
Abstract
Apoptosis or programmed cell death is a highly regulated process, which eliminates unwanted and damaged cells. Inhibition of apoptosis is a hallmark of cancer cells. BCL2 family proteins are known to play a vital role in the regulation of apoptosis. Overexpression of BCL2, an antiapoptotic protein, provides the advantage of prolonged survival to cancer cells. Over the years, several BCL2 inhibitors have been investigated extensively for their anticancer potential. However, most of them were abolished before clinical use due to their side effects. Previously, we had identified and characterized a novel BCL2 inhibitor, Disarib, with the potential to eliminate tumor cells in a BCL2 specific manner leading to reduction in tumor burden in multiple mouse models. Notably, a head-to-head comparison of Disarib to ABT199, the only FDA approved BCL2 inhibitor revealed that Disarib is as potent as ABT199. Recent studies using mice revealed that Disarib did not invoke significant side effects in mice. In the present study, we have investigated the acute toxicity of Disarib in Wistar rats. The bioavailability studies following exposure of Disarib in Wistar rats revealed its maximum availability in serum at 24 h following oral administration. Acute toxicity analysis revealed that even a dose as high as 2000 mg/kg of Disarib did not cause significant toxicity in rats. There was no significant variation in blood parameters or kidney and liver functions following administration of Disarib. Histological analysis of different tissues from Disarib treated groups revealed standard architecture with no observable cellular damage. Importantly, exposure to Diasrib did not result in genotoxicity as determined by micronucleus assay. Further, solubility assays revealed that besides DMSO, Disarib is also soluble in alcohol. While the high acidic condition can increase the solubility of Disarib, even a lower percentage of alcohol with acidic conditions can improve its solubility. Thus, the toxicological profile in the current study revealed no significant side effects when Disarib was administered orally to rats.
Similar content being viewed by others


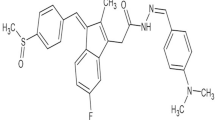
Introduction
Impaired apoptosis is one of the key strategies of cancer cells for survival1,2. BCL2 is an antiapoptotic protein known to be overexpressed in several cancers1,3,4,5. Since BCL2 family proteins have a vital role in the regulation of apoptosis, over the years, several strategies have been used to target BCL2 for anticancer therapies3,4,6,7,8,9. The use of small molecule inhibitors for disrupting the interaction between BCL2 with its proapoptotic partners (BAK/BAX) has become popular6,10,11,12. Several small molecule inhibitors that target BCL2 have been investigated extensively for their anticancer potential, such as Gossypol, YC137, HA14.1, TW37, Chelerythrine, ABT737, AT101, Obatoclax, ABT263 and ABT19911,13,14,15,16. Unfortunately, most BCL2 inhibitors failed in clinical trials due to their pan activity17,18. Some of the BCL2 inhibitors, such as ABT737, ABT263, and ABT199, had efficient clinical relevance; however, ABT737 and ABT263 were eliminated due to dose-limiting toxicities, including neutropenia and thrombocytopenia4,19. Only one BCL2 inhibitor, ABT199 (Venetoclax), has been approved by the Food and Drug Administration (FDA) to treat Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma and Acute Myeloid Leukemia6,11,20.
Previously, we reported the identification of a novel BCL2 inhibitor, Disarib, which efficiently eradicated cancer cells with high BCL2 levels21. We showed that Disarib administration via intraperitoneal route in mice led to efficient tumor regression comparable to that of ABT199, without causing significant side effects21,22,23. Since ABT199 is the only FDA approved drug for anti-tumor effects, we were interested in pursuing further studies to facilitate clinical translation of Disarib. Importantly, toxicological studies of Disarib in mice revealed LD50 of > 2000 mg/kg when administered via oral route22. Moreover, Disarib specifically interacted with BCL2, but not with other antiapoptotic proteins such as BCL-xL, which is one of the reasons for its platelet sparing property, as BCL-xL is the primary survival factor for platelets12,23. We also observed that treatment of Disarib resulted in disruption of the interaction of BCL2 with its proapoptotic partner BAK. However, it did not disrupt the interaction of BCL2 with BAX or with other members of the pro-apoptotic family, which finally resulted in the activation of the intrinsic pathway of apoptosis followed by cell death12, suggesting that Disarib is a promising BCL2 inhibitor and thus underlining the need for further studies.
Based on the guidelines by Central Drugs Standard Control Organization (CDSCO), India on the preclinical evaluation of any chemical compound intended for human use, in our previous study we had performed single dose toxicity studies of Disarib by oral administration in mice22. Results revealed that even the highest dose of Disarib, 2000 mg/kg, did not cause any visible toxicity in mice. The behavior of treated animals and food and water consumption were comparable to that of control animals. Moreover, oral administration in mice led to significant reduction in tumor progression in multiple mice tumor models. Further, oral Disarib treatment did not affect the platelet count even at the highest dose, unlike several other BCL2 inhibitors7,22.
In the present study we have reported the preclinical acute toxicity, pharmacokinetics, and pharmacodynamics of BCL2 inhibitor, Disarib, in Wistar rats. As per CDSCO, India and FDA, USA guidelines, systemic toxicity in mice should be confirmed in another rodent species (preferably rats) in order to establish linear relationship between toxicity and body surface area, so as to further determine the starting dose for Phase I trial. Our study showed that oral administration of Disarib in rats did not lead to any significant behavioral or body weight changes. Analysis of biochemical, histological, blood parameters, and genotoxic effects revealed that even at the highest dose (limiting dose) of Disarib, there was no significant toxic effect in Wistar rats. Importantly, HPLC studies revealed bioavailability up to 24 h following oral administration of Disarib. Thus, the toxicological profile described in this study underlines the potential clinical relevance of the new BCL2 inhibitor.
