
Abstract
Genetic mapping is used in forward genetics to narrow the list of candidate mutations and genes corresponding to the mutant phenotype of interest. Even with modern advances in biology such as efficient identification of candidate mutations by whole-genome sequencing, mapping remains critical in pinpointing the responsible mutation. Here we describe a simple, fast, and affordable mapping toolkit that is particularly suitable for mapping in Caenorhabditis elegans. This mapping method uses insertion-deletion polymorphisms or indels that could be easily detected instead of single nucleotide polymorphisms in commonly used Hawaiian CB4856 mapping strain. The materials and methods were optimized so that mapping could be performed using tiny amount of genetic material without growing many large populations of mutants for DNA purification. We performed mapping of previously known and unknown mutations to show strengths and weaknesses of this method and to present examples of completed mapping. For situations where Hawaiian CB4856 is unsuitable, we provide an annotated list of indels as a basis for fast and easy mapping using other wild isolates. Finally, we provide rationale for using this mapping method over other alternatives as a part of a comprehensive strategy also involving whole-genome sequencing and other methods.
Similar content being viewed by others


Introduction
Forward genetics using randomly generated mutations and mutants has traditionally driven genetic analysis in Caenorhabditis elegans1. Establishing linkage between a mutation and the corresponding mutant phenotype is important in forward genetics. To help establish linkage, mapping could be done to place a mutation within a small interval, and rescue experiments2 could further narrow the interval. Sanger DNA sequencing3 has long been used to identify candidate mutations while whole-genome sequencing4,5 has simplified the identification of candidate mutations. A candidate mutation needs to be confirmed, which could involve reiteration of the same mutant phenotype by mutations in the same gene using mutagenesis perhaps CRISPR/Cas9 or possibly by RNA-mediated interference (RNAi), complementation test, and rescue experiments. Even with increasing popularity of reverse genetic engineering methods, such as CRISPR/Cas9 in its early6,7,8,9,10,11,12,13,14 and later iterations15,16,17,18,19 and RNAi20,21,22,23,24, forward genetics is a key to new discoveries and remains important.
Preferred methods of mapping have changed with emerging technologies. With advances in large-scale DNA sequencing, use of DNA sequence polymorphisms have supplemented and largely replaced many traditional methods25, including deficiency mapping and classic 2-point and 3-point mapping using mutations that cause a visible mutant phenotype. Many popular methods use wild isolate CB485626 from Hawaii (HA) with a large number of single nucleotide polymorphisms (SNPs) between CB4856 HA and the standard wild-type strain N2 Bristol. Initially, a special class of SNPs that lead to the creation or destruction of restriction sites were exploited27, and this method was simplified further by only using 48 SNPs that affect DraI restriction sites throughout the genome28. SNP-comparative genome hybridization (SNP-CGH) method29 uses ~ 76,000 SNP probes and allows a very high-resolution mapping, but this method is expensive and requires specialized material, equipment, and highly purified genomic DNA. Alternatively, whole-genome sequencing could be combined with mapping30, which could involve the use of CB4856 HA strain31 or could involve outcrossing mutations introduced by mutagenesis and dispense with CB485632.
Here, we describe a distinct group of insertion-deletion polymorphisms (indels), specifically indels with length change from 40 to 699 base pairs (bp). While indels have been used for mapping in the past with C. elegans33,34 and with C. briggsae35, indels of 40 to 699 bp (i40-699) provide notable advantages. There are many i40-699 indels, which allows fairly high-resolution mapping, in 40 wild isolates whole-genome sequenced by the C. elegans million mutation project (MMP)36. Next, i40-699 indels could be identified quickly and easily by polymerase chain reaction (PCR) simply followed by agarose gel electrophoresis. Furthermore, we optimized the primers for detecting i40-699 indels so that PCR could be performed without DNA purification. This optimization improves the speed and ease of mapping because there is no need to spend several days growing many large populations of worms. No need for many worms also allows easier mapping with mutations causing severe biological consequences such as sterility. We describe examples of mapping using previously known and unknown mutations. While we focus on i40-699 indels between N2 wild type and CB4856 HA, i40-699 indel mapping could be done instead using similar indels present in other C. elegans wild isolates sequenced by the MMP. Finally, we present ideas for the use of i40-699 indel mapping in conjunction with current techniques in forward genetics.
Methods and materials
C. elegans strains were cultured using standard methods1. Strains used to initiate this study are: CB1033 che-2(e1033), CB3329 che-10(e1809), IW523 che-10(iw109); daf-3(iw108), CB3241 clr-1(e1754), CB1376 daf-3(e1376), VC20208 without daf-3(gk269916), VC20379 daf-3(iw108), RB2589 daf-3(ok3610), SD378 dpy-17(e164) unc-79(e1068) / mpk-1(ga117), JN554 dyf-11(pe554), PR813 osm-5(p813), VC40961 rund-1(gk901813), MT7554 sqv-3(n2842) unc-69(e587)/qC1, MT9647 unc-29(e1072) sqv-5(n3039) / hT2, PR691 tax-2(p691); che-2(iw107), RB1546 tmc-1(ok1859), VC40425 tmc-1(gk631913), CB4856 Hawaiian HA wild isolate, and N2 wild-type strain. Additional strains were made using these strains.
The i40-699 indels from WormBase37 version WS256 natural variant data were extracted using custom R38 scripts in a separate study39. We also examined the WormBase version WS276 data to ensure that WS256 data is up-to-date. Using additional custom R scripts, we identified the nearest gene corresponding to each i40-699 indel along with the genetic and physical position of the gene using WormBase annotated gene dataset. A list of 11,556 annotated i40-699 indels are in Supplementary Table S1 with the physical position and the size of the indels, nearest gene with their physical and genetic position, frequency of appearance among the 40 wild isolates, and wild isolates with the i40-699 indel.
