from 12.87 to 123.11 mg/g when the temperature decreases from 80 to − 20 ℃. A novel low-temperature adsorption process is developed to simultaneously remove SO2 and NOx from flue gas. A pilot scale test platform is built and the low-temperature adsorption process is tested. Near-zero emission of both SO2 and NOx is achieved during a 72 h performance validation test.
References
Srivastava, R. & Jozewicz, W. Flue gas desulfurization: the state of the art. J. Air Waste Manag. Assoc. 2002(51), 1676–1688 (1995).
Wang, E., Lei, S., Zhong, L. & Zhang, S. Review of advanced technology of flue gas desulphurization. Adv. Mater. Res. 852, 86–91 (2014).
Flagiello, D. Seawater desulphurization of simulated flue gas in spray and packed columns: an experimental and modelling comparison. Chem. Eng. Trans. https://doi.org/10.3303/CET1869134 (2018).
Flagiello, D. et al. Seawater desulphurization scrubbing in spray and packed columns for a 4.35 MW marine diesel engine. Chem. Eng. Res. Des. 148, 56–67 (2019).
Guo, L., Shu, Y. & Gao, J. Present and future development of flue gas control technology of DeNO_X in the world. Energy Proc. 17, 397–403 (2012).
Tsuji, K. & Shiraishi, I. Combined desulfurization, denitrification and reduction of air toxics using activated coke: 1. Activity of activated coke. Fuel 76(6), 549–553 (1997).
Tsuji, K. & Shiraishi, I. Combined desulfurization, denitrification and reduction of air toxics using activated coke: 2. Process applications and performance of activated coke. Fuel 76(6), 555–560 (1997).
Olson, D., Tsuji, K. & Shiraishi, I. The reduction of gas phase air toxics from combustion and incineration sources using the MET-Mitsui-BF activated coke process. Fuel Process. Technol. 65(66), 393–405 (2000).
Sun, F., Gao, J., Zhu, Y. & Qin, Y. Mechanism of SO2 adsorption and desorption on commercial activated coke. Korean J. Chem. Eng. 28, 2218–2225 (2011).
Wang, X., Piao, G., Xie, H. & Zhao, X. Selective catalytic reduction of NOx by activated carbon. J. Southeast Univ. (Nat. Sci. Edn.) 41, 145–149 (2011).
Zhu, T., Xu, W., Guo, Y. & Li, Y. Pollutants emission and control for sintering flue gas. In Ironmaking and Steelmaking Processes (ed. Cavaliere, P.) (Springer, Cham, 2016).
Jiang, J. C., Jiang, X. & Yang, Z. S. Flue gas desulfurization and denitrification by activated coke: a mini-review. Rec. Patents Chem. Eng. 6(3), 143–151 (2013).
Xu, Z. et al. An efficient and sulfur resistant K-modified activated carbon for SCR denitrification compared with acid- and Cu-modified activated carbon. Chem. Eng. J. 395, 125047 (2020).
Zuo, Y., Yi, H. & Tang, X. Metal-modified active coke for simultaneous removal of SO2 and NOx from sintering flue gas. Energy Fuels 29, 377–383 (2015).
Mochida, I. et al. Removal of SOx and NOx over activated carbon fibers. Carbon 38, 227–239 (2013).
Chengxue, W. & Zhenheng, D. Studies on HZSM-5 zeolite catalysts for desulfurization and denitrification. In 2011 International Conference on Consumer Electronics, Communications and Networks (CECNet) 4429–4432. IEEE (2011).
Penkova, A. et al. FTIR spectroscopic study of low temperature NO adsorption and NO+O2 coadsorption on H-ZSM-5. Langmuir ACS J. Surf. Colloids 20, 5425–5431 (2004).
Tseng, H.; Haslbeck, J.; Neal, L., Evaluation of the NOXSO combined NOxSO flue gas treatment process: process chemistry, reaction kinetics, sorbent performance, process design and cost analysis. Final report. [Alumina substrate impregnated with sodium carbonate]. 2020.
Liu, D., Chen, S., Fei, X., Huang, C. & Zhang, Y. Regenerable CuO-based adsorbents for low temperature desulfurization application. Ind. Eng. Chem. Res. 54, 3556–3562 (2015).
Mochida, I., Ogaki, M., Fujitsu, H., Komatsubara, Y. & Ida, S. Catalytic activity of coke activated with sulphuric acid for the reduction of nitric oxide. Fuel 62, 867–868 (1983).
Condon, J. B. An overview of physisorption. Surf. Area Porosity Determ. Physisorp. Meas. Theory 1, 1–27 (2006).
Fang, Z., Yu, X. & Tu, S.-T. Catalytic oxidation of NO on activated carbons. Energy Proc. 158, 2366–2371 (2019).
Dastgheib, S. A., Salih, H., Ilangovan, T. & Mock, J. NO oxidation by activated carbon catalysts: impact of carbon characteristics, pressure, and the presence of Water. ACS Omega 5(33), 21172–21180 (2020).
Guo, Z.-C., Xie, Y., Hong, I. & Kim, J. Catalytic oxidation of NO to NO2 on activated carbon. Energy Convers. Manage. 42, 2005–2018 (2001).
Sousa, J., Pereira, M. & Figueiredo, J. Catalytic oxidation of NO to NO2 on N-doped activated carbons. Catal. Today 176, 383–387 (2011).
Xu, X. et al. Method for the control of NOx emissions in long-range space travel. Energy Fuels Am. Chem. Soc. J. 17, 1303–1310 (2003).
Kong, Y. & Cha, C. NOx adsorption on char in presence of oxygen and moisture. Carbon 34, 1027–1033 (1996).
Park, D., Ju, Y., Kim, J. H., Ahn, H. & Lee, C. H. Equilibrium and kinetics of nitrous oxide, oxygen and nitrogen adsorption on activated carbon and carbon molecular sieve. Sep. Purif. Technol. 223, 63–80 (2019).
Tsukahara, H., Ishida, T. & Mayumi, M. Gas-phase oxidation of nitric oxide: chemical kinetics and rate constant. Nitric Oxide Biol. Chem. 3, 191–198 (1999).
Sager, U. et al. Differences between the adsorption of NO2 and NO on modified activated carbon. Gefahrstoffe Reinhalt. Luft 74, 181–184 (2014).
Acknowledgements
This study was supported by the China Huaneng Group (Grant Nos. TY-19-HJK01 and TY-19-HJK05).
Author information
Authors and Affiliations
Contributions
S.W. discovered the phenomenon and organized this study; S.X., S.G., P.X., M.J. and B.H. contributed significantly on the process design and experiment design. H.Z. L.L., H.N., J.W. and D.G. contributed significantly on the pilot platform design and pilot test. S.W. wrote the manuscript. All authors discussed and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, S., Xu, S., Gao, S. et al. Simultaneous removal of SO2 and NOx from flue gas by low-temperature adsorption over activated carbon. Sci Rep 11, 11003 (2021). https://doi.org/10.1038/s41598-021-90532-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90532-9
This article is cited by
-
A sustainable solution: mitigating sulphur dioxide emissions through adsorption on chemically modified iron oxide nanoparticles
Clean Technologies and Environmental Policy (2024)
-
Reduction of sulphur dioxide emission into the environment by adsorption on enhanced α-hematite surface
Environmental Monitoring and Assessment (2022)
-
Adsorption behaviour of molecular sieve and activated carbon for CO2 adsorption at cold temperatures
Carbon Neutrality (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