Results
Disarib is soluble in DMSO and alcohol
It is crucial to evaluate the solubility potential of drugs in their early stage of development. Drug activity can be masked by low solubility24, leading to reduced absorption25. The solubility of Disarib was investigated by dissolving a fixed quantity of Disarib (100 µM) in increasing concentrations of DMSO, alcohol, and HCl (Fig. 1). Solubility potential was studied by HPLC analysis. Results showed that complete dissolution of Disarib was obtained with > 75% DMSO (Fig. 1A) and alcohol (75% or above) (Fig. 1B). Disarib was soluble only in higher acidic conditions such as 0.3 N HCl (Fig. 1C) when increasing acidic conditions were used. However, in combination with alcohol and acids, Disarib was soluble in 40% alcohol and 0.3 and 0.6 N HCl (Fig. 1D).

Evaluation of solubility of Disarib in different solvents. (A–C) Solubility of Disarib in different concentration of DMSO (A), alcohol (B) and HCl (C). Concentrations used were 25, 50, 70 and 100% for DMSO, 40, 60, 70, 80, and 100% for alcohol and 0.1, 0.3 and 0.6 N for HCl. (D) Analysis of solubility in alcohol (40, 60%) in combination with HCl. In each panel, HPLC analyses were performed at three independent times at 232 nm wavelength. The area under the curve (AUC) was calculated to obtain the solubility of Disarib.
Disarib is stable when dissolved in PBS
The compound's stability plays an essential role in the early stage of drug development26,27. It will also help rationalize the dose of a compound used for clinical trials24,25. The stability of Disarib in plasma and PBS was investigated by dissolving Disarib (54.76 mg/ml) in either PBS or 80% plasma (diluted in PBS), and HPLC analysis was performed at different time points to evaluate the reduction in Disarib peak. Our results revealed that while Disarib was stable in PBS up to 6 h (Fig. 2A), the stability of Disarib gradually decreased in plasma with increasing time (Fig. 2B).

In vitro stability and in vivo bioavailability of Disarib in rats. (A,B) Stability analysis of Disarib at different time points (0, 2, 4 and 6 h) in PBS (A) and plasma (B). (C) In vivo bioavailability analysis of Disarib in the serum of Wistar rats at 0.25, 0.50, 1, 2, 4, 6, 8, 10, 12, 24, 48, 72 h after treatment. Two animals each were treated with Disarib orally and sera collected at each time points were subjected to HPLC analysis. The area under the curve (AUC) was calculated to obtain bioavailability. HPLC analysis were done at 232 nm wavelength.
Pharmacokinetic profile of Disarib revealed maximum bioavailability at 24 h post-administration in rats
Investigating the pharmacokinetic profile of a compound can minimize the time taken for drug development as it eases the way for further early clinical trials24. In our previous study on intraperitoneal administration of Disarib (10 mg/kg) in mice maximum circulating concentration (Cmax) was obtained at 30 min, suggesting faster absorption in vivo23. We have also reported a t1/2 of 2.3 h and a clearance rate (CL) of 6.3 ml/min/kg for Disarib in mice. In the present study, serum samples were collected from the Wistar rats at different time points (0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, 48 and 72 h) after oral Disarib treatment. Since oral route is the preferred route of administration for intended clinical use, the pharmacokinetics was determined upon Disarib administration (50 mg/kg b. wt) via oral route. The concentration of Disarib in serum was determined by calculating the area under the curve (AUC) using HPLC LabSolutions software. HPLC analysis post Disarib oral administration revealed that the maximum concentration of Disarib (Cmax) was ~ 18 μg/ml, time to Cmax (Tmax) was 24 h, and t1/2 was determined as 36 h in the serum of rats (Fig. 2C). Moreover, clearance rate and steady state volume of distribution (Vss) were estimated to be 0.44 ml/min/kg and 4.16 l/kg based on the rate of drug elimination and amount of drug in the plasma in equilibrium condition.
Acute toxicity studies in rat revealed no visible toxicity following Disarib treatment
To investigate toxic effects following administration of Disarib in a rodent model system, Wistar rats were selected for the present study based on the guidelines from CDSCO, India (Fig. 3). Previously, acute toxicity studies in mice did not reveal any significant toxic effects22. In the current study, three different dose ranges, low (100 mg/kg), medium (1000 mg/kg), and high (2000 mg/kg), were selected. Disarib was orally administered (n = 5) after preparing a solution using carboxymethyl cellulose, which also served as vehicle control.

Schematic representation showing route followed for acute dose toxicity analysis following Disarib treatment in Wistar rats. 100, 1000 and 2000 mg/kg dose grades represents low, medium, and higher dose, respectively. Mortality, body weight, general cage side examinations, food and water consumption were analyzed everyday during the experiment. Haematology, serum biochemistry, and histopathology were analyzed after 14 days of dose administration.
Treated groups of rats and control animals were observed for mortality up to 14 days after Disarib oral administration as per the guidelines of CDSCO, India. All the experimental animals were alive throughout the experiment, and importantly, they were as healthy as control animals. Experimental animals were daily examined for signs of toxicity caused by oral administration of Disarib. Interestingly, we observed that all parameters analyzed, such as behavior, appearance, lachrymation, changes in locomotion, hair, and skin, were comparable to the vehicle control group.