In designing primers, we aimed for uniform PCR condition and easy distinction of N2 and CB4856 HA DNA. For example, we chose i40-699 indels with longer N2 DNA than CB4856 for easy distinction. For identical annealing temperature, optimum melting temperature Tm was set at 60 °C. For identical elongation time, we kept the sizes of N2 and CB4856 products within a narrow range. With the final primer set, N2 product sizes are between 500 and 1500 bp whereas CB4856 product sizes are between 400 and 1300 bp. For easier separation of PCR products, we usually picked indels of > 100 bp length change, but we picked few indels of < 100 bp length change to have a representative i40-699 indel in most megabase (Mb) intervals. Otherwise, we picked i40-699 indels arbitrarily. To minimize confusing PCR products amplified from another part of the genome, primer-BLAST40 was performed initially using the six C. elegans chromosome sequences as the template with specificity check against the C. elegans non-redundant (nr) nucleotide database. Additional primers were selected using primer-BLAST again or using primer341 with the appropriate cosmid, fosmid or YAC clone sequence, and we tested the primers until a satisfactory primer pair was identified. A total of 584 primers were tested, and the best primer pairs are listed in Supplementary Table S2 with expected sizes of the PCR products, PCR success rates under two different conditions, annotations of the position in 96-well plate where applicable, and wild isolates with the i40-699 indel.
Working stocks of premixed primer pairs were stored in 96-well plates frozen because long-term storage at 4 °C led to evaporation and possible degradation. Unless indicated otherwise, adult hermaphrodites were lysed individually in 10 μl of 1 × lysis buffer (40 mM KCl, 10 mM Tris pH 8.3, 2.5 mM MgCl2, 0.45% IGEPAL, 0.45% Tween 20) with proteinase K added prior to lysis at 60 μg/ml final concentration. Lysis was performed at 65 °C for 1 h with inactivation at 95 °C for 15 min using 8-strip tubes. Lysed worms were often mixed or pooled, and combined samples were often diluted with water or 1 × lysis buffer lacking proteinase K. PCR was performed in 25 µl volume using standard conditions without detergent for Taq polymerase. Either 8-strip tubes or 96-well plates were used for PCR with aluminum foil seal for 96-well plates to reduce evaporation. The PCR temperature cycler condition was: 5′ 95 °C, 35 cycles of (30″ 95 °C, 30″ 60 °C, 2′ 72 °C), 5′ 72 °C. PCR products were visualized after separation by electrophoresis for 40 min at 120 V on 2% agarose gel with ~ 1.5 l of Tris–acetate-EDTA running buffer unless indicated otherwise. Recurring poor PCR results were usually rectified by new preparation of 1 × lysis buffer and discarding old stocks of 1 × lysis buffer, which could become less effective after many months.
To obtain F2 mutants for mapping, CB4856 males were mated with mutant hermaphrodites of interest. Next, F1 heterozygous hermaphrodites were placed in new plates as L4 larvae to reproduce F2 progeny by self-fertilization. Finally, F2 homozygous mutants were identified by their mutant phenotype, which could be clear (Clr) and blistering body42, squashed vulva (Sqv) at L4 larval stage43 and associated sterility with a stereotypical morphology of embryos incapable of cytokinesis44, or a temperature-dependent constitutive dauer mutant phenotype45. Collection of temperature-dependent dauer mutants involved placing a mixed population of embryos and L1 larvae at 28 °C for 10 days in the presence of plenty of E. coli bacteria serving as food prior to a return to 20 °C to allow mutant dauer survivors to become mutant fertile adults.
Complementation tests using the temperature-dependent constitutive dauer mutant phenotype were performed using a few different approaches. For iw108 located in chromosome X, either daf-3(ok3610) or dyf-11(pe554) mutant hermaphrodites were mated with N2 males, and F1 hemizygous mutant XO males were mated with either daf-3(iw108) or dpy-17(e164) unc-79(e1068); daf-3(iw108) mutant hermaphrodites. Mixed populations of F2 progeny were moved to 28 °C for 10 days and subsequently were moved to 20 °C, and non-Dpy non-Unc fertile adult hermaphrodites were sought in the presence of alive males. Dauer survivors that became adult males were used to assess successful mating with the absence of fertile hermaphrodites indicating no complementation. With tests using daf-3(iw108) hermaphrodite mutants without dpy-17 or unc-79 mutations, dauer survivors that became fertile adult hermaphrodites were examined using PCR to confirm the presence or absence of the mutations of interest. Similar approach was used with iw107 also located in chromosome X. Here, one of che-2(e1033), daf-3(ok3610), dyf-11(pe554), rund-1(gk901813), osm-5(p813), or iw107 mutant hermaphrodites were mated with N2 males, and F1 hemizygous mutant males were mated with one of che-2(iw107), dpy-17(e164) unc-79(e1068); che-2(iw107), dpy-17(e164) unc-79(e1068); che-2(e1033), dpy-17(e164) unc-79(e1068); dyf-11(pe554), or dpy-17(e164) unc-79(e1068); osm-5(p813) hermaphrodites. A different approach was used with iw109 located in chromosome II. Here, che-10(e1809) mutant hermaphrodites were mated with N2 males, and heterozygous F1 males were mated with either che-10(iw109) or dpy-17(e164) unc-79(e1068); che-10(iw109) mutant hermaphrodites. After 10 days at 28 °C, the presence of dauer survivors that became males was used as evidence of complementation.
Whole genome sequencing of che-10(iw109) mutant strain was performed with Novogene (en.novogene.com), using their Plant and Animal Whole Genome Sequencing service with 2 Gb data output for 20 × coverage of the genome by paired-end 150 bp sequencing. Sanger sequencing was performed with Genewiz (www.genewiz.com).
Results
Easily distinguished indels in CB4856
Indels of a certain range of DNA length change, for example from 40 to 699 bp, could serve as excellent mapping resource. Mapping with i40-699 indels could be done with PCR followed by gel electrophoresis without restriction enzyme digest. From over 230,000 indels identified by the MMP in 40 wild isolates36, we picked and annotated over 11,000 i40-699 indels (Supplementary Table S1). CB4856 HA wild isolate, which is commonly used by many laboratories for mapping, contains over 1,600 i40-699 indels. These i40-699 indels are distributed throughout the genome in an uneven manner with central regions of autosomal chromosomes having far fewer i40-699 indels than autosomal chromosome arms (Fig. 1), as is typical in wild populations for all SNPs and indels39. Unique among the 40 wild isolates, CB4856 has at least one i40-699 indel within every interval of one Mb. Given these advantages, we picked i40-699 indels in CB4856 for mapping.

