
Abstract
Shell-color polymorphism is a common phenomenon in several mollusk species and has been associated with thermal capacity, developmental stability, shell strength, and immunity. Shell-color polymorphism has been related to the differential expression of genes in several signal transduction pathways; however, the functions of micro-RNAs (miRNAs) in shell-color formation remain unclear. In the present study, we compared high-quality, small-RNA transcriptomes in three strains of the Manila clam Ruditapes philippinarum with specific shell-color patterns, artificially selected for six generations. Totals of 114 known and 208 novel miRNAs were identified by high-throughput sequencing, of which nine known and one novel miRNA were verified by stem-loop quantitative real time-polymerase chain reaction. Predicted miRNA targets were subjected to Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses. miR-137 and miR-216b and the Hedgehog signaling pathway and Wnt signaling pathway were identified as being potentially involved in pigment formation and regulation in R. philippinarum. These results may help to clarify the role of miRNAs in shell coloration and shed light on the mechanisms regulating color formation in bivalve shells.
Similar content being viewed by others

Introduction
Shell-color polymorphism is a common phenomenon among mollusks. Shell pigmentation has been potentially associated with various factors, including predation1, climatic effects2,3, thermal capacity2,4,5, developmental stability6, shell strength7, and immunity8,9. Mollusk shells are multi-layered structures consisting of calcium carbonate crystals, together with proteinaceous material and pigments10. The shell is generated by the outer fold of the mantle, and shell growth and pigmentation are regulated by neurosecretory mechanisms11,12. Shells grow in a linear fashion by adding new material to the growing edge; pigments are thus incorporated into the shell along this growing edge, and change their position continually as new shell is added13. Pigments may be laid down in the outer or other layers of mollusk shells10,12. To date, three common classes of pigments including melanin, carotenoids and tetrapyrroles was identified in mollusk10. Melanin is thought to be responsible for dark colors of mollusks10. The molecular pathway for the synthesis of melanin was considered important to contribute the shell (and pearl) pigmentation14,15, and the tyrosinase genes, the most important enzymes in the regulation and production of melanin has been identified in Pinctada fucata14, Mizuhopecten yessoensis16, R. philippinarum17, Crassostrea gigas18 and Amusium pleuronectes19. Addition, microphthalmia-associated transcription factor (MITF), a positively regulated gene of tyrosinase was also cloned in Patinopecten yessoensis20 and Meretrix petechialis21. Carotenoids is thought to be related to the yellow and orange pigment of the shells22. In Hyriopsis cumingii, the apolipoprotein (Apo) gene which related to carotene metabolism was found to be expressed differently in purple and white inner-shell color23. In Patinopecten yessoensis, GWAS identified three genes (LDLR, FRIS, and FRIY) involved in the carotenoid metabolism were responsible for the shell colors of this species24. In Chlamys nobilis, the SRB-like-3 gene which responsible for carotenoid deposition was believe associated with the orange color25. Transcriptome analysis of Mercenaria mercenaria showed the genes related to Carotenoid and Porphyrin and chlorophyll metabolism appeared to be associated with shell pigmentation26. Porphyrins were often associated with red, brown or purple shell coloration27. In Calliostoma zizyphinum, the genes associated with the synthesis of porphyrins were proved to participate in the synthesis of shell color28. Shell coloration has been shown to involve a complex bioprocess involving the original pigment and the chemical interactions between the pigments and the shell matrix10. To clarify the mechanism of shell color formation requires more research data. However, no miRNAs have yet been demonstrated to be involved in the determination of mollusk shell color. MicroRNAs (miRNAs) are a class of non-coding RNAs of approximately 20 nucleotides (nt) long that play important roles in regulating gene expression in biological processes29. miRNAs have also been suggested to play a crucial role in shell biomineralization. The miRNAs pm-miR-2386 and pm-miR-13b were predicted to participate in biomineralization in the pearl oyster Pinctada martensii by regulating the formation of the organic matrix or the differentiation of mineralogenic cells during shell formation30, while miR-2305 was also shown to participate in nacre formation in the shell31. These results suggest that miRNAs may play a role in the formation of shell color; however, no miRNAs have yet been demonstrated to be involved in the determination of mollusk shell color.