We observed that even at the highest dose tested (limiting dose of 2000 mg/kg b. wt.) Disarib did not cause mortality or any visible sign of toxicity in animals. In the absence of Disarib induced mortality in the highest dose group, the LD50 for oral dose of Disarib in Wistar rats was determined as > 2000 mg/kg b. wt, which was consistent with the LD50 determined in mice22. Maximum Tolerated Dose (MTD) was estimated to be > 2000 mg/kg since the animals did not show any observable body weight changes. Further, to investigate the impact of Disarib on body weight, rats were weighed periodically every 4 days up to 32 days, from the day of administration of the compound (Fig. 4). There was a gradual increase in body weight of the animals, both in control and treated groups (Fig. 4A–F). Further, evaluation of the effect of Disarib on food and water ingestion was assessed daily. The results revealed no difference in eating and drinking habits in treated groups and control animals, suggesting no alteration in food and water consumption following treatment of Disarib.

Single-dose toxicity studies of Disarib in Wistar rats. (A–D) Body weight analysis of rats on 0, 8, 16, 24, 32 days after oral administration of vehicle control (carboxymethyl cellulose) (A), low dose (100 mg/kg) (B), intermediate dose (1000 mg/kg) (C) and high dose (2000 mg/kg) (D) of Disarib. (E,F) Comparison of change in body weight among control and treated groups after 16 days (E) and 32 days (F). Five rats per group were used for the study. Error bars denote mean + SEM (ns not significant).
Haematological studies show normal blood parameters following Disarib treatment in rats
Blood was collected by heart puncture from treated and control Wistar rats after the 14th day of administration of Disarib in anticoagulant coated vials, and total blood count was evaluated. Results showed no significant difference in the total number of RBC and HGB content between control and treated groups, and the values were within the normal range (Fig. 5)28,29,30. The decrease in platelet count upon higher doses is commonly reported for most BCL2 inhibitors4,19. Although a marginal variation in platelet count was observed in the case of 1000 mg/kg b. wt. Disarib treated group; such a difference was not observed in lower and higher dose treated groups (100 and 2000 mg/kg, respectively) (Fig. 5). Therefore, these results suggested that Disarib treatment in rats did not result in dose-limiting toxicity such as thrombocytopenia (Fig. 5). The analysis also revealed that there was no significant difference in WBC counts between control and treated groups. Further, no significant difference was observed between the neutrophils and lymphocytes count in control and treated animals (Fig. 5). Although there were some variations among different groups and animals within the group, all the values were within the normal physiological range (Fig. 5)28,30. Analyses of PCV, MCV, MCH, MCHC, eosinophils, and monocytes suggested no toxic effects caused by Disarib as all these parameters were comparable to that of the control group (Table 1).

Blood parameter analysis following oral administration of Disarib in rats. Analysis of red blood corpuscles (RBC), white blood corpuscle (WBC), haemoglobin (HGB), Platelets, Neutrophils, Lymphocytes count among control and Disarib treated groups (100, 1000, 2000 mg/kg b. wt) after 14 days of dose administration. Error bars denote mean + SEM (ns not significant).
Administration of Disarib did not affect hepatic or renal functions
After acute dose administration of Disarib in rats, serum was collected 14 days post treatment and analyzed for factors that indicate kidney and liver functions. Hepatic function tests for SGPT, ALP, SGOT, total protein, Albumin, and Bilirubin suggested that Disarib did not cause any adverse effect on liver function (Fig. 6A). Although, ALP levels observed were higher in 100 and 1000 mg/kg Disarib treated groups compared to control, the increase was limited and was not significant. Moreover, the group treated with 2000 mg/kg Disarib showed an ALP level similar to that of the control (Fig. 6A)28,30,31. Consistent with this, the level of SGOT was marginally higher in the maximal dose treated group; however, the increase was within the normal range and was not significant compared to control28 (Fig. 6A). Renal function analysis showed that BUN levels, Creatinine, Phosphorous, and Uric acid did not have any appreciable variation between treated and control groups (Fig. 6B). Thus, the above results suggested that tested doses of Disarib, including the highest (2000 mg/kg), showed neither mortality nor deviation from normal hepatic and renal function.

Analysis of liver and kidney function following treatment with Disarib in rats. (A) Analysis of serum parameters for liver function among control and different Disarib treated rats (100, 1000, 2000 mg/kg b. wt.) after 14 days of dose administration. SGPT serum glutamic pyruvic transaminase, SGOT serum glutamic-oxaloacetic transaminase, ALP alkaline phosphatase. (B) Analysis of serum parameters for kidney function among control and different doses of Disarib (100, 1000, 2000 mg/kg b. wt.) after 14 days of dose administration. BUN Blood urea nitrogen. Error bars denote mean + SEM (ns not significant).
Histopathological analysis does not reveal a significant change in the structure of tissues
Since we did not observe any significant difference in blood and serum parameters between control and Disarib treated groups of rats, we performed histological analysis of liver, kidney, intestine, spleen, lung, and heart following Disarib administration to check for morphological changes. Results showed no cellular change in lobular liver texture, hepatocytes and sinusoidal spaces between control and treated animals (Fig. 7A,B). The kidneys of control and treated groups of animals showed a normal distribution of Malpighian corpuscles. No glomerular dilation and tubular necrosis were seen in the case of Disarib treated tissues (Fig. 7A,B). Histology of vital organs such as lungs and heart did not show difference in the structure of alveoli and cardiac muscles, respectively, between control and treated groups. Intact cardiac muscle cells in heart and intact alveoli and bronchiole structure in lungs were observed in control and treated groups (Fig. 7A,B). Normal structure and organization of the intestine were seen after treatment of Disarib (Fig. 7A,B). Similarly, no difference in spleen architecture was observed when the histological evaluation was performed (Fig. 7A,B). Thus, histological analyses and other studies revealed that the Disarib treatment did not cause any toxicity in rats.