Distribution of i40-699 indels in CB4856. Shown is distribution of i40-699 indels in 1 Mb intervals across six chromosomes of CB4856. Red bars indicate i40-699 indels that are also present in at least one of the other 39 wild isolates. Yellow bars indicate i40-699 indels that are unique to CB4856 among the 40 wild isolates.
To confirm the presence of i40-699 indels, we examined 124 i40-699 indels by PCR with 586 primers (Supplementary Table S3). All PCR was performed using lysed worms without DNA purification. We obtained PCR products of expected sizes for all but three loci with both CB4856 and N2, thus confirming the existence of most i40-699 indels. With two unconfirmed i40-699 indels WBVar02075736 and WBVar02075902, PCR products corresponding to N2 were obtained but not CB4856. We do not consider the absence of CB4856 products as a proof that i40-699 indel is absent because PCR failure could be caused by altered primer-binding site, which could be a consequence of reported or unreported mutation. We subsequently examined six primer binding sites used for successful PCR of these two indel loci and identified two SNPs affecting 5′ end and middle of two binding sites for WBVar02075902 in CB4856, but PCR is often possible with such minor DNA changes. As for the third problematic locus with two closely neighboring i40-699 indels, the larger of the two indels WBVar02076224 appeared to be absent while the status of the smaller indel WBVar02091533 was inconclusive. Thus, false positive rate for i40-699 indel calling by the MMP seems low (< 1%) with CB4856, but reported and unreported differences in the DNA sequences flanking the i40-699 indel could pose problems with primer design for indel detection.
Next, we selected primers capable of detecting i40-699 indels reliably with a fraction of a worm. Here, 0.1 adult hermaphrodites were used for each PCR sample to test the primers using 96-well PCR plates, and unsatisfactory primers with a variety of complications were abandoned. Targeting 102 different i40-699 indel loci, PCR products of expected sizes were obtained at > 90% rate with 95 primer pairs (Supplementary Table S2). To serve as a convenient reference, a set of 2% agarose gel images of PCR products using either N2 or an equal mix of N2 and CB4856 with 96 primer pairs is shown in Fig. 2. In another test, PCR was performed using a mix of 0.01 CB4856 adults and 0.09 N2 adults per sample. Here, PCR products corresponding to CB4856 were obtained at ≥ 75% rate with 82 primer pairs, and 100% success rate associated with nearly half of the primer pairs (Supplementary Table S2). A set of 2% agarose gel images of PCR products using 1-to-9 mix of CB4856 and N2 is provided in Supplementary Figure S1. Thus, our primers could be reliably used without needing several days to grow large populations of worms and without DNA purification.

PCR using the primer set for detecting 96 i40-699 indels. Pairs of PCR products using DNA from N2 wild type on left and from equal parts N2 and CB4856 on right. Each PCR sample used 0.1 adults as template. The samples are arranged in 8 rows from A to H with each row containing 12 different primer pairs. Shorthand designations from A1 to A12 through from H1 to H12 indicate primer pair positions in a 96-well plate. Chromosomes are indicated as I, II, III, IV, V, and X on top. 1 kb Plus Ladder Invitrogen.
Validation mapping with known and unknown mutations
Common 96-well plate format was used for ease in quickly performing all experimental steps under identical conditions. We arranged the placement of primers in a 96-well plate from chromosome I to chromosome X and from left to right end of each chromosome (Fig. 3). Equipment needed is same as or less stringent than the older SNP mapping methods27,28 prior to the SNP-CGH method29, including multi-channel pipettor, temperature cycler capable of using 96-well plates, and agarose gel electrophoresis apparatus. As is standard, genetic manipulations start with CB4856 Hawaiian males mating with mutant hermaphrodites to produce heterozygous F1 animals. Homozygous F2 mutants from F1 heterozygotes are identified based on their mutant phenotypes. To make analysis easy, we performed PCR and gel electrophoresis using lysed N2 wild type worms in parallel with pooled F2 mutants. With an i40-699 indel closely linked to the mutation of interest, we expect only one PCR product corresponding to N2 in most trials. On the other hand with an unlinked i40-699 indel, we expect two PCR products corresponding to CB4856 and to N2. Because we designed CB4856 products to be smaller than the corresponding N2 products, it is easy to identify a set of i40-699 indels that are closely linked to the mutation of interest by spotting a set of neighboring PCR products without smaller CB4856 products. Finally, we used crude lysates using as little as 0.001 adult hermaphrodites for each PCR sample to demonstrate the ease of mapping using these primers.

Location of 102 i40-699 indels with good primers. Six C. elegans chromosomes are depicted with their left ends on top. Each i40-699 indel is marked by their position in the 96-well plate (e.g. A1 to H12) used for most mapping experiments or is marked as NU if not a part of the oft-used 96-well plate. Physical location of indel is marked with a notch, and genetic position is indicated with a number. Indels on the left side of each chromosome have negative number for genetic position. Indels with primer pairs showing 100% PCR success rate with 1:9 dilution of CB4856 and N2 are marked with star(*).
First, to validate the primers and i40-699 indels for mapping, we mapped three known mutations clr-1(e1754)42, sqv-3(n2842)46, and sqv-5(n3039)47. These mutations were chosen in part because the corresponding homozygous mutants are sterile, which makes it difficult to obtain large amounts of genomic DNA from homozygous mutants. As is standard, all mutants were originally isolated from mutagenized N2 and subsequently were outcrossed with non-mutagenized N2 by others. From heterozygous F1 progeny derived from mating of CB4856 males with mutant hermaphrodites of interest, we obtained individual F2 adult hermaphrodites with Clr or Sqv mutant phenotype. Ten F2 mutants were lysed and pooled for mapping each mutation using 96 primer pairs. Here, we mapped all three mutations to within 6 to 8 Mb intervals by identifying neighboring sets of PCR samples without smaller CB4856 products (Supplementary Figure S2). We did not pursue use of lysates with 0.001 adult hermaphrodites in these examples unlike in the next set of mapping of previously unknown mutations.