In this study, we constructed high-quality small-RNA transcriptomes for three shell-color lines of the Manila clam Ruditapes philippinarum, to provide basic data to further our understanding of the mechanisms regulating shell-color formation in this species.
Results
Sequence analysis of miRNAs
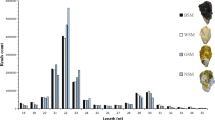
To characterize small RNA transcripts, we constructed three small-RNA libraries for the three shell-color morphs of R. philippinarum using Illumina high-throughput sequencing technology. Totals of 22,042,778 (Wh), 22,357,681 (Or), and 21,442,934 (Zs) raw reads were obtained. After removing low-quality reads, adaptors, and small reads (length < 16), 3,891,284, 10,769,530, and 12,561,241 clean reads, respectively, were obtained, and 1,323,648, 16,671,16, and 1,140,082 unique sequences were obtained by clustering. The sequence lengths of the clean reads ranged from 16 to 30 nt, with most miRNAs being 21–23 nt long (Fig. 1).

Length distribution of sequencing reads in three libraries.
Differential expression of miRNAs in R. philippinarum with different shell colors
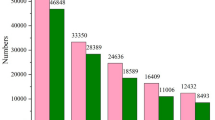
A total of 114 miRNAs were identified that were the same as or similar to known mature miRNAs (Supplementary Table S1), while 208 miRNAs were predicted to be novel miRNAs with hairpin structures (Supplementary Table S2). There were 10 significantly DEMs in Wh compared with Or (six upregulated and four downregulated), 104 significant DEMs in Wh compared with Zs (51 upregulated and 53 downregulated), and 52 significant DEMs in Or compared with Zs (22 upregulated and 30 downregulated) (Fig. 2; Supplementary Table S3). Moreover, only 2 known miRNAs including miR-137 and miR-216 were EEMs between in Wh and Or groups, there another 14 and 36 know DEMs between Or and Zs shell -color strains and between the Wh and Zs shell -color strains respectively. Several miRNAs previously identified as related to pigmentation synthesis, including MiR-13732,33 and miR-7-5p, were included among the DEMs34. The expression levels of ten selected miRNAs were validated by qRT-PCR, and all showed a consistent pattern with those from small-RNA sequencing analysis (Fig. 3), indicating reliability of the analysis.

Differentially expressed miRNAs (DEMs) in three pairwise comparison groups. Red and green indicate up- and down-regulated miRNAs, respectively.

Expression of 10 selected miRNAs determined by qRT-PCR.
Target-gene prediction and functional analysis of DEMs
The 10, 104, and 52 DEMs from the above three comparisons corresponded to 6228, 14,793, and 10,479 target genes, which were successfully mapped to existing gene categories and categorized into 66, 47, and 43 functional groups, respectively (Supplementary Table S4). KEGG pathway analysis was used to estimate the biochemical metabolic pathways and functions of the candidate target genes, and showed that these target genes could be classified into several pigmentation-related pathways, including the Wnt, Notch, and dopaminergic synapse signaling pathways (Supplementary Table S5). Illustration of the regulatory network between the DEMs and their targets (Fig. 4) indicated that miR-137 and miR-216b might play central roles in the interacting network. These results suggested a complicated regulatory network between miRNAs and their targets in pigmentation differentiation in R. philippinarum.

Potential regulatory networks between DEMs and their target genes. (a) Wh vs Or, (b) Wh vs Zs, (c) Or vs Zs. The regulation network of miRNAs and their target genes was illustrated by Cytoscape software. Red Hexagons and blue circles represent miRNAs and target genes, respectively.