Histopathological analyses of different tissues of rat following administration of Disarib. (A,B) HE staining of tissues from control and 2000 mg/kg Disarib treated rats. Images shown are with a magnification of 10 × (A) or 20 × (B). DCT distal convoluted tubule, PCT proximal convoluted tubule. Scale bar 100 µm.
Disarb does not induce genotoxicity in rats
The pharmacokinetic profile of Disarib reveals the maximum bioavailability at 24 h post Disarib oral administration. Thus, bone marrow cells were collected from rats to evaluate genotoxicity after 24 h, 48 h, and 72 h post-treatment with Disarib (50 mg/kg b. wt.). Bone marrow cells were used for micronucleus assay after fixing the cells in 2% paraformaldehyde32,33,34. Results revealed no DNA damage induced by Disarib in rats following treatment with Disarb in any of the time points investigated (Fig. 8A,B). Micronucleus formation in treated rats was comparable to that of control animals. In contrast, there was a significant increase in the number of micronuclei when rats were exposed to γ-radiation, which served as a positive control (Fig. 8A,B) for the assay. Thus in vivo genotoxicity analysis suggested that Disarib did not cause DNA damage in rats.

Micronucleus assay to evaluate genotoxicity of Disarib. (A) Representative image of vehicle control-treated and Disarib treated (24, 48 and 72 h) bone marrow cells (at 100 ×) after staining the nucleus with DAPI. In each case rats were treated with Disarib (50 mg/kg). 4 Gy IR irradiated cells served as positive control. (B) Bar diagram showing quantitation of micronucleus present in bone marrow cells of vehicle control (CNT), and Disarib treated animals (24, 48 and 72 h). Positive control, irradiated samples (4 Gy) were also quantitated and presented. Two animals at each time point were analyzed, and 500 cells per animal were counted for the analysis. Error bars denote mean + SEM (ns not significant, ***p < 0.0001).
Discussion
In our previous studies, we have identified a novel BCL2 specific inhibitor, Disarib7,12,21,23. Studies revealed that Disarib showed better efficacy when compared head to head with the FDA approved drug ABT199 in terms of cytotoxicity in cancer cell lines and tumor regression in mice23. When administered via oral route in mice, Disarib (50 mg/kg b. wt) showed promising anti-tumor efficacy, without any acute toxicity in mice22. Guidelines from CDSCO, India and FDA, USA on preclinical analysis of drug of interest suggest that systemic toxicity of any intended drug should be compared in two different rodent species (preferably mice and rats). This would help to establish linear relationship between toxicity and body surface area, so as to further determine the starting dose for future Phase I trial of Disarib. In the present study, we have performed pharmacokinetics, pharmacodynamics, and acute toxicity analysis of Disarib in another rodent species, Wistar rats, facilitating early stages of Disarib development as a potent anticancer agent.
Our studies revealed that LD50 of Disarib in Wistar rats is > 2000 mg/kg b. wt., which is indeed promising for further studies, and was the maximum allowed dose as per the guidelines of CDSCO, India. Importantly, this was also consistent with the results observed in mice22. Further, we noted that Disarib did not cause any toxicity when administered in rats, even at the highest dose.
Some of the BCL2 inhibitors failed miserably in previous studies, mainly due to dose-limiting toxicity such as neutropenia and thrombocytopenia4,19. Studies from the current investigation suggested no significant reduction in platelets, even when the highest dose of Disarib was orally administrated to rats. This is promising and comparable to ABT199, the only clinically approved BCL2 inhibitor in the world. In contrast, preclinical studies of BCL2 inhibitors, such as ABT737 and ABT263, showed a rapid and concentration-dependent decrease in the number of circulating platelets20,35,36. This was also seen when these molecules were used for clinical trials in myelodysplastic syndromes, acute myeloid leukemia and ovarian cancers. (https://www.clinicaltrials.gov/ct2/show/NCT00413114?term=obatoclax&draw=2&rank=10)13,20,35,36. Studies of ABT199 on the other hand showed no toxic effect on platelets in clinical trials20. All the other blood parameters analyzed, such as RBC, PCV, MCV, MCH, MCHC, haemoglobin, neutrophils, lymphocytes, eosinophils, monocytes, suggested that treatment with Disarib did not have any impact on the number of blood cell types.
Liver and kidney play important roles in maintaining the metabolic activity of the system. Hence, it is crucial to analyze drug-induced toxic effects on these organs. In the current study, we have analyzed the liver and kidney function by evaluating the levels of SGOT, SGPT, ALP, bilirubin, total protein, creatinine, BUN, phosphorous, and uric acid in the serum. Interestingly, we observed that treatment of Disarib did not have any ill effects on normal liver and kidney function, suggesting that Disarib did not cause toxicity to the liver and kidney. Further, histopathological analysis of tissues from organs such as liver, kidney, intestine, lung, heart, and spleen revealed no change in normal architecture and organization upon administration of Disarib than control tissues, affirming that Disarib has no deleterious effect on organ structure and histology. Similar results were also seen when Disarib was administered in mice22.