We mapped a previously unknown iw108 mutation present in an MMP strain VC20379. VC20379 is a part of 2,007 whole-genome sequenced strains that had been inbred following mutagenesis36, and we found an unknown mutation that is associated with a constitutive temperature-dependent dauer mutant phenotype at 28 °C45. Using pooled and individual F2 mutants, we mapped the mutation to the left side of chromosome X using the i40-699 indel detection primer set (Supplementary Figure S3). Within the mapped interval according to MMP sequencing data, there are two point mutations in introns of tmc-1 and fbxa-40 along with five point mutations in intergenic regions without any interesting nearby genes. Subsequently, we obtained an outcrossed constitutive dauer mutant isolate with the tmc-1 mutation but without five of the seven mutations including the fbxa-40 mutation. We examined tmc-1(ok1859) 2025 bp deletion and tmc-1(gk631913) Q578amber mutants, but these two mutants were without dauer mutant phenotype. After exhausting other possibilities including genome-wide examination of all mutations with moderate or strong effect on protein coding in VC20379 as reported by MMP, we performed de novo analysis of the raw VC20379 MMP sequencing data using a standard pipeline of bowtie248, samtools49, vcftools50, and snpEff51. To the left of the aforementioned seven mutations, we identified an unreported mutation iw108, which is predicted to cause a G-to-T missense mutation causing C848F change in the transcript F25E2.5a.1 of daf-3 SMAD4. The aforementioned outcrossed mutant isolate with the tmc-1 mutation but not fbxa-40 mutation contains daf-3(iw108). Curiously, daf-3(iw108) mutation is identical to daf-3(gk269916) reported to be present in another MMP strain VC20208. The daf-3(gk269916) mutation is absent in VC20208, and our best guess is that this is a WormBase annotation error. Mutations in daf-352 are associated with dauer defective phenotype at standard growth temperatures53 and with dauer constitutive phenotype at temperatures above 25 °C54. We also have examined the canonical daf-3(e1376) mutant and a previously uncharacterized daf-3(ok3610) mutant, both of which have a similar temperature-dependent dauer mutant phenotype as daf-3(iw108) and support the notion that daf-3 is the gene of interest. Finally, we found that daf-3(ok3610) and daf-3(iw108) mutations complement each other to show a temperature-dependent dauer constitutive mutant phenotype. Investigation of daf-3(iw108) is an example of mapping of unknown mutation, mutation identification, and confirmation with additional mutant alleles with the same mutant phenotype and complementation test.
Next in a daf-3(iw108) mutant strain, we identified a previously unknown iw109 mutation associated with a temperature-dependent constitutive dauer mutant phenotype at 28 °C. We found the iw109 mutation while trying to obtain nice images of daf-3 pooled mapping, which did not result in the expected mapping of daf-3(iw108) to the left side of chromosome X. Instead, we mapped iw109 to chromosome II using an outcrossed iw109 strain without daf-3(iw108) (Supplementary Figure S4). The iw109 mutation is associated with a stronger dauer constitutive mutant phenotype as compared to daf-3(iw108), whose molecular lesion had been known to us for two years by the time of our discovery of iw109. We presume that iw109 mutation arose spontaneously and became fixed during two years of culturing of daf-3(iw108) mutants. Whole-genome sequencing of iw109 mutants revealed a 29 bp deletion mutation with the loss of DNA sequence TTTTTGCAAAGTTGAAAGAGGAATTGTTC in the second exon of che-10 ciliary rootlet coiled-coil (CROCC) or rootletin55 in the mapped region. Here we expect only 168 amino acids of four isoforms of full-length CHE-10 proteins ranging from 1575 to 1995 amino acids to be retained. The sequencing also revealed a ~ 1.3 kb deletion removing a part of nearby hmg-11 gene and upstream DNA, but we think that this deletion is unlikely to be responsible for the dauer mutant phenotype. Notably, che-10(e1809) mutants are defective in differentiation of amphid neurons and dauer formation at normal temperature under 25 °C56, and these mutant phenotypes are associated with dauer constitutive mutant phenotype only at above 25 °C54,57. Furthermore, we used the che-10(e1809) mutation for a complementation test with iw109, and these two mutations complement each other to show a dauer constitutive mutant phenotype at 28 °C. Our studies starting with mapping suggest that iw109 could be a che-10 null mutation.
Third, we identified previously unknown iw107 mutation associated with a temperature-dependent constitutive dauer mutant phenotype at 28 °C. We found iw107 mutation in PR691 tax-2(p691), which is called RS1 in older manuscripts58,59,60, during outcrossing. Specifically, we obtained iw107 mutants without tax-2(p691) mutation as well as tax-2(p691) mutants by screening for temperature-dependent dauer mutant descendants following mating of PR691 with N2 wild type. We think that iw107 mutation may have arisen spontaneously and become fixed in PR691, possibly prior to our receipt of PR691 from the Caenorhabditis Genetics Center. We mapped iw107 to the left side of chromosome X using pooled and individual F2 mutants (Supplementary Figure S5). Within the mapped interval, there are five genes che-261, daf-352, dyf-1162,63, osm-564, and rund-165, which by our assessment could have a temperature-dependent constitutive dauer mutant phenotype. We performed complementation tests using mutants with lesions in these genes, and among these only che-2(e1033) Q601ochre61 and iw107 complemented each other to show a lasting dauer constitutive mutant phenotype at 28 °C. Sanger sequencing of iw107 mutant did not reveal any mutation in the coding region of che-2, but we identified mutations in two different intervening introns. Specifically in the che-2 intraflagellar transport 80 (IFT8066) transcript F38G1.1.1 unspliced and untranslated (UTR) sequence of 5055 bp, the two mutations are G-to-A at positions 2750 and 3864 with the latter mutation near the splice donor of exon 11 (GA/gtgagt to GA/gtgaat with slash denoting exon/intron boundary and underline denoting the site of sequence change). In addition to che-2(iw107) mutants having fewer dauer survival at 28 °C as compared to che-2(e1033) mutants, we saw that che-2(iw107) mutants do not show strong male mating defect observed with che-2(e1033) mutants67,68, which is consistent with iw107 being a weak loss-of-function mutation. In summary, iw107 appears to be a weak che-2 loss-of-function mutation as shown by mapping and other studies.