Discussion
miRNAs represent a large class of small noncoding RNAs that bind to the 3′-untranslated region of target genes, thus affecting cleavage or translational repression to regulate gene expression at the posttranscriptional level. miRNAs play important roles in diverse biological processes, including cell proliferation, differentiation, and apoptosis. miRNAs have also recently been suggested to play a key role in the formation of body color in animals. For example, miR-137 was shown to influence the phenotype of coat color in transgenic mice by down-regulating microphthalmia-associated transcription factor (MITF)33, while miR-8 was required for the proper spatial patterning of pigment on the dorsal abdomen of the fruit fly35.
Several miRNAs have been reported to be involved in skin-color formation in fish. miR-429 silencing resulted in a visible change in skin pigmentation in the common carp (Cyprinus carpio)36, and miR-138-5p and miR-722 were predicted to play potential roles in skin-color differentiation in red tilapia37. However, reports on the functions of miRNAs in relation to shell color in mollusks remain scarce. In the present study, we compared the miRNAs from three shell-color strains of R. philippinarum and analyzed them using high-throughput sequencing technology, which identified a total of 322 miRNAs, including 114 known and 208 novel miRNAs. The numbers of DEMs differed significantly between clams with different shell colors.
It is worth noting that the expression levels of miR-137 and miR-216b differed significantly among the three shell-color strains, with miR-137 being significantly downregulated in Or compared with Wh and Zs. One of the miR-137 target gene- MITF mRNAs were previously reported to be mostly upregulated in the Or strain in R. philippinarum38. And the MITF target gene-tyrosinase gene were also found highest expression level in Or compare with Wh and Zs shell color strains of R. philippinarum39. These results suggest that miR-137 can also affect shell color in R. philippinarum by affecting the formation of melanin. In addition, only these two known miRNA shows different expression between the Or and Wh strains. There are more DEMs between Or and Zs, and between the Wh and Zs shell -color strains. Compared with the simpler shell color of the Or and Wh strains, the Zs strains exhibited more complex stripes and shell colors. This result is consistent with the transcriptome analysis in the four different shell color strains R. philippinarum17. These results indicate that more regulatory pathways are needed to regulate the formation of complex shell colors in this species. More studies were needed to confirm this conclusion. However, there have been no reports on the role of miR-216b in pigmentation to date.
Shell pigmentation is a complex and precise process. To obtain a better understanding of the mechanism of pigmentation in R. philippinarum, we predicted and analyzed the target genes of DEMs based on the clam’s genomic data. KEGG pathway analysis showed that DEM target genes including casein kinase I, glycogen synthase kinase 3, protein kinase A and Jun-N-terminal kinase were mainly involved in the Hedgehog and Wnt signaling pathways, which might be involved in pigment formation and regulation40,41,42,43. In Mollusca, Hedgehog signaling was shown to be involved in muscle differentiation in Sepia officinalis and in myogenesis in Crassostrea gigas44,45. The Hedgehog signaling pathway was also involved in the differentiation of retinal pigment epithelium cells in Xenopus46. Hedgehog signaling also plays a critical role in neural development47, including pattering of the neural tube in mice and chicks48,49. The neural system of the mantle has been suggested to be involved in pigmentation in mollusks by sensing the current pigmentation pattern and governing the formation of new pigment and shell material at a fine positional level6. The patterns of pigment on mollusk shells have been suggested to arise from the stimulation of secretory cells in the mantle by the animal’s central nervous system50. Shell pigmentation in Haliotis asinine was considered to be controlled by numerous cells and the secretion of pigments via tubule-based secretory tubules12. These results suggest a correlation between shell-color formation and nervous system development. The Wnt signaling pathway plays important roles in embryonic development and in adult tissue homeostasis and regeneration51. Wnt signaling was also shown to regulate pigment-cell differentiation in zebrafish40 and to promote the differentiation of neural crest cells into melanocytes in mice52. Wnt genes were also differentially expressed in mantle regions underlying shell with different pigmentation in the Yesso scallop53. We also previously found an expansion of Wnt genes and their potential roles in shell-color pattern in R. philippinarum54. Furthermore the relationship between Wnt/β-catenin signaling and MITF has been reported to be a critical feature in melanocyte development and subsequent pigmentation55. These results suggest that the Wnt signaling pathway might play a key role in pigmentation, especially in the formation of melanin, in R. philippinarum.