No detectable genotoxicity was observed in vivo when Disarib treated rat bone marrow was analyzed using micronucleus assay, indicating that Disarib did not induce DNA damage in treated animals. Therefore, these studies suggested that the administration of Disarib is safe and does not induce DNA damage.
Solubility studies revealed that Disarib was soluble in DMSO (> 75%), alcohol (> 70%), and acids (0.3 N HCl). Further, combining acidic conditions with a lower percentage of alcohol could increase the solubility in alcohol. The majority of BCL2 inhibitors, including ABT199, ABT737, and ABT263, also showed solubility in organic solvents such as DMSO and Dimethylformamide while their solubility was minimal in aqueous buffers11,20. ABT199 and ABT263 were also soluble in absolute ethanol20,37.
Disarib is stable in PBS for 6 h while in vitro plasma stability of Disarib decreased with time. Other BCL2 inhibitors, such as ABT263 and ABT737, have low plasma clearance value, which was one of the limitations in their clinical development16.
Pharmacokinetic studies were used to determine the bioavailability of Disarib in rat serum. The pharmacokinetics results suggested that the maximum concentration of Disarib in serum is ~ 18 µg/ml, which was seen even up to 24 h of oral administration. While ABT737 is not orally bioavailable, ABT199 and ABT263 were orally bioavailable. ABT263 showed ~ 20% of bioavailability in rats16. However, preclinical data of ABT263 suggested dose-limiting thrombocytopenia, which was overcome by the development of selective BCL2 inhibitor ABT199. Importantly, ABT199 also exhibited high bioavailability with half life ranging from 12 h in dogs and 2.2 h in monkey (https://www.ema.europa.eu/en/documents/assessment-report/venclyxto-epar-public-assessment-report_en.pdf)16. Thus, in conjunction with the previous investigation, our current study makes Disarib a potential candidate for further studies and future clinical trials.
Methods
Chemical synthesis
Synthesis and characterization of Disarib was reported previously21.
Pharmacokinetics analysis of Disarib
Wistar rats (Rattus norvegicus) were administered with 50 mg/kg Disarib orally, and blood was collected by heart-puncture method following sacrifice at different time points (15 min, 30 min, 1, 2, 4, 6, 8, 10, 12, 24, 48 and 72 h) following treatment. Blood was allowed to clot, and serum was separated by centrifugation (900×g, 10 min) and deproteinized by adding acetonitrile. Samples for standard calibration curve were prepared by spiking 100 μM Disarib in rat plasma. The supernatant was loaded onto a C18 column, and HPLC analysis was performed in acetonitrile:water gradient, as described before23,38 (Shimadzu, Kyoto, Japan). Disarib specific peak were acquired at 232 nm wavelength23. At least two mice were sacrificed at each time point for the study. Pharmacokinetic parameters were analysed by using LabSolutions software (Shimadzu, Japan), and the values obtained were plotted with GraphPad Prism (ver5.1) software, where C is predicted concentration and t is time. Data was analysed using nonlinear regression analysis. Maximum drug plasma concentration (Cmax) and Time to reach Cmax was determind by area under the curve versus time curve. Clearance (Cl) and steady state volume of distribution (Vss) were calculated respectively by rate of drug elimination and amount of drug in the body at equilibrium condition vs steady state drug concentration in plasma.
Disarib solubility assay
The solubility of Disarib was determined by the shake flask method, as described earlier39. Briefly, Disarib (100 μM) was dissolved in various solvents such as alcohol (40, 60, 80 and 100%), acid (0.1, 0.3 and 0.6 N HCl) and DMSO (25, 50, 75 and 100%). Further, solubility of Disarib was also tested in a combination of alcohol (40, 60%) and acid (0.1, 0.3 N). The suspension was vortexed 2–3 min and kept in a water bath for 5 min, as described before25,26,39. In each case, the samples were diluted in acetonitrile and analyzed using HPLC in acetonitrile:water gradient27,40. The area under the curve (AUC) was determined by using the software LabSolutions, Shimadzu (Japan). The values used for plotting in GraphPad Prism (ver 5.1) softwarewere the mean of three independent experiments.
Determination of stability of Disarib
The stability of Disarib (100 μM) was investigated in phosphate-buffered saline (PBS) and in plasma. The blood was collected from rats by heart puncture, and plasma was separated for the study. After the addition of Disarib into plasma or PBS, the mixture was incubated at 37 °C in water bath25,26. The samples were then collected at different time points, 0, 2, 4, and 6 h, mixed manually with acetonitrile (1:1), and analyzed on HPLC27,40. The values plotted represent the mean of three independent experiments.
Animals, grouping, and dose administration
Wistar rats, Rattus norvegicus (~ 175 g) were purchased from Central Animal Facility, IISc, Bangalore. All the animals were housed in polypropylene cages and kept in controlled lighting of 12 h light/dark cycle throughout the experiment. Standard pellet diet (Agro Corporation Pvt. Ltd. India) and water ad libitum have been provided to the animals. Maintenance and handling of the animals were according to the guidelines of the animal ethical committee. The experimental design and methods followed institutional guidelines and were approved by Institutional Animal Ethics Committee of Indian Institute of Science, Bangalore (Ethical committee approval No: CAF/Ethics/551/2017 and CAF/Ethics/744/2020).
All the studies were designed according to the guidelines of the Central Drug Standard Control Organization (Schedule-Y-CDSCO, Appendix III), India (https://cdsco.gov.in/opencms/opencms/en/Drugs/New-Drugs/). Single-dose toxicity analysis was performed in male Wistar rats. Animals were segregated in groups of five, and Disarib was administered using oral gavage (100, 1000, 2000 mg/kg b. wt.). Carboxymethyl cellulose, used for preparing Disarib, served as vehicle control to feed control animals. Mortality, general appearance, and behavior were initially observed for 24 h. Animals were further observed for 14 days and were further subjected to toxicity analysis.