Easily distinguished indels in other wild isolates
While neither CB4856 nor N2 wild-type strains shows constitutive dauer mutant phenotype, hybrid progeny resulting from mating between CB4856 and N2 could survive as dauer larvae for a long time at high temperature45. The dauer survival phenotype of the hybrid is very weak and usually is not reproducible using the progeny of the survivors, and thus CB4856 still could be used for mapping with sufficient caution. In general, however, a common phenotype shared between the mutant of interest and CB4856 could render CB4856 unsuitable for use in mapping. For example, CB4856 displays social feeding behavior similar to mutants of neuropeptide receptor npr-169 and thus would be inappropriate for mapping mutations associated with such feeding behavior. Here, another wild isolate without the same social feeding behavior is needed. To assess other wild isolates for use with i40-699 indel mapping, we examined the 40 wild isolates sequenced by the MMP36 with the vast majority of reported i40-699 indels. While there is whole-genome sequence data of over 300 wild isolates available elsewhere in the C. elegans natural diversity resource CeNDR70, bigger indels were identified at low rate by CeNDR. For example with CB4856, only two indels of > 40 bp were identified by CeNDR using higher threshold for quality control as opposed to over 1,600 i40-699 indels according to MMP. Thus to identify more useful wild isolates for mapping, we only examined the distribution of over 11,000 i40-699 indels in the 40 MMP wild isolates.
A suitable alternate wild isolate should be evolutionarily divergent from CB4856 and therefore is less likely to share a common phenotype with CB4856. Also, a good alternate should contain many i40-699 indels throughout the genome. We discarded 34 of the 40 wild isolates from consideration because their genomes have too many 1 Mb intervals without any i40-699 indels (Supplementary Figure S6). We note that these 34 wild isolates contain over 6000 unique i40-699 indels combined, and some of these wild isolates could be used for mapping small parts of the genome. Of the others, three isolates JU1171, MY2, and MY14 share most of their i40-699 indels evenly throughout the genome (Supplementary Figure S7), meaning they are closely related to each other as also noted previously39,71. Such closely related wild isolates are likely to share common phenotypes, and thus we chose MY2 to represent this group of three. This leaves us with the three best wild isolates JU258, JU775, and MY2 all with i40-699 indels in most but not all 1 Mb intervals (Fig. 4). We recommend using JU258, JU775, and MY2 for building a similar mapping toolkit using a different wild isolate, and we provide a list of over 11,000 potentially useful i40-699 indels with annotations in Supplementary Table S1.

Three more wild isolates with many i40-699 indels. Shown are distributions of i40-699 indels in 1 Mb intervals of wild isolates (A) JU258, (B) JU775, and (C) MY2. Orange bars indicate i40-699 indels that are unique among the four wild isolates JU258, JU775, MY2, and CB4856. Red bars indicate i40-699 indels that are also present in other wild isolates.
Discussion
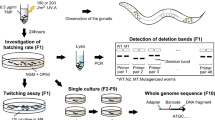
Using i40-699 indels, mapping could be done quickly and easily. Pooled mapping could be completed on the day of F2 mutant collection, and additional fine mapping could be completed the next day if needed. The amount of time required on temperature cyclers and electrophoresis apparatus include 1 h 20 min for single worm lysis, 2 h 20 min for PCR, and 40 min for agarose gel electrophoresis. Time needed for transfer of 10 N2 and 10 mutant adults for lysis, preparation of two sets of 96 PCR reactions for whole-genome mapping, and gel loading of PCR products could be under one hour with experienced hands and good equipment. With other mapping methods, five or more days are needed to collect large populations of worms derived from F1 mutant worms before starting subsequent procedures. No need to grow many worms is a big advantage with the primers we designed for the i40-699 indel mapping. Thus mapping using i40-699 indels is faster than all existing mapping methods.
Mapping is a part of many experiments necessary to link a mutation in a gene to a mutant phenotype. Other experiments could involve complementation test, rescue, or identification of many mutant alleles in the same gene with the same mutant phenotype. A key requirement is the identification of the molecular lesion in the DNA, which requires DNA sequencing. Of existing mapping methods, combining whole-genome-sequencing and mapping is especially attractive, and a comparison with i40-699 indel mapping could be worthwhile. With speed, i40-699 indel mapping is faster. With resolution, mapping with whole-genome-sequencing is usually better. With cost, we discuss the parameters only because the exact cost is neither static nor global. Here, a consideration is that whole-genome-sequencing mapping could involve different modern approaches72,73, which may or may not involve pooling. With pooling, 20 to 50 populations derived from F2 mutants need to be grown before pooling for DNA purification, library construction, and sequencing run in addition to extra reagents for culturing worms. Without pooling, there is a need for separate DNA purifications, library constructions, and more sequencing runs or space. With i40-699 indel mapping, there are extra supplies and reagents, such as PCR plates, pipette tips, Taq polymerase, dNTPs, and primers. Assuming that whole-genome sequencing is needed, combining whole-genome sequencing and mapping using pooling probably would be the cheapest option with a single mutation. We note that with i40-699 indel mapping and with many mutations, prior complementation tests could reduce the number of needed whole-genome sequencing runs, and the use of Sanger sequencing to identify mutations could lower the cost. In our opinion, i40-699 indel mapping is a valid fast option.
We also discuss a special circumstance, which should be more common in our opinion. Pre-existing whole-genome-sequencing data exists with the 2007 MMP strains36, which we think are a very useful resource for the C. elegans scientific community. In this manuscript with i40-699 indel mapping, we did not need a new whole-genome sequencing to identify daf-3(iw108) with MMP strain VC20208. Similarly, 2007 MMP strains could be used for future genetic screens and identification of interesting mutations without new whole-genome sequencing. Notably following mutagenesis and genetic screen, new mutants with whole-genome sequencing data could have the same utility as the MMP strains. Here prior to sequencing, outcrosses removing unlinked mutations is counterproductive, but inbreeding could be useful. With i40-699 indel mapping, whole-genome-sequenced mutants could be added to the useful existing scientific resource consisting of the 2007 MMP strains. On the other hand with the use of whole-genome sequencing for mapping, the sequenced worms are not useful because they have either too few mutations or a confounding mix of CB4856 and N2 DNA. In summary, the use of i40-699 mapping could be used as a part of a general effort to improve the C. elegans community resource for forward genetics (Fig. 5).