Materials and methods
Sample collection and RNA extraction
Three shell-color lines of R. philippinarum, zebra stripe (Zs), orange (Or), and white (Wh) (Fig. 5), established by self-copulation for six generations, were collected from a farm in Dalian, China, and transported to the laboratory. Mantles were dissected from six clams from each strain, immediately ground into a powder in liquid nitrogen, and then stored at − 80 °C before processing for RNA extraction. Each sample was duplicated.

The three shell color lines of R. philippinarum used to construct small RNAs transcriptomes: zebra stripe (a), white (b), and orange (c).
Total RNA was extracted from the mantle using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. RNAs of 15–30 nt long were purified by 15% denaturing polyacrylamide gel electrophoresis, ligated to 3ʹ and 5ʹ adapters, and reverse transcribed using an Illumina sequencing kit (Illumina, San Diego, CA, USA). The polymerase chain reaction (PCR) products were purified using phenol/chloroform extraction and ethanol precipitation and sequenced by Beijing Biomarker Technologies (Beijing, China) using an Illumina HiSeq 2000 Genome Analyzer (Illumina).
Small-RNA sequencing and read alignment
Raw sequence reads for the Wh, Or, and Zs strains were analyzed as described previously56. Briefly, reads with excessively small tags or from contaminating adapter-adapter ligation were removed. Clean reads were then aligned to the RepBase (v15, http://www.girinst.org), Rfam (http://www.sanger.ac.uk/Software/Rfam/ftp.shtml), and GenBank databases to remove small RNAs that mapped to annotated exons, repeats, rRNAs, tRNAs, or small nuclear RNAs. Unique sequences were obtained for further analysis.
Identification of conserved and novel miRNAs
Clean reads were mapped to the clam genome38 using SOAP57 and their expression and distribution were analyzed. To identify conserved miRNA sequences, reads were aligned using miRBase (release 20.0) to search for perfectly matched short reads. Reads that did not match the databases were marked as unannotated, and unannotated sequences were aligned with the clam transcriptomic sequences to predict potential novel candidate miRNAs. The secondary structures of these candidate miRNAs were further analyzed using miREv and miRDeep2.0 software58,59.
Differential expression analysis of miRNAs
We compared miRNA expression among the three shell-color groups by normalizing the read counts for each identified miRNA to the total number of reads in each given sample. Differential expression between two shell-color groups was analyzed using the DEGseq package, and the P-value was adjusted using the q-value. Fold-changes in miRNAs were calculated as the ratio of read counts in different shell-color groups followed by log2 transformation. The q value < 0.05 and log2 (fold-change) ≥ 1.0 were considered as the thresholds for significant differential expression.
Validation of R. philippinarum miRNAs by stem-loop quantitative real-time-PCR (qRT-PCR)
Ten significantly differentially expressed miRNAs (DEMs) including miR-137 and miR-216b that expressed differed significantly among the three shell-color strains and other five known miRNAs and one putative novel miRNA were selected randomly for analysis by RT-PCR, with the U6 fragment as an internal control (Table 1). miRNA was extracted from mantle tissue using a miRcute microRNA isolation kit (Tiangen, China) and reverse transcribed using a miRcute miRNA first-strand cDNA kit (Tiangen). qRT-PCR was performed using a LightCycler 480 II Real-time PCR Instrument (Roche, Switzerland) with a miRcute miRNA qPCR Detection Kit (SYBR Green) (Tiangen). The reactions were carried out in a total volume of 20 μl, including 10 µl 2 × miRcute Plus miRNA PreMix, 0.4 µl each of sense and antisense primers (10 µM), 6.6 µl diethylpyrocarbonate-treated water, and 2 µl of DNA template according to the following steps: initial denaturation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 15 s, 94 °C for 20 s, and 60 °C for 34 s. For every miRNA, qRT-PCR reactions for each sample were repeated three times. The relative expression levels were calculated using 2−△△Ct method.