Determination of LD50
The median lethal dose was determined in male Wistar rats. Adult male rats of comparable body weight (~ 175 g) were divided into four groups (n = 5). Disarib was orally administered after making solution using carboxymethyl cellulose (100, 1000, 2000 mg/kg), and lethality was evaluated over 14 days after treatment. Carboxymethyl cellulose treated animals served as vehicle control.
General observation
General cage-side examination of the treated and control animals were performed every day during the experiment. General behavior, appearance, skin, hairs, secretion (lachrymation), and death were evaluated daily, according to the guidelines of CDSCO, India41,42.
Body weight was recorded on every 4th day from the date of administration of Disarib up to 32 days. Food and water consumption were monitored every day from the date of administration of the compound. During the present study, on every alternate days ~ 250 g food pellet and ~ 500 ml water were given to each cage (control and treatment groups). There were no significant differences observed in food and water consumption of animals in each cage compared to control.
Haematological analysis
After 14 days of Disarib treatment, two animals from each group were sacrificed using CO2 asphyxiation, and blood was collected in EDTA-coated vials by heart puncture. Red blood cells (RBC), white blood cells (WBC), platelet counts (PLT), packed cell volume (PCV), hemoglobin (HGB), mean cell volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), lymphocytes, neutrophils, eosinophils and monocytes were analyzed as described before28,31,43. Analysis of blood samples was performed at Rohana Veterinary Diagnostic Lab, Bangalore, India.
Serum biochemistry
After 14 days of Disarib administration, two animals were sacrificed using CO2 asphyxiation from each group, and blood was collected in tubes without using anticoagulant and centrifuged (900×g, 10 min). Serum glutamic pyruvic transaminase (SGPT), serum glutamic oxaloacetic transaminase (SGOT), alkaline phosphatase (ALP), albumin, total protein, bilirubin, blood urea nitrogen (BUN), creatinine, uric acid, and phosphorous were analyzed as described earlier28,29. Analysis of serum samples was performed at Rohana Veterinary Diagnostic Lab, Bangalore, India.
Histology
At the end of the experimental period (14 days), two animals from each group were sacrificed following CO2 asphyxiation. Liver, kidney, intestine, spleen, lungs, and heart were collected, fixed in 4% paraformaldehyde (PFA) and processed for histology, as described before44,45,46. For histological studies, tissues from rats treated with the highest dose (2000 mg/kg) and the vehicle control group were selected. Paraffin blocks were prepared and sections were taken using a rotary microtome (Leica Biosystems, Buffalo Grove, IL, USA) with a thickness of 5 µm. Following deparaffination, sections were stained with hematoxylin and eosin (H&E), mounted in DPX, and imaged under a bright-field microscope (Carl Zeiss AxioVision, Oberkochen, Germany).
Genotoxicity evaluation
To evaluate genotoxicity caused by Disarib at the effective dose, micronucleus assay was performed32,33,34. Wistar rats (n = 6) were orally administered with Disarib (50 mg/kg), and bone marrow cells were collected after 24, 48, and 72 h post-treatment. Bone marrow cells were flushed out in 1X PBS containing EDTA, centrifuged, fixed (2% PFA) and stained with DAPI. Cells exposed to γ-irradiation (4 Gy) served as a positive control for the assay and were imaged using a fluorescence microscope (Nikon, Tokyo, Japan). At least 500 cells were counted from each animal for each time point, and data was plotted and represented as a bar diagram47.
HPLC analysis
The samples were analyzed by HPLC using the Shimadzu HPLC system. LC was carried out on the C18 analytical column. The mobile phase was the gradient of acetonitrile and water. Disarib specific peaks were detected at 232 nm. The injection volume was 20 μl, and the flow rate was 1.2 ml/min. The standard curves were constructed by plotting the peak area vs. concentration of Disarib40.
Statistical analysis
Statistical analysis was performed in GraphPad Prism (ver5.1) software (GraphPad Software, Inc.) using one way ANOVA. In case of analysis of body weight and blood parameters, non-parametric Kruskal–Wallis test was performed. Analysis of effects of genotoxicity between positive control and treated samples were analysed using one way ANOVA (post hoc Dunnett test).
Ethical approval
This is to confirm that the study decribed in the current manuscript was carried out in compliance with the ARRIVE guidelines. All the procedures, experimental design and methods performed in the study were in accordance with institutional guidelines and were approved by Institutional Animal Ethics Committee, Indian Institute of Science, Bangalore. Ethical committee approval No: CAF/Ethics/551/2017 and CAF/Ethics/744/2020.
References
Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 35, 495–516. https://doi.org/10.1080/01926230701320337 (2007).
Wong, R. S. Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 30, 87–87. https://doi.org/10.1186/1756-9966-30-87 (2011).
Perini, G. F., Ribeiro, G. N., Pinto Neto, J. V., Campos, L. T. & Hamerschlak, N. BCL-2 as therapeutic target for hematological malignancies. J. Hematol. Oncol. 11, 65. https://doi.org/10.1186/s13045-018-0608-2 (2018).
R, S. S. & Eastman, A. BCL2 inhibitors as anticancer drugs: A plethora of misleading BH3 mimetics. Mol. Cancer Ther. 15, 2011–2017. https://doi.org/10.1158/1535-7163.mct-16-0031 (2016).