Using i40-699 indel mapping with outcrossing and sequencing for gene identification, mutant characterization, and reusable forward genetics resource. Outcrossing could eliminate most background mutations, which could interfere with the mutant phenotype. Mapping and sequencing also could help pinpoint a mutation to a gene. Validations could be provided by complementation test, rescue experiments, or many mutations in the same gene, which phenocopy the mutation of interest. We recommend performing whole-genome sequencing using non-outcrossed mutant strain, which could be re-used for more forward genetics. No new whole-genome sequencing is needed with MMP strains. White worm is mutant, black worm is CB4856 HA, red worm is N2 wild type, and gray worm is heterozygous.
For efficient use of i40-699 indel primers with mutant lysates with a mix of N2 and CB4856 DNA, we recommend using 0.01 adults for each PCR reaction. While it is possible to use 0.001 adults per PCR sample (Supplementary Figures S3 to S5), PCR success rate is lower at such low template concentration, and there could be more off-target PCR products. Strong off-target PCR products could make the results more difficult to interpret. We also note that the use of a smaller subset of primer pairs (e.g. 48 instead of 96) for whole-genome mapping could be more cost-effective. Here, we suggest picking primer pairs based on high PCR success rate using less CB4856 DNA (Supplementary Table S2). Some of the alternative primer pairs described in Supplementary Table S2 other than the most extensively tested 102 pairs may prove to be more reliable. Also, we recommend using a small number (e.g. 10) of F2 mutant adults for pooling to avoid unnecessary dilution. With the mutation of interest mapped to a small interval, primer pairs for detecting a subset of 102 i40-699 indel loci could be used for further mapping using individual lysates of F2 mutants (Fig. 5).
To assess how much mapping may be needed, we examined MMP whole-genome sequencing data of 2,007 mutagenized strains36 for the genomic distribution of mutations. MMP used six mutagenesis methods, and MMP obtained the largest number of mutations with 50 mM EMS, which is commonly used in many laboratories, and with the combination of 50 mM EMS and 1 mM ENU (Supplementary Table S4). With 50 mM EMS alone (n = 656), MMP identified 376 ± 100 (mean ± s.d.) mutations, of which 86 ± 24 mutations are predicted to have an effect. Here, the following search terms were used as a prediction of an effect: nonsense, start ATG, missense, frame shift, coding exon, across exon boundary, splicing, miRNA, rRNA, scRNA, snRNA, snoRNA, and tRNA. While there are many mutations far away from exons known to have substantial biological effects, we expect that most mutations of intergenic region, intron, non-coding exon, UTR, and ncRNA have no effect. With the combination of 50 mM EMS and 1 mM ENU (n = 943), MMP identified 545 ± 149 mutations, of which 123 ± 34 mutations are predicted to have an effect. Laboratory-induced mutations are evenly distributed throughout the genome. Given the C. elegans genome of just over 100 Mb, a typical mutagenized strain using EMS alone has on average less than one mutation with a predicted effect per Mb. With primer pairs for i40-699 indel in most 1 Mb intervals, narrowing the number of strong candidates to one should be possible in many cases with i40-699 indels.
Mapping using i40-699 indels and small number of worms is fast and easy for a typical laboratory and does not need expensive specialty equipment. In planning a comprehensive strategy for research, optimal use of all necessary methods is desired. Among key techniques used in genetics, recently developed methods include whole-genome sequencing and CRISPR/Cas9. Combining i40-699 indel mapping with whole-genome sequencing and other methods as described here could be a part of comprehensive strategy for forward genetics (Fig. 5).
Data availability
Additional material including strains and scripts are available upon request.
References
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974).
Mello, C. C., Kramer, J. M., Stinchcomb, D. & Ambros, V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959–3970. https://doi.org/10.1002/j.1460-2075.1991.tb04966.x (1991).
Sanger, F. & Coulson, A. R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 94, 441–448. https://doi.org/10.1016/0022-2836(75)90213-2 (1975).
Hu, P. J. Whole genome sequencing and the transformation of C. elegans forward genetics. Methods 68, 437–440. https://doi.org/10.1016/j.ymeth.2014.05.008 (2014).
Smith, H. E., Fabritius, A. S., Jaramillo-Lambert, A. & Golden, A. Mapping challenging mutations by whole-genome sequencing. G3 (Bethesda) 6, 1297–1304. https://doi.org/10.1534/g3.116.028316 (2016).
Friedland, A. E. et al. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 10, 741–743. https://doi.org/10.1038/nmeth.2532 (2013).
Dickinson, D. J., Ward, J. D., Reiner, D. J. & Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods 10, 1028–1034. https://doi.org/10.1038/nmeth.2641 (2013).
Chen, C., Fenk, L. A. & de Bono, M. Efficient genome editing in Caenorhabditis elegans by CRISPR-targeted homologous recombination. Nucleic Acids Res. 41, e193. https://doi.org/10.1093/nar/gkt805 (2013).
Chiu, H., Schwartz, H. T., Antoshechkin, I. & Sternberg, P. W. Transgene-free genome editing in Caenorhabditis elegans using CRISPR-Cas. Genetics 195, 1167–1171. https://doi.org/10.1534/genetics.113.155879 (2013).
Cho, S. W., Lee, J., Carroll, D., Kim, J. S. & Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics 195, 1177–1180. https://doi.org/10.1534/genetics.113.155853 (2013).
Katic, I. & Grosshans, H. Targeted heritable mutation and gene conversion by Cas9-CRISPR in Caenorhabditis elegans. Genetics 195, 1173–1176. https://doi.org/10.1534/genetics.113.155754 (2013).
Lo, T. W. et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics 195, 331–348. https://doi.org/10.1534/genetics.113.155382 (2013).
Tzur, Y. B. et al. Heritable custom genomic modifications in Caenorhabditis elegans via a CRISPR-Cas9 system. Genetics 195, 1181–1185. https://doi.org/10.1534/genetics.113.156075 (2013).