Target gene prediction and analysis of potential mRNA–regulatory miRNA networks involved in shell-color formation
Target genes were predicted using the computational prediction programs RNAhybrid60, Miranda61, and TargetScan62. Genes supported by any two algorithms were considered to be likely potential miRNA targets. The target reference sequences were genome unigenes and annotation information for R. philippinarum. We further investigated the functions of the miRNAs in R. philippinarum pigmentation by enrichment analysis of the predicted target genes by Gene Ontology (GO) (http://www.geneontology.org/) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (http://www.genome.jp/kegg/) analyses. To identify key DEMs and their regulatory networks in shell-color formation, the predicted mRNA–miRNA regulatory interactions between the DEMs and their target genes in the top 10 pathways were visualized using Cytoscape version 3.6.163.
Data availability
All the sequencing reads were deposited in the NCBI Short Read Archive database (http://www.ncbi.nlm.nih.gov/sra/) and are retrievable under the accession numbers SAMN10337891, SAMN10337892, SAMN10337893, SAMN10337894, SAMN10337895, and SAMN10337896. The R. philippinarum assembly was deposited with the accession number QUSP00000000 under PRJNA479743. All sequence data for the R. philippinarum genome were deposited in the NCBI Sequence Read Archive under the accession numbers SRR7716263–SRR7716297.
References
Heller, J. & Gadot, M. Shell polymorphism of Theba pisana-the effects of rodent distribution. Malacologia 25, 349–354 (1984).
Mitton, J. B. Shell color and pattern variation inMytilus edulis and its adaptive significance. Chesap. Sci. 18, 387–390 (1977).
Heller, J. Visual versus climatic selection of shell banding in the landsnail Theba pisana in Israel. J. Zool. 194, 85–101 (1981).
Scheil, A. E., Gärtner, U. & Köhler, H.-R. Colour polymorphism and thermal capacities in Theba pisana (O.F. Müller 1774). J. Therm. Biol. 37, 462–467. https://doi.org/10.1016/j.jtherbio.2012.03.006 (2012).
Etter, R. J. Physiological stress and color polymorphism in the intertidal snail Nucella lapillus. Evolution, 660–680 (1988).
Bauchau, V. Developmental stability as the primary function of the pigmentation patterns in bivalve shells?. Belg. J. Zool. 131, 23–28 (2001).
Cain, A. The colours of marine bivalve shells with special reference to Macoma balthica. Malacologia 28, 289–318 (1988).
Scheil, A. E., Hilsmann, S., Triebskorn, R. & Köhler, H.-R. Shell colouration and parasite tolerance in two helicoid snail species. J. Invertebr. Pathol. 117, 1–8. https://doi.org/10.1016/j.jip.2014.01.003 (2014).
Scheil, A. E., Hilsmann, S., Triebskorn, R. & Kohler, H. R. Shell colour polymorphism, injuries and immune defense in three helicid snail species, Cepaea hortensis, Theba pisana and Cornu aspersum maximum. Results Immunol. 3, 73–78. https://doi.org/10.1016/j.rinim.2013.06.002 (2013).
Williams, S. T. Molluscan shell colour. Biol. Rev. 92, 1039–1058 (2017).
Boettiger, A., Ermentrout, B. & Oster, G. The neural origins of shell structure and pattern in aquatic mollusks. Proc. Natl. Acad. Sci. 106, 6837–6842 (2009).