Skommer, J., Brittain, T. & Raychaudhuri, S. Bcl-2 inhibits apoptosis by increasing the time-to-death and intrinsic cell-to-cell variations in the mitochondrial pathway of cell death. Apoptosis Int. J. Program. Cell Death 15, 1223–1233. https://doi.org/10.1007/s10495-010-0515-7 (2010).
Bate-Eya, L. T. et al. High efficacy of the BCL-2 inhibitor ABT199 (venetoclax) in BCL-2 high-expressing neuroblastoma cell lines and xenografts and rational for combination with MCL-1 inhibition. Oncotarget 7, 27946–27958. https://doi.org/10.18632/oncotarget.8547 (2016).
Radha, G. & Raghavan, S. C. BCL2: A promising cancer therapeutic target. Biochim. Biophys. Acta Rev. Cancer 309–314, 2017. https://doi.org/10.1016/j.bbcan.2017.06.004 (1868).
Vogler, M., Dinsdale, D., Dyer, M. J. S. & Cohen, G. M. Bcl-2 inhibitors: Small molecules with a big impact on cancer therapy. Cell Death Differ. 16, 360–367. https://doi.org/10.1038/cdd.2008.137 (2009).
Zinkel, S., Gross, A. & Yang, E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ 13, 1351–1359. https://doi.org/10.1038/sj.cdd.4401987 (2006).
Klenke, S. et al. The small molecule Bcl-2/Mcl-1 inhibitor TW-37 shows single-agent cytotoxicity in neuroblastoma cell lines. BMC Cancer 19, 243. https://doi.org/10.1186/s12885-019-5439-1 (2019).
Souers, A. J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208. https://doi.org/10.1038/nm.3048 (2013).
Vartak, S. V. et al. Novel BCL2 inhibitor, Disarib induces apoptosis by disruption of BCL2-BAK interaction. Biochem Pharmacol 131, 16–28. https://doi.org/10.1016/j.bcp.2017.02.015 (2017).
Broecker-Preuss, M., Becher-Boveleth, N., Müller, S. & Mann, K. The BH3 mimetic drug ABT-737 induces apoptosis and acts synergistically with chemotherapeutic drugs in thyroid carcinoma cells. Cancer Cell Int. 16, 27. https://doi.org/10.1186/s12935-016-0303-8 (2016).
Manero, F. et al. The small organic compound HA14-1 prevents Bcl-2 interaction with bax to sensitize malignant glioma cells to induction of cell death. Can. Res. 66, 2757. https://doi.org/10.1158/0008-5472.CAN-05-2097 (2006).
Real, P. J. et al. Breast cancer cells can evade apoptosis-mediated selective killing by a novel small molecule inhibitor of Bcl-2. Can. Res. 64, 7947. https://doi.org/10.1158/0008-5472.CAN-04-0945 (2004).
Tse, C. et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 68, 3421–3428. https://doi.org/10.1158/0008-5472.can-07-5836 (2008).
Harazono, Y., Nakajima, K. & Raz, A. Why anti-Bcl-2 clinical trials fail: A solution. Cancer Metastasis Rev. 33, 285–294. https://doi.org/10.1007/s10555-013-9450-8 (2014).
Waites, G. M., Wang, C. & Griffin, P. D. Gossypol: reasons for its failure to be accepted as a safe, reversible male antifertility drug. Int. J. Androl. 21, 8–12. https://doi.org/10.1046/j.1365-2605.1998.00092.x (1998).
Vogler, M. et al. BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood 117, 7145–7154. https://doi.org/10.1182/blood-2011-03-344812 (2011).
Cang, S., Iragavarapu, C., Savooji, J., Song, Y. & Liu, D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 8, 129. https://doi.org/10.1186/s13045-015-0224-3 (2015).
Iyer, D. et al. Identification of a novel BCL2-specific inhibitor that binds predominantly to the BH1 domain. FEBS J. 283, 3408–3437. https://doi.org/10.1111/febs.13815 (2016).
Sharma, S. et al. Acute toxicity analysis of Disarib, an inhibitor of BCL2. Sci. Rep. 10, 15188. https://doi.org/10.1038/s41598-020-72058-8 (2020).
Vartak, S. V. et al. A novel inhibitor of BCL2, Disarib abrogates tumor growth while sparing platelets, by activating intrinsic pathway of apoptosis. Biochem. Pharmacol. 122, 10–22. https://doi.org/10.1016/j.bcp.2016.09.028 (2016).
Veseli, A., Žakelj, S. & Kristl, A. A review of methods for solubility determination in biopharmaceutical drug characterization. Drug Dev. Ind. Pharm. 45, 1717–1724. https://doi.org/10.1080/03639045.2019.1665062 (2019).
Savjani, K. T., Gajjar, A. K. & Savjani, J. K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 195727–195727, 2012. https://doi.org/10.5402/2012/195727 (2012).
Di, L., Kerns, E. H., Hong, Y. & Chen, H. Development and application of high throughput plasma stability assay for drug discovery. Int J Pharm 297, 110–119. https://doi.org/10.1016/j.ijpharm.2005.03.022 (2005).
Konsoula, R. & Jung, M. In vitro plasma stability, permeability and solubility of mercaptoacetamide histone deacetylase inhibitors. Int. J. Pharm. 361, 19–25. https://doi.org/10.1016/j.ijpharm.2008.05.001 (2008).