Waaijers, S. et al. CRISPR/Cas9-targeted mutagenesis in Caenorhabditis elegans. Genetics 195, 1187–1191. https://doi.org/10.1534/genetics.113.156299 (2013).
Paix, A. et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 Sites in Caenorhabditis elegans. Genetics 198, 1347–1356. https://doi.org/10.1534/genetics.114.170423 (2014).
Dickinson, D. J., Pani, A. M., Heppert, J. K., Higgins, C. D. & Goldstein, B. Streamlined genome engineering with a self-excising drug selection cassette. Genetics 200, 1035–1049. https://doi.org/10.1534/genetics.115.178335 (2015).
Norris, A. D., Kim, H. M., Colaiacovo, M. P. & Calarco, J. A. Efficient genome editing in Caenorhabditis elegans with a toolkit of dual-marker selection cassettes. Genetics 201, 449–458. https://doi.org/10.1534/genetics.115.180679 (2015).
Paix, A., Schmidt, H. & Seydoux, G. Cas9-assisted recombineering in C. elegans: genome editing using in vivo assembly of linear DNAs. Nucleic Acids Res. 44, e128. https://doi.org/10.1093/nar/gkw502 (2016).
Au, V. et al. CRISPR/Cas9 methodology for the generation of knockout deletions in Caenorhabditis elegans. G3 (Bethesda) 9, 135–144. https://doi.org/10.1534/g3.118.200778 (2019).
Fire, A. et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. https://doi.org/10.1038/35888 (1998).
Tabara, H., Grishok, A. & Mello, C. C. RNAi in C. elegans: soaking in the genome sequence. Science 282, 430–431. https://doi.org/10.1126/science.282.5388.430 (1998).
Timmons, L. & Fire, A. Specific interference by ingested dsRNA. Nature 395, 854. https://doi.org/10.1038/27579 (1998).
Kamath, R. S. et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237. https://doi.org/10.1038/nature01278 (2003).
Conte, D. Jr., MacNeil, L. T., Walhout, A. J. M. & Mello, C. C. RNA Interference in Caenorhabditis elegans. Curr Protoc Mol Biol 109:26.3.1–26.3.30. https://doi.org/10.1002/0471142727.mb2603s109 (2015).
Fay, D. S. Classical genetic methods. In WormBook: The Online Review of C. elegans Biology, 1–58, https://doi.org/10.1895/wormbook.1.165.1 (2013).
Hodgkin, J. & Doniach, T. Natural variation and copulatory plug formation in Caenorhabditis elegans. Genetics 146, 149–164 (1997).
Wicks, S. R., Yeh, R. T., Gish, W. R., Waterston, R. H. & Plasterk, R. H. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat. Genet. 28, 160–164. https://doi.org/10.1038/88878 (2001).
Davis, M. W. et al. Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics 6, 118. https://doi.org/10.1186/1471-2164-6-118 (2005).
Flibotte, S. et al. Rapid high resolution single nucleotide polymorphism-comparative genome hybridization mapping in Caenorhabditis elegans. Genetics 181, 33–37. https://doi.org/10.1534/genetics.108.096487 (2009).
Sarin, S., Prabhu, S., O’Meara, M. M., Pe’er, I. & Hobert, O. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat. Methods 5, 865–867. https://doi.org/10.1038/nmeth.1249 (2008).
Doitsidou, M., Poole, R. J., Sarin, S., Bigelow, H. & Hobert, O. C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS ONE 5, e15435. https://doi.org/10.1371/journal.pone.0015435 (2010).
Zuryn, S., Le Gras, S., Jamet, K. & Jarriault, S. A strategy for direct mapping and identification of mutations by whole-genome sequencing. Genetics 186, 427–430. https://doi.org/10.1534/genetics.110.119230 (2010).
Williams, B. D., Schrank, B., Huynh, C., Shownkeen, R. & Waterston, R. H. A genetic mapping system in Caenorhabditis elegans based on polymorphic sequence-tagged sites. Genetics 131, 609–624 (1992).
Zipperlen, P. et al. A universal method for automated gene mapping. Genome Biol. 6, R19. https://doi.org/10.1186/gb-2005-6-2-r19 (2005).
Koboldt, D. C. et al. A toolkit for rapid gene mapping in the nematode Caenorhabditis briggsae. BMC Genomics 11, 236. https://doi.org/10.1186/1471-2164-11-236 (2010).
Thompson, O. et al. The million mutation project: a new approach to genetics in Caenorhabditis elegans. Genome Res. 23, 1749–1762. https://doi.org/10.1101/gr.157651.113 (2013).
Harris, T. W. et al. WormBase 2014: new views of curated biology. Nucleic Acids Res. 42, D789-793. https://doi.org/10.1093/nar/gkt1063 (2014).
R: A language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna, Austria, 2014).
Hwang, H. Y. & Wang, J. Effect of mutation mechanisms on variant composition and distribution in Caenorhabditis elegans. PLoS Comput. Biol. 13, e1005369. https://doi.org/10.1371/journal.pcbi.1005369 (2017).
Ye, J. et al. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 13, 134. https://doi.org/10.1186/1471-2105-13-134 (2012).
Untergasser, A. et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. 40, e115. https://doi.org/10.1093/nar/gks596 (2012).
Kokel, M., Borland, C. Z., DeLong, L., Horvitz, H. R. & Stern, M. J. clr-1 encodes a receptor tyrosine phosphatase that negatively regulates an FGF receptor signaling pathway in Caenorhabditis elegans. Genes Dev. 12, 1425–1437 (1998).
Herman, T., Hartwieg, E. & Horvitz, H. R. sqv mutants of Caenorhabditis elegans are defective in vulval epithelial invagination. Proc. Natl. Acad. Sci. U.S.A. 96, 968–973. https://doi.org/10.1073/pnas.96.3.968 (1999).
Hwang, H. Y. & Horvitz, H. R. The SQV-1 UDP-glucuronic acid decarboxylase and the SQV-7 nucleotide-sugar transporter may act in the Golgi apparatus to affect Caenorhabditis elegans vulval morphogenesis and embryonic development. Proc. Natl. Acad. Sci. U.S.A. 99, 14218–14223. https://doi.org/10.1073/pnas.172522199 (2002).