Budd, A., McDougall, C., Green, K. & Degnan, B. M. Control of shell pigmentation by secretory tubules in the abalone mantle. Front. Zool. 11, 9. https://doi.org/10.1186/s12983-014-0062-0 (2014).
Fowler, D. R., Meinhardt, H. & Prusinkiewicz, P. in ACM SIGGRAPH Computer Graphics. 379–387 (ACM).
Lemer, S., Saulnier, D., Gueguen, Y. & Planes, S. Identification of genes associated with shell color in the black-lipped pearl oyster Pinctada margaritifera. BMC Genom. 16, 568. https://doi.org/10.1186/s12864-015-1776-x (2015).
Palumbo, A. Melanogenesis in the ink gland of Sepia officinalis. Pigment Cell Res. 16, 517–522 (2003).
Sun, X., Yang, A., Wu, B., Zhou, L. & Liu, Z. Characterization of the mantle transcriptome of yesso scallop (Patinopecten yessoensis): Identification of genes potentially involved in biomineralization and pigmentation. PLoS ONE 10, e0122967 (2015).
Nie, H. et al. Transcriptome analysis reveals the pigmentation related genes in four different shell color strains of the Manila clam Ruditapes philippinarum. Genomics 112, 2011–2020 (2020).
Feng, D., Li, Q., Yu, H., Zhao, X. & Kong, L. Comparative transcriptome analysis of the Pacific oyster Crassostrea gigas characterized by shell colors: identification of genetic bases potentially involved in pigmentation. PLoS ONE 10, e0145257 (2015).
Huang, R.-L. et al. Mantle branch-specific RNA sequences of moon scallop Amusium pleuronectes to identify shell color-associated genes. PLoS ONE 10, e0141390 (2015).
Mao, J. et al. Genome-wide identification, characterization and expression analysis of the MITF gene in Yesso scallops (Patinopecten yessoensis) with different shell colors. Gene 688, 155–162. https://doi.org/10.1016/j.gene.2018.11.096 (2019).
Zhang, S. et al. Identification of a gene encoding microphthalmia-associated transcription factor and its association with shell color in the clam Meretrix petechialis. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 225, 75–83. https://doi.org/10.1016/j.cbpb.2018.04.007 (2018).
Hedegaard, C., Baeseau, J.-F. & Chateigner, D. Molluscan shell pigments: an in situ resonance Raman study. J. Molluscan Stud. 72, 157–162 (2006).
Li, X. et al. Cloning, differential tissue expression of a novel hcApo gene, and its correlation with total carotenoid content in purple and white inner-shell color pearl mussel Hyriopsis cumingii. Gene 538, 258–265 (2014).
Zhao, L. et al. A genome-wide association study identifies the genomic region associated with shell color in Yesso Scallop, Patinopecten yessoensis. Mar. Biotechnol. 19, 301–309. https://doi.org/10.1007/s10126-017-9751-y (2017).
Liu, H. et al. A de novo transcriptome of the noble scallop, Chlamys nobilis, focusing on mining transcripts for carotenoid-based coloration. BMC Genomics 16, 1–13 (2015).
Hu, Z. et al. De novo assembly transcriptome analysis reveals the preliminary molecular mechanism of pigmentation in juveniles of the hard clam Mercenaria mercenaria. Genomics 112, 3636–3647. https://doi.org/10.1016/j.ygeno.2020.04.020 (2020).
Comfort, A. Acid-soluble pigments of shells. 1. The distribution of porphyrin fluorescence in molluscan shells. Biochem. J. 44, 111 (1949).
Williams, S. T. et al. Colorful seashells: Identification of haem pathway genes associated with the synthesis of porphyrin shell color in marine snails. Ecol. Evol. 7, 10379–10397. https://doi.org/10.1002/ece3.3552 (2017).
Bartel, D. P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004).