Delwatta, S. L. et al. Reference values for selected hematological, biochemical and physiological parameters of Sprague-Dawley rats at the Animal House, Faculty of Medicine, University of Colombo, Sri Lanka. Anim. Models Exp. Med. 1, 250–254. https://doi.org/10.1002/ame2.12041 (2018).
Ochiai, Y. et al. Blood biochemistry and hematological changes in rats after administration of a mixture of three anesthetic agents. J. Vet. Med. Sci. 80, 387–394. https://doi.org/10.1292/jvms.17-0497 (2018).
Serfilippi, L. M., Pallman, D. R. & Russell, B. Serum clinical chemistry and hematology reference values in outbred stocks of albino mice from three commonly used vendors and two inbred strains of albino mice. Contemp. Top Lab. Anim. Sci. 42, 46–52 (2003).
Asano, Y., Susami, M., Honda, K. & Serikawa, T. Haematological and serum biochemical values in spontaneously epileptic male rats and related rat strains. Lab Anim. 32, 214–218. https://doi.org/10.1258/002367798780600052 (1998).
Hayashi, M. The micronucleus test-most widely used in vivo genotoxicity test. Genes Environ. 38, 18–18. https://doi.org/10.1186/s41021-016-0044-x (2016).
Iz, S. G., Gürhan, S. & en, B. Comparison of in vitro cytotoxicity and genotoxicity of MMA-based polymeric materials and various metallic materials. Turkish J. Med. Sci. 40, 905–916 (2010).
Jha, A. M., Singh, A. C., Sinha, U. & Kumar, M. Genotoxicity of crotonaldehyde in the bone marrow and germ cells of laboratory mice. Mutat. Res. 632, 69–77. https://doi.org/10.1016/j.mrgentox.2007.04.008 (2007).
Wilson, W. H. et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 11, 1149–1159. https://doi.org/10.1016/s1470-2045(10)70261-8 (2010).
Zhang, H. et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 14, 943–951. https://doi.org/10.1038/sj.cdd.4402081 (2007).
Del Gaizo Moore, V. et al. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J. Clin. Invest. 117, 112–121. https://doi.org/10.1172/JCI28281 (2007).
Srivastava, M. et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 151, 1474–1487. https://doi.org/10.1016/j.cell.2012.11.054 (2012).
Baka, E., Comer, J. E. A. & Takács-Novák, K. Study of equilibrium solubility measurement by saturation shake-flask method using hydrochlorothiazide as model compound. J. Pharm. Biomed. Anal. 46, 335–341. https://doi.org/10.1016/j.jpba.2007.10.030 (2008).
Petrova, O. E. & Sauer, K. High-performance liquid chromatography (HPLC)-based detection and quantitation of cellular c-di-GMP. Methods Mol. Biol. 1657, 33–43. https://doi.org/10.1007/978-1-4939-7240-1_4 (2017).
Bonthagarala, B. et al. Regulatory process and ethics for clinical trials in India (CDSCO). Pharma Innov. J. 6, 165–169 (2017).
Saxena, P. & Saxena, R. Clinical trials: changing regulations in India. Indian J. Community Med. 39, 197–202. https://doi.org/10.4103/0970-0218.143018 (2014).
Wolford, S. T. et al. Reference range data base for serum chemistry and hematology values in laboratory animals. J. Toxicol. Environ. Health 18, 161–188. https://doi.org/10.1080/15287398609530859 (1986).
Sebastian, R. & Raghavan, S. C. Endosulfan induces male infertility. Cell Death Dis. 6, e2022–e2022. https://doi.org/10.1038/cddis.2015.368 (2015).
Srivastava, S. et al. Quercetin, a natural flavonoid interacts with DNA, arrests cell cycle and causes tumor regression by activating mitochondrial pathway of apoptosis. Sci. Rep. 6, 24049–24049. https://doi.org/10.1038/srep24049 (2016).
Thomas, E., Gopalakrishnan, V., Somasagara, R. R., Choudhary, B. & Raghavan, S. C. Extract of vernonia condensata, inhibits tumor progression and improves survival of tumor-allograft bearing mouse. Sci. Rep. 6, 23255–23255. https://doi.org/10.1038/srep23255 (2016).
Frieauff, W., Martus, H. J., Suter, W. & Elhajouji, A. Automatic analysis of the micronucleus test in primary human lymphocytes using image analysis. Mutagenesis 28, 15–23. https://doi.org/10.1093/mutage/ges047 (2013).
Acknowledgements
We thank Namrata M Nilavar, Dipayan Ghosh, Sumedha Dahal and members of SCR laboratory for critical reading of the manuscript and their comments.This work was supported by grants from Glue Grant, Department of Biotechnology, India (BT/PR23078/MED/29/1253/2017) to SCR and BC; IISc-DBT partnership programme [BT/PR27952-INF/22/212/2018] to SCR. SS is supported by Junior Research fellowship (JRF) from DBT, India, KKV acknowledges funding from SERB-NPDF (PDF/2017/000785), UR is supported by Research Associateship from IISc and HS is supported by Senior Research fellowship (SRF) from UGC.
Author information
Authors and Affiliations
Contributions
S.C.R. and B.C. coordinated the study and provided the reagents; S.C.R., B.C. and S.S. designed the experiments, interpreted results, and wrote the manuscript; S.S., K.K.V., U.R., H.S., S.M. and A.E.J. conducted the experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sharma, S., Varsha, K.K., Ray, U. et al. Acute toxicity analysis of an inhibitor of BCL2, Disarib, in rats. Sci Rep 11, 9982 (2021). https://doi.org/10.1038/s41598-021-89387-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89387-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