Hwang, H. Y., Dankovich, L. & Wang, J. Thermotolerance of tax-2 is uncoupled from life span extension and influenced by temperature during development in C. elegans. Front. Genet. 11, 2–8. https://doi.org/10.3389/fgene.2020.566948 (2020).
Herman, T. & Horvitz, H. R. Three proteins involved in Caenorhabditis elegans vulval invagination are similar to components of a glycosylation pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 974–979. https://doi.org/10.1073/pnas.96.3.974 (1999).
Hwang, H. Y., Olson, S. K., Esko, J. D. & Horvitz, H. R. Caenorhabditis elegans early embryogenesis and vulval morphogenesis require chondroitin biosynthesis. Nature 423, 439–443. https://doi.org/10.1038/nature01634 (2003).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. https://doi.org/10.1093/bioinformatics/btp352 (2009).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158. https://doi.org/10.1093/bioinformatics/btr330 (2011).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. https://doi.org/10.4161/fly.19695 (2012).
Patterson, G. I., Koweek, A., Wong, A., Liu, Y. & Ruvkun, G. The DAF-3 Smad protein antagonizes TGF-beta-related receptor signaling in the Caenorhabditis elegans dauer pathway. Genes Dev. 11, 2679–2690. https://doi.org/10.1101/gad.11.20.2679 (1997).
Riddle, D. L., Swanson, M. M. & Albert, P. S. Interacting genes in nematode dauer larva formation. Nature 290, 668–671. https://doi.org/10.1038/290668a0 (1981).
Ailion, M. & Thomas, J. H. Dauer formation induced by high temperatures in Caenorhabditis elegans. Genetics 156, 1047–1067 (2000).
Mohan, S., Timbers, T. A., Kennedy, J., Blacque, O. E. & Leroux, M. R. Striated rootlet and nonfilamentous forms of rootletin maintain ciliary function. Curr. Biol. 23, 2016–2022. https://doi.org/10.1016/j.cub.2013.08.033 (2013).
Starich, T. A. et al. mutations affecting the chemosensory neurons of Caenorhabditis elegans. Genetics 139, 171–188 (1995).
Ailion, M. & Thomas, J. H. Isolation and characterization of high-temperature-induced Dauer formation mutants in Caenorhabditis elegans. Genetics 165, 127–144 (2003).
Dusenbery, D. B., Sheridan, R. E. & Russell, R. L. Chemotaxis-defective mutants of the nematode Caenorhabditis elegans. Genetics 80, 297–309 (1975).
Hedgecock, E. M. & Russell, R. L. Normal and mutant thermotaxis in the nematode Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 72, 4061–4065. https://doi.org/10.1073/pnas.72.10.4061 (1975).
Dusenbery, D. B. Chemotactic behavior of mutants of the nematode Caenorhabditis elegans that are defective in their attraction to NaCl. J. Exp. Zool. 198, 343–352. https://doi.org/10.1002/jez.1401980307 (1976).
Fujiwara, M., Ishihara, T. & Katsura, I. A novel WD40 protein, CHE-2, acts cell autonomously in the formation of C. elegans sensory cilia. Development 126, 4839–4848 (1999).
Bacaj, T., Lu, Y. & Shaham, S. The conserved proteins CHE-12 and DYF-11 cilium function in are required for sensory Caenorhabditis elegans. Genetics 178, 989–1002. https://doi.org/10.1534/genetics.107.082453 (2008).
Kunitomo, H. & Iino, Y. Caenorhabditis elegans DYF-11, an orthologue of mammalian Traf3ip1/MIP-T3, is required for sensory cilia formation. Genes Cells 13, 13–25. https://doi.org/10.1111/j.1365-2443.2007.01147.x (2008).
Haycraft, C. J. et al. elegans homolog of the murine cystic kidney disease gene Tg737 functions in a ciliogenic pathway and is disrupted in osm-5 mutant worms. Development 128, 1493–1505 (2001).
Ailion, M. et al. Two Rab2 interactors regulate dense-core vesicle maturation. Neuron 82, 167–180. https://doi.org/10.1016/j.neuron.2014.02.017 (2014).
Beales, P. L. et al. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 39, 727–729. https://doi.org/10.1038/ng2038 (2007).
Lewis, J. A. & Hodgkin, J. A. Specific neuroanatomical changes in chemosensory mutants of nematode Caenorhabditis elegans. J. Comp. Neurol. 172, 489–510. https://doi.org/10.1002/cne.901720306 (1977).
Perkins, L. A., Hedgecock, E. M., Thomson, J. N. & Culotti, J. G. Mutant sensory cilia in the nematode Caenorhabditis elegans. Dev. Biol. 117, 456–487 (1986).
de Bono, M. & Bargmann, C. I. Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans. Cell 94, 679–689 (1998).
Cook, D. E., Zdraljevic, S., Roberts, J. P. & Andersen, E. C. CeNDR, the Caenorhabditis elegans natural diversity resource. Nucleic Acids Res. 45, D650–D657. https://doi.org/10.1093/nar/gkw893 (2017).
Thompson, O. A. et al. Remarkably divergent regions punctuate the genome assembly of the Caenorhabditis elegans Hawaiian strain CB4856. Genetics 200, 975–989. https://doi.org/10.1534/genetics.115.175950 (2015).
Doitsidou, M., Jarriault, S. & Poole, R. J. Next-generation sequencing-based approaches for mutation mapping and identification in Caenorhabditis elegans. Genetics 204, 451–474. https://doi.org/10.1534/genetics.115.186197 (2016).
Joseph, B. B., Blouin, N. A. & Fay, D. S. Use of a sibling subtraction method for identifying causal mutations in Caenorhabditis elegans by whole-genome sequencing. GBethesda 8, 669–678. https://doi.org/10.1534/g3.117.300135 (2018).
Funding
National Institutes of Health NS079324.
Author information
Authors and Affiliations
Contributions
H.H. and J.W. designed research and wrote the paper; H.H. performed research and analyzed data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hwang, HY., Wang, J. Fast genetic mapping using insertion-deletion polymorphisms in Caenorhabditis elegans. Sci Rep 11, 11017 (2021). https://doi.org/10.1038/s41598-021-90190-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90190-x
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