Zheng, Z. et al. Computational prediction of candidate miRNAs and their potential functions in biomineralization in pearl oyster Pinctada martensii. Saudi J. Biol. Sci. 23, 372–378. https://doi.org/10.1016/j.sjbs.2015.04.001 (2016).
Jiao, Y. et al. MicroRNA, Pm-miR-2305, participates in nacre formation by targeting pearlin in pearl oyster Pinctada martensii. Int. J. Mol. Sci. 16, 21442–21453. https://doi.org/10.3390/ijms160921442 (2015).
Hao, S. et al. miR-137 inhibits proliferation of melanoma cells by targeting PAK2. Exp. Dermatol. 24, 947–952. https://doi.org/10.1111/exd.12812 (2015).
Dong, C. et al. Coat color determination by miR-137 mediated down-regulation of microphthalmia-associated transcription factor in a mouse model. RNA 18, 1679–1686. https://doi.org/10.1261/rna.033977.112 (2012).
Giles, K. M., Brown, R. A. M., Epis, M. R., Kalinowski, F. C. & Leedman, P. J. miRNA-7-5p inhibits melanoma cell migration and invasion. Biochem. Biophys. Res. Commun. 430, 706–710. https://doi.org/10.1016/j.bbrc.2012.11.086 (2013).
Kennell, J. A., Cadigan, K. M., Shakhmantsir, I. & Waldron, E. J. The microRNA miR-8 is a positive regulator of pigmentation and eclosion in Drosophila. Dev. Dyn. 241, 161–168. https://doi.org/10.1002/dvdy.23705 (2012).
Yan, B. et al. microRNA regulation of skin pigmentation in fish. J. Cell Sci. 126, 3401–3408. https://doi.org/10.1242/jcs.125831 (2013).
Wang, L. et al. Comparative microRNA-seq analysis depicts candidate miRNAs involved in skin color differentiation in Red Tilapia. Int. J. Mol. Sci. 19. https://doi.org/10.3390/ijms19041209 (2018).
Yan, X. et al. Clam genome sequence clarifies the molecular basis of its benthic adaptation and extraordinary shell color diversity. Iscience 19, 1225–1237 (2019).
Jiang, K., Jiang, L., Nie, H., Huo, Z. & Yan, X. Molecular cloning and expression analysis of tyrosinases (tyr) in four shell-color strains of Manila clam Ruditapes philippinarum. PeerJ 8, e8641. https://doi.org/10.7717/peerj.8641 (2020).
Dorsky, R. I., Raible, D. W. & Moon, R. T. Direct regulation of nacre, a zebrafish MITF homolog required for pigment cell formation, by the Wnt pathway. Genes Dev. 14, 158–162 (2000).
Jiang, J. & Struhl, G. Protein kinase A and hedgehog signaling in Drosophila limb development. Cell 80, 563–572 (1995).
Price, M. A. & Kalderon, D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 108, 823–835 (2002).
Rosso, S. B., Sussman, D., Wynshaw-Boris, A. & Salinas, P. C. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat. Neurosci. 8, 34–42 (2005).
Grimaldi, A. et al. A hedgehog homolog is involved in muscle formation and organization of Sepia officinalis (mollusca) mantle. Dev. Dyn. 237, 659–671 (2008).
Li, H., Li, Q. & Yu, H. Molecular characterization of the hedgehog signaling pathway and its necessary function on larval myogenesis in the Pacific oyster Crassostrea gigas. Front. Physiol. 9, 1536 (2018).
Perron, M. A novel function for Hedgehog signalling in retinal pigment epithelium differentiation. Development 130, 1565–1577. https://doi.org/10.1242/dev.00391 (2003).
Gulino, A., Di Marcotullio, L., Ferretti, E., De Smaele, E. & Screpanti, I. Hedgehog signaling pathway in neural development and disease. Psychoneuroendocrinology 32, S52–S56 (2007).
Echelard, Y. et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75, 1417–1430 (1993).
Riddle, R. D., Johnson, R. L., Laufer, E. & Tabin, C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 75, 1401–1416 (1993).
Ermentrout, B., Campbell, J. & Oster, G. A model for shell patterns based on neural activity. Veliger 28, 369–388 (1986).
Nusse, R. & Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017).
Dunn, K. J., Williams, B. O., Li, Y. & Pavan, W. J. Neural crest-directed gene transfer demonstrates Wnt1 role in melanocyte expansion and differentiation during mouse development. Proc. Natl. Acad. Sci. 97, 10050–10055 (2000).
Mao, J. et al. Histological and expression differences among different mantle regions of the Yesso Scallop (Patinopecten yessoensis) provide insights into the molecular mechanisms of biomineralization and pigmentation. Mar. Biotechnol. 21, 683–696. https://doi.org/10.1007/s10126-019-09913-x (2019).
Bai, Y., Nie, H., Wang, Z. & Yan, X. Genome-wide identification and transcriptome-based expression profiling of Wnt gene family in Ruditapes philippinarum. Compar. Biochem. Physiol. Part D Genom. Proteom. 35, 100709. doi:https://doi.org/10.1016/j.cbd.2020.100709 (2020).
Gajos-Michniewicz, A. & Czyz, M. WNT Signaling in Melanoma. Int. J. Mol. Sci. 21, 1. https://doi.org/10.3390/ijms21144852 (2020).
Reddy, A. M. et al. Cloning, characterization and expression analysis of porcine microRNAs. BMC Genomics 10, 65. https://doi.org/10.1186/1471-2164-10-65 (2009).
Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 24, 713–714 (2008).
Wen, M., Shen, Y., Shi, S. H. & Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinformatics 13, 10. https://doi.org/10.1186/1471-2105-13-140 (2012).
Friedlander, M. R. et al. Discovering microRNAs from deep sequencing data using miRDeep. Nature Biotechnol. 26, 407–415. https://doi.org/10.1038/nbt1394 (2008).
Krüger, J. & Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 34, W451–W454 (2006).
Betel, D., Wilson, M., Gabow, A., Marks, D. S. & Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 36, D149–D153 (2008).
Lewis, B. P., Burge, C. B. & Bartel, D. P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 (2005).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Acknowledgements
This work was supported by the Chinese Ministry of Science and Technology through the National Key Research and Development Program of China (2018YFD0901400) and the Natural Science Foundation of Liaoning Province (2020-MS-279).
Author information
Authors and Affiliations
Contributions
Q.W. did the experiment, J.D. analyzed the data and drafted the manuscript. X.Y. conceived of the study, participated in experimental design and coordination, and contributed to the manuscript preparation. Z.H., H.N. and Y. Q. participated in the data analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ding, J., Wen, Q., Huo, Z. et al. Identification of shell-color-related microRNAs in the Manila clam Ruditapes philippinarum using high-throughput sequencing of small RNA transcriptomes. Sci Rep 11, 8044 (2021). https://doi.org/10.1038/s41598-021-86727-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-86727-9
This article is cited by
-
The lgi-miR-2d is Potentially Involved in Shell Melanin Synthesis by Targeting mitf in Manila Clam Ruditapes philippinarum
Marine Biotechnology (2024)
-
microRNA regulation of skin pigmentation in golden-back mutant of crucian carp from a rice-fish integrated farming system
BMC Genomics (2023)
-
In the Sea of Japan and the Adriatic Sea, a COX1 DNA test revealed genetic similarity of Manila clams, although shell parameters, spermatogenesis patterns, and sperm characteristics are area specific
Biologia (2023)
-
The first high-density genetic map of common cockle (Cerastoderma edule) reveals a major QTL controlling shell color variation
Scientific Reports (2022)
-
MiRNA-mRNA Integration Analysis Reveals the Regulatory Roles of MiRNAs in Shell Pigmentation of the Manila clam (Ruditapes philippinarum)
Marine Biotechnology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
